

Abstract
HIV-1 is an enveloped virus that enters target cells by fusion either directly at the plasma membrane or at the endosomal membrane. The latter mechanism follows a rapid engulfment of HIV-1 after its receptor engagement at the cell surface, and its scale depends on cellular endocytosis/degradation rates and virus fusion kinetics. HIV-1 has recently been shown to exploit a novel Pak1-dependent macropinocytosis mechanism as a way to productively infect macrophages. However, macrophages are highly heterogeneous cells that can adapt functionally to their changing environment, and their endosomal/lysosomal pathway is highly regulated upon cell activation. These changes might impact the ability of HIV-1 to exploit endocytosis as a way to productively infect macrophages. In this study, we compared HIV-1 endocytosis/degradation rates in nonactivated, M1-activated, and M2a-activated monocyte-derived macrophages (MDMs). We found that the rate of HIV-1 endocytosis was increased in M1-activated but decreased in M2a-activated MDMs. However, both M1 and M2a activations of MDMs led specifically to a greater clathrin-mediated endocytosis of HIV-1, which was independent of CD4 and CCR5 binding. Furthermore, clathrin-mediated endocytosis is unlikely to result in productive HIV-1 infection, given that it leads to increased viral degradation. Therefore, we suggest that viral fusion following endocytosis is restricted in activated macrophages.
INTRODUCTION
The productive entry of enveloped viruses involves lipid mixing between host cell membranes and the viral envelope (a phenomenon better known as fusion), which occurs following conformational changes in the viral fusion protein (33). Such events are subsequent to receptor engagement at the plasma membrane or in endosomes, with the latter case possibly requiring the acidification of the endosomal milieu. Early studies showed that the HIV-1 fusion process is pH independent and thus is observed at a neutral pH directly at the plasma membrane (plasma membrane fusion [PMF]) (21, 35). Such findings are further supported by the formation of syncytia upon the expression of viral glycoproteins at the surface of infected cells (16). However, HIV-1 can also traffic within the endosomal apparatus (27). Although the majority of engulfed virions are degraded during their transit (19), HIV-1 endocytosis can lead to enhanced pathogenesis via transfer enhancement and in certain cases can allow for productive infection (fusion after endocytosis [FAE]) (22, 37, 40). HIV-1 fusion with endosomal membranes is pH independent and has been suggested as an outcome following the rapid removal of virus-engaged receptors from the cell surface (28). According to this model, the occurrence of FAE depends on the balance between the cellular endocytosis/lysosomal degradation rates and virus fusion kinetics. It should therefore be highly cell type dependent. Indeed, endocytosis has proven to be a major, if not the only, productive entry pathway for HIV-1 in transformed cell lines such as trophoblasts (36) and epithelial cells of intestinal (4) or cervical (10, 22) origin. However, Schaeffer and colleagues showed previously that both PMF and FAE occur in primary human CD4+ T cells (30), the principal cellular targets of HIV-1 in peripheral blood, whereas it was reported that only PMF occurs in dendritic cells (15). In macrophages, a stable cellular reservoir of HIV-1 in tissues, a novel Rac1- and Pak1-dependent but phosphatidylinositol 3-kinase-independent macropinocytosis pathway was shown to allow for productive HIV-1 entry, although the importance of this process compared to the importance of PMF has yet to be more clearly defined (6, 20).
Peripheral blood monocytes exhibit a versatile and flexible potential for differentiation. The immediate consequence of this flexibility of the monocyte differentiation capability is a great functional heterogeneity of the resulting macrophage populations. This unique characteristic allows macrophages to adapt rapidly to their changing environment (13, 24). Following the exposure of human resting macrophages (unpolarized/M0 phenotype) to different activating stimuli, such as cytokines, they may functionally polarize into M1- or M2-activated macrophages. Such an activation leads to differences in susceptibility to intracellular pathogens (32) and the potential for antigen presentation (26), among others. M1 (or classically activated) macrophages are highly microbicidal; secrete proinflammatory mediators such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and IL-6; and are important producers of reactive nitrogen and oxygen intermediates (18). They are generally induced by gamma interferon (IFN-γ) and microbial products such as lipopolysaccharide (LPS) (17). M2 (or alternatively activated) macrophages are involved in the resolution of inflammation and tissue repair and secrete anti-inflammatory mediators such as IL-10 (13). Different subgroups of M2-activated macrophages have been suggested, based on their activating stimuli. M2a macrophages are induced by IL-4 or IL-13, M2b macrophages are induced by immune complexes and Toll-like receptor (TLR) or IL-1 receptor (IL-1R) agonists, and M2c macrophages are induced by IL-10 or glucocorticoid hormones (18).
M1- and M2a-activated monocyte-derived macrophages (MDMs) have both been shown to be restrictive for HIV-1 replication in vitro. It has been established that HIV-1 infection of M1-activated MDMs is blocked at an early step in the virus replication cycle, through the downregulation of CD4 at the cell surface and CCR5-binding chemokine production (7). M2a MDMs also exhibit diminished HIV-1 production, although a level of viral protein production similar to that of virus-infected resting cells was observed previously, suggesting a block in an HIV-1 late/postintegration event (7). Given that the antigen presentation process is highly linked to the endosomal system, macrophage activation has also been shown to induce changes in this crucial biological function. Phagocytosis, pinocytosis, and endolysosomal trafficking were all shown previously to be modulated after IFN-γ or IL-4 treatment of MDMs (12, 23), although these changes seem to be cytokine and cargo dependent. Thus, macrophage activation might affect the ability of HIV-1 to infect macrophages via FAE.
To our knowledge, so far, no study has addressed the impact of macrophage activation on HIV-1 endocytosis and trafficking in primary human macrophages. We thus evaluated the impact of M1 (treated with IFN-γ and LPS) and M2a (treated with IL-4) activation on HIV-1 endocytosis and fate in MDMs and investigated how activation-induced changes in their endosomal system contribute to the restriction of HIV-1 infection. We report here that M1 activation is associated with an increase in HIV-1 endocytosis, the opposite of what is seen in M2a-activated MDMs. However, both treatments are associated with increases in virus degradation and clathrin-mediated endocytosis (CME) of HIV-1, which we show to be independent of CD4 and CCR5 binding. Therefore, FAE is only very limited in activated macrophages.
MATERIALS AND METHODS
Reagents and antibodies.
The anti-p24 hybridomas 31-90-25 and 183-H12-5C were obtained from the American Type Culture Collection (Manassas, VA) and the NIH AIDS Research and Reference Reagent Program (Germantown, MD), respectively, and were used in our in-house sandwich enzyme-linked immunosorbent assay (ELISA) (3). Both antibodies were purified by using MabTrap protein affinity columns (Pharmacia Biotech, Piscataway, NJ) according to the manufacturer's instructions. IFN-γ was obtained from Peprotech (Rocky Hill, NJ). Both LPS and IL-4 were purchased from Cedarlane Laboratories Ltd. (Burlington, Ontario, Canada). Chlorpromazine (CPZ) was obtained from Calbiochem (EMD Millipore, Toronto, Ontario, Canada) and was kept as a stock solution of 10 mg/ml in ethanol. Dimethylamiloride (DMA), 5-(N-ethyl-N-isopropyl)amiloride (EIPA), and bafilomycin A1 were obtained from Sigma-Aldrich (St. Louis, MO) and were all suspended in dimethyl sulfoxide (DMSO) at concentrations of 100 mM, 25 mM, and 100 μM, respectively.
Preparation and activation of macrophages.
Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by Ficoll-Hypaque gradient centrifugation. Monocytes were separated from other cells, mostly lymphocytes, by adhesion for 2 h at 37°C in 150- by 20-mm tissue culture dishes, followed by washing with endotoxin-free phosphate-buffered saline (PBS; Sigma-Aldrich). Monocytes were then allowed to differentiate for 7 days in RPMI 1640 medium supplemented with 5% autologous plasma and antibiotics and then recovered by scraping following incubation with Accutase (eBioscience, San Diego, CA). MDMs were allowed to rest for at least 24 h before stimulation for 18 or 72 h either with IFN-γ (20 ng/ml) and LPS (100 ng/ml) or with IL-4 (20 ng/ml), to obtain M1 and M2a MDMs, respectively. Untreated MDMs were cultured in parallel for the same period of time as controls. Experiments were performed with RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics in the absence of cytokines.
Virus production.
Viruses were produced by the calcium phosphate transfection of HEK 293T cells, as described previously (3). The R5-tropic pNL4-3Balenv molecular clone was a kind gift of R. Pomerantz (Thomas Jefferson University, Philadelphia, PA) (11). The pNL4-3env− molecular clone was obtained by introducing a −1 frameshift in the envelope (Env) precursor and was a kind gift from D. E. Ott (National Cancer Institute, Frederick, MD). JRFLenv-pseudotyped (R5-tropic) and vesicular stomatitis virus G protein (VSV-G)-pseudotyped viruses were obtained by the cotransfection of pNL4-3env− and pJRFLenv (obtained from N. R. Landau, New York University School of Medicine, New York, NY) or pHCMV-G (obtained from J. C. Burns, University of California at San Diego, San Diego, CA), respectively. To obtain NL4-3luc+env−R+/JRFLenv viruses, cells were cotransfected with pNL4-3luc+env−R+, also kindly provided by N. R. Landau (8), and pJRFLenv. A sandwich ELISA previously developed in our laboratory was used to quantify the amount of p24 in each viral preparation (3). Virus infectivity was determined by infection of the TZM-bl reporter cell line (29, 39).
Intracellular p24 assay.
Activated MDMs (105 cells/well in 24-well plates) were washed and then put in contact with different virus preparations (20 ng of p24/105 cells) for 2.5 h at 37°C to allow for their internalization (endocytosis and fusion). Cells were then washed again and treated with trypsin for 5 min to remove bound viruses from their surface. This treatment efficiently removed 75 to 80% of the viruses present at the MDM cell surface after a 2-h binding experiment (20 ng of p24/105 cells) at 4°C (data not shown). After additional washes, cells were disrupted in lysis buffer (i.e., PBS supplemented with 0.05% Tween 20, 2.5% Triton X-100, and 0.02% thimerosal), and the amount of intracellular p24 was evaluated by an ELISA (3). In some experiments, cells were preincubated with bafilomycin A1, CPZ, EIPA, DMA, or their respective vehicles for 30 min before allowing for virus internalization. Final drug concentrations were 100 nM bafilomycin A1, 10 μg/ml CPZ, 25 μM EIPA, and 100 μM DMA. The amounts of intracellular p24 detected in cells varied from 0.2 to 2.4 ng/ml (NL4-3Balenv), 0.2 to 5 ng/ml (pNL4-3env−/VSV-G), 0.1 to 1.9 ng/ml (pNL4-3env−), and 0.2 to 3.2 ng/ml (pNL4-3env−/JRFLenv) and varied depending on the activation state and origin (PBMCs/blood donor) of the MDMs and the drug treatment, if applicable. All viral entry assays were normalized by using a cellular viability assay.
Cellular viability test.
Cellular metabolism was monitored by incubating untreated or vehicle-alone- or drug-treated cells with CellTiter 96 Aqueous One Solution cell proliferation assay reagent (Promega, Madison, WI) for 1 h at 37°C, according to the manufacturer's instructions. The absorbance of the supernatant at 490 nm was read by using an ELx808 Absorbance microplate reader (BioTek, Winooski, VT).
Quantification of HIV-1 infection.
Resting MDMs either left untreated (M0 phenotype) or treated for 72 h with IFN-γ and LPS (M1 phenotype) or IL-4 (M2a phenotype) (105 cells/well in 24-well plates) were put in contact with NL4-3Balenv (20 ng of p24/105 cells) for 2 h at 37°C to allow for viral infection and then washed. Virus production was assessed by measuring the extracellular p24 release in the cell culture supernatant at days 0, 3, 6, and 10 postinfection by using our in-house ELISA (3). Alternatively, the various macrophage populations were put in contact with recombinant luciferase-encoding viruses (NL4-3luc+env−R+/JRFLenv). Luciferase activity (expressed in relative light units [RLU]) was quantified at day 10 postinfection, as previously described (25), by using an MLX microtiter luminometer (Dynex Technologies, Chantilly, VA).
Viral degradation assay.
The various macrophage populations (M0, M1, and M2a) (105 cells/well in 24-well plates) were put in contact with NL4-3Balenv or VSV-G-pseudotyped particles (20 ng of p24/105 cells) at 37°C to allow for virus internalization. Following 2 h of incubation, cells were washed and treated with trypsin for 5 min to remove bound viruses from their surface (time zero) and then incubated at 37°C to allow for virus degradation. Cell lysis was performed at time zero and at 1.5 and 3 h, and the intracellular p24 content was measured for each time point by using our in-house ELISA (3). Alternatively, MDMs were preincubated with bafilomycin A1 (100 nM) for 30 min before HIV-1 was added and were left in contact with the drug for the duration of the experiment.
Statistical analyses.
Paired Student t tests were performed by using GraphPad Prism 5 software.
RESULTS
HIV-1 internalization is differentially modulated in M1- and M2a-activated MDMs.
In order to study the putative impact of macrophage activation on HIV-1 uptake in human macrophages, we compared resting MDMs (M0 phenotype) to M1- and M2a-activated MDMs, given that these populations of activated MDMs have been studied previously in the context of HIV-1 infection (7). We chose to derive our resting population in the absence of macrophage colony-stimulating factor (M-CSF), as this cytokine has been shown to induce M2c polarization of macrophages (13). Resting, 7-day-differentiated MDMs, here called M0 MDMs, were left untreated or treated either with a combination of IFN-γ (20 ng/ml) and LPS (100 ng/ml) or with IL-4 (20 ng/ml), to obtain M1 and M2a MDMs, respectively.
We first determined if cellular activation had an impact on the overall HIV-1 internalization (i.e., fusion and endocytosis processes) in MDMs. We therefore activated M0 MDMs for either 18 h or 72 h with the different cytokine combinations. We then compared the amounts of intracellular p24 by an ELISA following 2.5 h of contact with NL4-3Balenv at 37°C in our different MDM populations. It is important to underline that the measured intracellular p24 level represents the total amount of vesicular and cytosolic p24 in MDMs at the moment of cell lysis. According to data reported previously by Maréchal and coworkers (19), vesicular and cytosolic p24 should contribute 45 and 55% of the measured intracellular p24, respectively, and therefore, our methodology should allow for the detection of significant variations in both overall HIV-1 fusion and endocytosis. To take into account any modulatory impact on MDM metabolism mediated by long cytokine treatments, we normalized the determined intracellular p24 level by the cellular metabolic activity, as measured by an in vitro proliferation assay (see Materials and Methods). As shown in Fig. 1, after an 18-h cytokine treatment, we could not observe a significant difference between the mean amounts of NL4-3Balenv intracellular p24 detected in M0, M1, or M2a MDMs. However, following a 72-h cytokine treatment, the mean amount of intracellular p24 was 1.7-fold higher in M1 MDMs than in their M0 counterparts, whereas it was reduced to 0.6-fold in M2a MDMs. It is noteworthy that we observed a great variation in p24 fold increases in M1 MDMs derived from the PBMCs of different blood donors. As the quantity of intracellular p24 may be influenced by the rate of virus degradation, we repeated the previous experiment using bafilomycin A1, a well-known inhibitor of vesicular acidification (5). Briefly, MDMs treated for 72 h with the above-described cytokines were preincubated with 100 nM bafilomycin A1 for 30 min before NL4-3Balenv viruses were added, and the experiment was then performed as described above. As shown in Fig. 2, in the presence of bafilomycin A1, the previously obtained differences in intracellular p24 levels in M1- and M2a-activated MDMs were again observed, suggesting that the differences in the rates of p24 degradation between resting and activated MDMs are not solely responsible for the different amounts of p24 obtained in activated cells. Taken together, these results suggest that the internalization of NL4-3Balenv is affected by the type of MDM activation.
Fig 1.
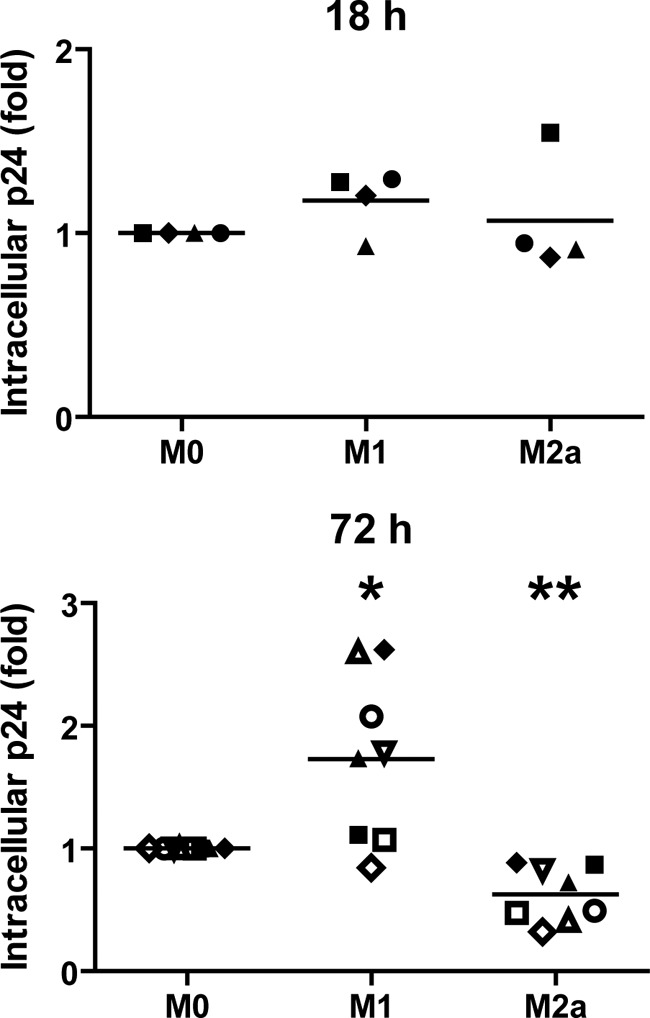
HIV-1 internalization is differentially modulated in M1- and M2a-activated MDMs. MDMs were first differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 18 h (top) or 72 h (bottom); washed; and then allowed to internalize equal amounts of NL4-3Balenv virus (standardized in terms of p24) for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. The total intracellular p24 content was quantified by an ELISA and normalized for cell viability (Cellular viability test on uninfected cells; see Materials and Methods). Results are expressed as fold changes compared to M0 and represent mean values (triplicate samples) of several independent experiments (*, P < 0.05; **, P < 0.01). In each graph, one symbol represents MDMs from one specific blood donor.
Fig 2.
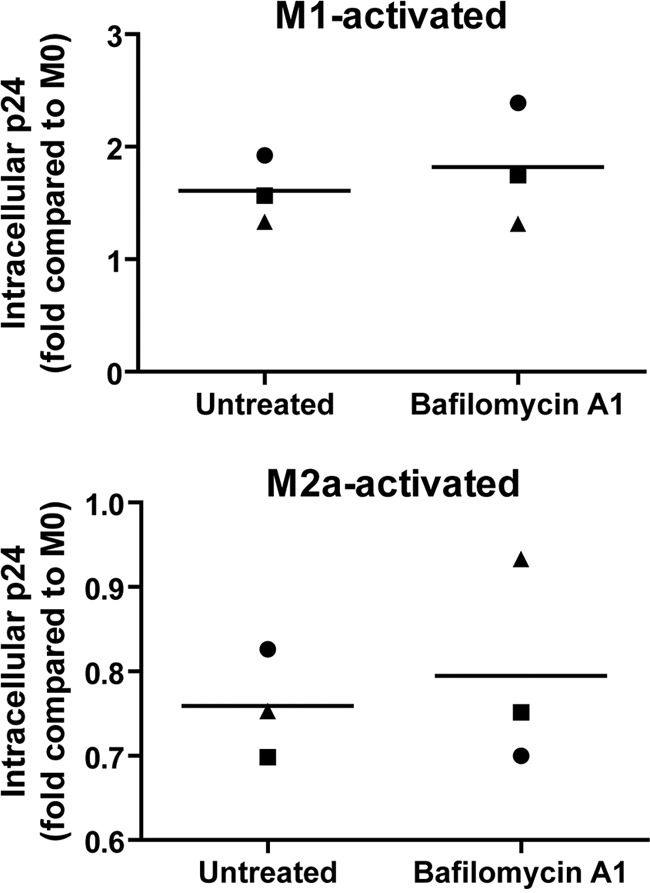
Differences in viral degradation are not solely responsible for modulation of HIV-1 internalization in activated MDMs. MDMs were first differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then pretreated or not with bafilomycin A1 (final concentration of 100 nM) for 30 min. Cells were than allowed to internalize equal amounts of NL4-3Balenv virus (standardized in terms of p24), in the presence of the drug, for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. The total intracellular p24 content was quantified by an ELISA and normalized for cell viability (Cellular viability test on uninfected cells). Results are expressed as fold changes compared to M0 for M1-activated (top) and M2a-activated (bottom) MDMs and represent mean values (triplicate samples) of several independent experiments. In each graph, one symbol represents MDMs from one specific blood donor.
MDM activation acts on a CD4/CCR5-independent HIV-1 endocytosis pathway.
In order to measure the contribution of either viral fusion or endocytosis to the different HIV-1 internalization rates observed for M1- and M2a-activated MDMs, we repeated the experiments described in the legend of Fig. 1 (72-h activation) but this time with a glycoprotein-deficient fusion-incompetent virus. As shown in Fig. 3A, we observed 2.4- and 0.5-fold differences in intracellular p24 levels in M1 and M2a MDMs, respectively, compared to levels in M0 cells. These data were similar to those obtained with fully infectious NL4-3Balenv virions (Fig. 1), therefore suggesting that the observed differences in HIV-1 intracellular p24 levels in activated MDMs involve primarily endocytosis rather than fusion. As endocytosis pathways are highly cargo and receptor dependent, and to further assess the involvement of HIV-1 receptor and coreceptor binding by gp120 in this process, we compared intracellular p24 levels using R5-tropic JRFLenv- and VSV-G-pseudotyped HIV-1 viruses (Fig. 3B and C, respectively). We determined that the mean amounts of intracellular p24 following internalization increased for both pseudotyped viruses in M1-activated macrophages (1.8- and 1.5-fold, respectively) and decreased in M2a-activated macrophages (0.7- and 0.6-fold, respectively). This finding suggests that the differences in intracellular p24 levels observed for activated MDMs are independent of CD4 and CCR5, given that they are similar to our previous results with either the NL4-3Balenv or the glycoprotein-deficient virus. Interestingly, MDMs that were derived from specific blood donors reacted similarly upon endocytosis for the different viruses examined, showing a similar increase/decrease for all of the viruses tested. Overall, these results suggest that a CD4/CCR5-independent mechanism of HIV-1 endocytosis is occurring in M1- and M2a-activated MDMs.
Fig 3.
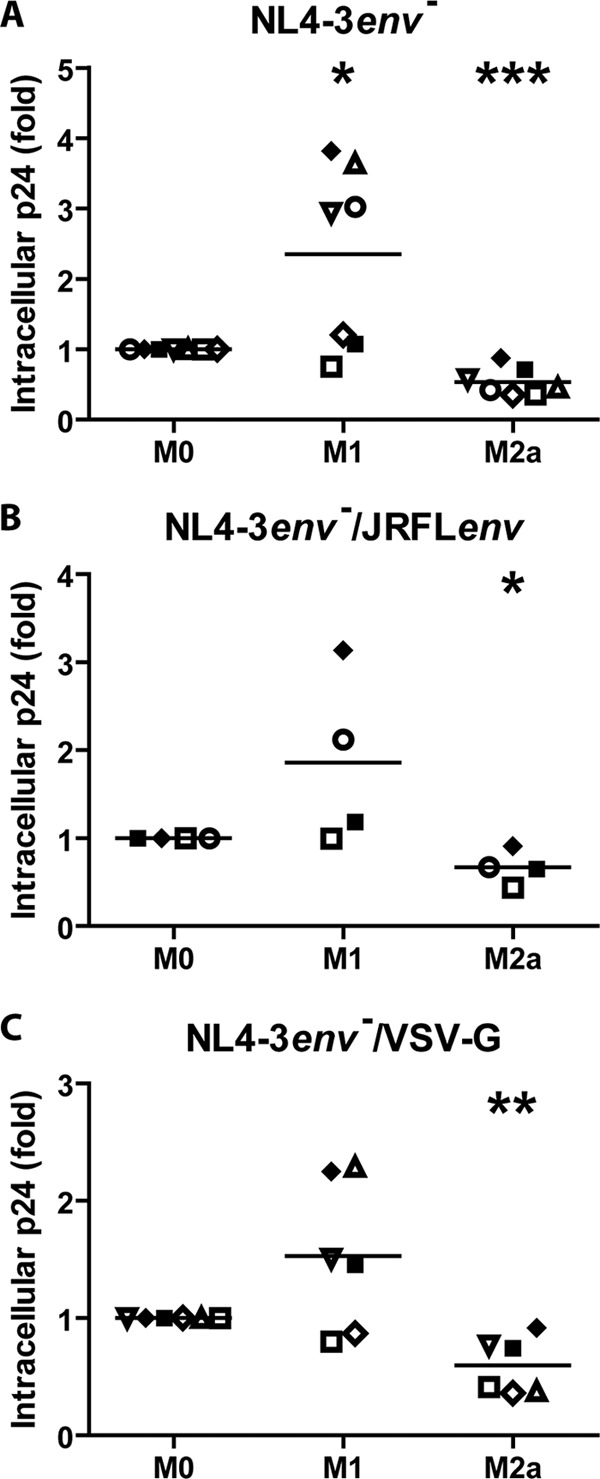
MDM activation acts on a CD4/CCR5-independent HIV-1 endocytosis pathway. MDMs were differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then allowed to internalize equal amounts of NL4-3env− (A), NL4-3env−/JRFLenv (B), or NL4-3env−/VSV-G (C) (standardized in terms of p24) for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. The total intracellular p24 content was quantified by an ELISA and normalized for cell viability (Cellular viability test on uninfected cells). Results are expressed as fold changes compared to M0 and represent mean values (triplicate samples) of several independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Each symbol represents MDMs from one specific blood donor, and identical symbols are linked to the same donor in panels A to C.
Macropinocytosis is not involved in HIV-1 endocytosis in both resting and activated MDMs.
As macropinocytosis has been shown to be involved in HIV-1 endocytosis in MDMs (6, 20), we set out to determine if HIV-1 macropinocytosis was modulated in activated MDMs. In order to assess this possibility, we pretreated the studied MDM populations derived from the same blood donor for 30 min with inhibitors specific for macropinocytosis before allowing for virus internalization at 37°C. The intracellular p24 content was next quantified following 2.5 h of incubation with NL4-3Balenv, glycoprotein-deficient, or pseudotyped virus, and the results were normalized to the cellular metabolic activity. Surprisingly, both EIPA (Fig. 4) and DMA (data not shown) failed to significantly reduce intracellular p24 levels in M0 MDMs, as opposed to what was previously reported (6, 20). Macropinocytosis is therefore unlikely to be involved in HIV-1 endocytosis in nonactivated macrophages. Furthermore, the activation of MDMs with IFN-γ and LPS (M1) or IL-4 (M2a) did not induce HIV-1 macropinocytosis, as we observed no significant difference in intracellular p24 levels in EIPA- or DMA-treated M1 and M2a MDMs (Fig. 4 and data not shown).
Fig 4.

Macropinocytosis is not involved in HIV-1 endocytosis in both resting and activated MDMs. MDMs were differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then pretreated with EIPA or the equivalent amount of the drug vehicle alone (i.e., DMSO) for 30 min. Cells were then allowed to internalize equal amounts of NL4-3Balenv, NL4-3env−, NL4-3env−/VSV-G, or NL4-3env−/JRFLenv virus (standardized in terms of p24) for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. The total intracellular p24 content was quantified by an ELISA and normalized for cell viability (Cellular viability test on uninfected drug-treated cells). Results are expressed as percentages of intracellular p24 levels compared to that of transporter-treated cells (100%) and represent mean values (triplicate samples) of several independent experiments. Each symbol represents MDMs from one specific blood donor, and identical symbols are linked to the same donor in the different panels.
Clathrin-mediated endocytosis is involved in HIV-1 internalization in activated MDMs.
As clathrin-mediated endocytosis has been shown to be involved in HIV-1 entry in a variety of cells (2, 9), we determined whether this pathway impacted HIV-1 uptake in activated macrophages. We therefore repeated the same experimental design as that described in the legend of Fig. 4 using CPZ, a specific inhibitor of clathrin-mediated endocytosis. As illustrated in Fig. 5, CPZ did not significantly affect NL4-3Balenv endocytosis in M0 MDMs, whereas this treatment efficiently reduced HIV-1 internalization in M1- and M2a-activated MDMs, suggesting that clathrin-mediated endocytosis of HIV-1 is prevalent in activated cells. Furthermore, this was also the case with NL4-3env−/JRFLenv-pseudotyped virus as well as with NL4-3env− or NL4-3env−/VSV-G virus, suggesting that the clathrin-mediated endocytosis of HIV-1 does not require gp120 binding to CD4 or CCR5. These data therefore suggest that, in response to M1 and M2a polarization stimuli, HIV-1 uptake relies on clathrin-mediated endocytosis.
Fig 5.

Clathrin-mediated endocytosis is involved in HIV-1 internalization in activated MDMs. MDMs were differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then pretreated with CPZ or the equivalent amount of the drug vehicle alone (i.e., ethanol) for 30 min. Cells were then allowed to internalize equal amounts of NL4-3Balenv, NL4-3env−, NL4-3env−VSV-G, or NL4-3env−/JRFLenv virus (standardized in terms of p24) for 2.5 h at 37°C. After a 5-min trypsin treatment to remove membrane-bound viruses, cell lysis was performed. The total intracellular p24 content was quantified by an ELISA and normalized for cell viability (Cellular viability test on uninfected cells). Results are expressed as percentages of the intracellular p24 level compared to that of transporter-treated cells (100%) and represent mean values (triplicate samples) of several independent experiments (*, P < 0.05; **, P < 0.01). Each symbol represents MDMs for one specific blood donor, and identical symbols are linked to the same donor in the different panels.
HIV-1 replication is restricted in both classically and alternatively activated MDMs.
Although it was previously shown that HIV-1 replication was restricted in M1- and M2a-activated MDMs (7), these restrictions were described only after an 18-h treatment with cytokines, and thus, we needed to determine the susceptibility to productive HIV-1 infection in MDM populations activated for 72 h, as was done in our current experiments.
In order to compare levels of HIV-1 production in the studied MDM populations, cells were treated for 72 h with cytokines, put in contact with NL4-3Balenv viruses for 2 h, washed, and then cultured for 10 days. The release of HIV-1 in the cell culture was monitored by measuring p24 levels in the supernatant at days 0, 3, 6, and 10 following virus infection. As previously reported (7), the level of HIV-1 production was reduced over time in M2a cells compared to their M0 counterparts, and p24 release was completely blocked in M1 MDMs (Fig. 6). Indeed, on the 10th day following virus infection, 3.6 to 7.3 ng/ml of HIV-1 p24 was detected in M0 MDMs, compared to 0.5 to 2 ng/ml in M2a MDMs and 0 ng/ml p24 in M1 MDMs. We also compared HIV-1 infection using reporter-pseudotyped viruses. Surprisingly, both M1 and M2a MDMs showed less virus-encoded luciferase activity, as opposed to what was observed by Cassol and colleagues, who reported previously that M2a activation was associated with a late posttranslational block (7). Nonetheless, these results confirm that HIV-1 infection/replication is indeed restricted in our M1- and M2a-activated MDMs.
Fig 6.
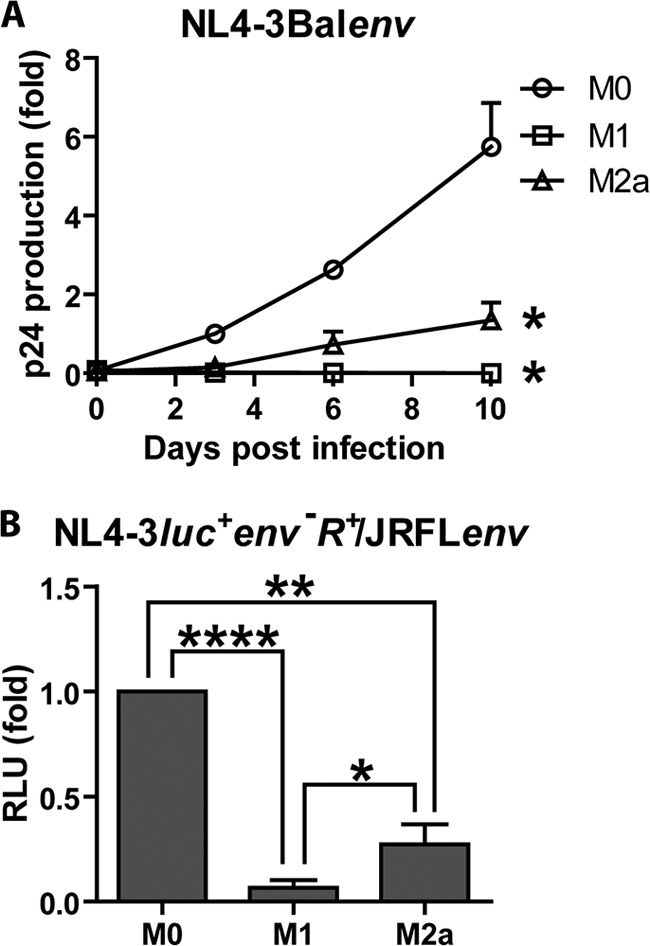
HIV-1 replication is restricted in both classically and alternatively activated MDMs. MDMs were differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then put in contact with similar amounts of NL4-3Balenv (A) or NL4-3luc+env−R+/JRFLenv (B) virus (standardized in terms of p24) for 2 h at 37°C to allow virus infection. (A) Virus in the cell culture supernatant was quantified over time for 10 days by measuring the p24 content with an in-house ELISA. Results are expressed as the fold p24 production compared to that of M0-infected MDMs on day 3 and represent means ± standard errors of the means from 3 independent experiments. (B) Luciferase activity (expressed in relative light units [RLU]) was quantified on day 10 following virus infection, as described in Materials and Methods. Results are expressed as fold changes compared to values for M0-infected MDMs and represent means ± standard errors of the means from 5 independent experiments performed in triplicate (*, P < 0.05; **, P < 0.01; ****, P < 0.0001).
The HIV-1 degradation rate is increased in both classically and alternatively activated macrophages.
To verify the impact of macrophage activation on HIV-1 following internalization by endocytosis, we compared the virus degradation rates of resting and cytokine-activated MDMs. We observed a significant drop of intracellular p24 levels over time for both NL4-3Balenv and NL4-3env−/VSV-G viruses in M1 MDMs (Fig. 7A), even though VSV-G-pseudotyped viruses should efficiently escape degradation by FAE following endosomal acidification. In order to confirm that the observed decrease in the intracellular p24 level was due to virus degradation, we repeated the same experimental procedure in the presence of bafilomycin A1. As shown in Fig. 7B, the decrease in the amount of intracellular p24 was completely reversed in the presence of the drug, confirming that it was indeed due to endosomal degradation. Overall, these results indicate that the occurrence of HIV-1 FAE may be decreased in M1 and M2a MDMs, as the rate of HIV-1 degradation is increased in these cells.
Fig 7.

HIV-1 degradation is increased in M1 and M2a MDMs. MDMs were differentiated for 7 days; either left untreated (M0), treated with IFN-γ (20 ng/ml) and LPS (100 ng/ml) (M1), or treated with IL-4 (20 ng/ml) (M2a) for 72 h; washed; and then left untreated (A) or pretreated with bafilomycin A1 (100 nM) (B) for 30 min at 37°C before allowing them to internalize NL4-3Balenv or NL4-3env−/VSV-G virus for 2 h at 37°C. Cells were then washed, trypsinized to remove bound viruses (time zero), and incubated at 37°C (in the presence of the drug or not) to allow for virus degradation. The intracellular p24 content was quantified over time by an ELISA after cell lysis. Results are expressed as fold changes compared to the initial amount of intracellular p24 at time zero and represent means ± standard errors of the means from 5 (A) and 2 (B) independent experiments performed in triplicate (*, P < 0.05).
DISCUSSION
Previous studies have shown that HIV-1 internalization by FAE leads to productive infection in several established cell lines (22, 37) and primary human CD4+ T cells or the CEM cell line (2, 27) as well as in MDMs (6, 20). However, macrophage endosomal functions have been shown to be highly regulated upon cellular activation, leading to resistance or the replication of intracellular pathogens such as Listeria monocytogenes (32) or Brucella abortus (1). In the current study, we show that these metabolic changes affect HIV-1 endocytosis and infection as well. Both HIV-1 internalization and degradation rates were modulated following macrophage activation in a cytokine-specific manner. Specific macrophage activation pathways are thus likely to have a different impact on HIV-1 infection.
The internalization process (fusion and endocytosis) of macrophage-tropic (R5-using) HIV-1 strains was significantly increased in activated M1 MDMs compared to inactivated M0 MDMs. This increase in the amount of intracellular p24 is explained by a higher endocytosis rate following M1 activation, as it was also observed for a viral glycoprotein-deficient and fusion-incompetent virus (i.e., NL4-3env−), thus excluding the possibility that M1-activated MDMs are more permissive to HIV-1 infection through PMF. According to a study reported previously by Permanyer et al., an increase in the rate of HIV-1 endocytosis should favor FAE over PMF (28). However, in this case, the endocytosis pathway that is upregulated upon M1 activation is unlikely to allow for FAE. First, an increase in the amount of intracellular p24 in M1 MDMs was observed with all the viruses tested, whether they carried the external envelope gp120 or not, suggesting that an unspecific surface receptor (other than CD4 or CCR5) is involved in this event and that the HIV-1 receptor and coreceptor are not present in the corresponding endosomal compartments. Second, M1 activation was coupled with a higher HIV-1 degradation rate, suggesting that the viral particle-receptor interaction in endosomal compartments would have less time to lead to fusion in M1 MDMs than in resting MDMs (i.e., M0). Furthermore, the efficiency of M1 MDMs to degrade incoming particles was striking, as even a VSV-G-pseudotyped virus lost its capacity to escape degradation in these cells. Taken together, these data suggest that M1 activation of macrophages induces changes in their endosomal pathways that limit both PMF and FAE. Our observations are in agreement with previous work showing that HIV-1 infection was restricted in M1 MDMs at early steps of the viral cycle (7). We therefore suggest that HIV-1 endocytosis contributes to restraining HIV-1 infection in M1 macrophages.
In contrast to our observations of M1 MDMs, M2a macrophages showed, after 2.5 h of HIV-1 internalization, lower intracellular p24 levels than their M0 counterparts due to a lower rate of endocytosis. Such a reduction in HIV-1 engulfment should favor PMF over FAE, as it would allow the virus to remain at the plasma membrane for a longer time before its removal from the cell surface. Furthermore, and although it was not as striking as that in M1-activated MDMs, M2a activation was coupled with an increased viral degradation rate, in keeping with the idea that FAE is less important in these cells. Recent studies have suggested that HIV-1 fusion at the plasma membrane does not allow for productive virus infection, as it does not permit proceeding beyond the lipid-mixing step (10, 22). According to those studies, fusion pore formation or enlargement is not possible at the plasma membrane. However, an unidentified factor could favor this step at the endosomal membrane, and such reports point to a role of dynamin in this process. However, such observations were obtained by using cell lines and primary human CD4+ T cells, and it would be interesting to see if the same conclusions apply to MDMs. In that case, the redirection of viral fusion toward the plasma membrane would not benefit the virus.
We used MDMs derived in the absence of M-CSF as resting MDMs (M0) in our experimental procedures, as M-CSF is known to induce M2c polarization in macrophages (13). Surprisingly, macropinocytosis was not found to be a major HIV-1 endocytosis pathway in any of our MDM populations (M0, M1, and M2a), as we did not significantly decrease NL4-3Balenv nor NL4-3env−/JRFLenv endocytosis using EIPA or another amiloride derivative, such as DMA (data not shown), in these cells, in contrast to what was previously reported (6, 20). This could reflect an involvement of M-CSF in macropinocytosis induction in MDMs. This induction of macropinocytosis by M-CSF was also reported by Zhao et al. in a previous study (41), in which the presence of M-CSF during macrophage differentiation induced the constitutive fluid-phase uptake of native low-density lipoprotein (LDL), as opposed to macrophages derived in the absence of M-CSF, which required phorbol myristate acetate (PMA) activation to exert the same function. As macropinocytosis has been shown to allow for productive HIV-1 infection by FAE, M2c-activated macrophages might represent a subpopulation of these cells that is particularly susceptible to HIV-1 infection in terms of FAE. Accordingly, M-CSF was shown previously to enhance HIV-1 entry and replication in cells of the monocyte/macrophage lineage (38). Indeed, we observed a higher level of p24 production after infection of MDMs derived in the presence of M-CSF than in MDMs derived without the cytokine (our unpublished data). In both M1- and M2a-activated macrophages, activation was coupled with HIV-1 internalization via CME. Although a higher CME efficiency parallels the upmodulation of HIV-1 endocytosis in M1-activated MDMs, it does not explain these observations entirely, as we did not find any correlation between the extent of HIV-1 engulfment and the efficiency of CME (data not shown), suggesting that other mechanisms are involved in HIV-1 endocytosis in these cells. As for M2a-activated MDMs, the increase in CME might reflect the shutdown of another pathway rather than true induction, as the overall rate of endocytosis was decreased in these cells.
Although CME was shown to allow for productive HIV-1 infection by FAE in the HeLa-CD4 cell line (9, 22) and CD4+ T cells (2), it is unlikely to allow for FAE in MDMs, as its occurrence does not require the binding of HIV-1 to its specific receptor and coreceptor. Additionally, given that we observed CME induction for all viruses tested, and it was coupled with increased HIV-1 degradation in both M1- and M2a-activated cells, it is possible that CME induction is a general mechanism to increase cargo degradation in activated macrophages. Although, to our knowledge, CME regulation in activated macrophages has not previously been reported, Souza et al. showed that the treatment of pancreatic cells with an IFN-γ-containing cocktail induces changes in mRNA expression which are likely to lead to upregulated CME (34). Those authors suggested that this general induction is a cell response for rapidly internalizing and degrading receptors in order to avoid cell damage and apoptosis in the case of a prolonged exposure to cytokines (34). This may also be the case for our data, as prolonged treatments with cytokines were required to observe an effect on HIV-1 endocytosis in MDMs. CME might therefore represent a dead end in HIV-1 infection in MDMs, as its use seems to be associated with a rapid degradation of internalized viruses, thus restricting FAE.
It was previously reported that HIV-1 infection is less effective in M1 and M2a macrophages activated for 18 h than in resting MDMs (7). This reduced susceptibility to HIV-1 infection involved early restriction factors and a diminution of CD4 expression at the cell surface in M1 MDMs, as opposed to a posttranslational restriction step in M2a-activated cells. This conclusion stems from the observation that the decrease in the overall reverse transcriptase activity in supernatants of infected cells was not associated with a decrease in viral protein accumulation in M2a MDMs (7). We have used two different methods to confirm the restriction of HIV-1 infection in macrophages following exposure to cytokines, i.e., the quantification of p24 production in the cell supernatant by an ELISA and reporter gene activity following infection with a recombinant luciferase-encoding virus. Although both methods confirmed the restriction of HIV-1 infection in M1 and M2a MDMs, the IL-4 activation of MDMs resulted in a reduction of luciferase activity, which rather suggests a blockade before or in translation. This reduction may be partly due to the reduced metabolic activity of MDMs following IL-4 treatment. However, this decrease in cellular activity does not account for the entire effect observed and could not be seen in all MDM populations derived from the different blood donors. Thus, it may also be the result of a longer exposure to IL-4 (72 h compared to 18 h [7]). Indeed, a block in HIV-1 infection at the reverse transcription step was previously reported, in which macrophages were differentiated in the presence of IL-4 for 5 days (31).
Taken together, our data suggest that different subtypes of activated macrophages, in modulating HIV-1 endocytosis, eventually lead to their different susceptibilities to HIV-1 infection in regard to PMF and FAE. These differences might also impact HIV-1 antigen presentation by MDMs and its transfer to other cellular targets and may therefore influence the overall pathogenesis of the disease. It was suggested that during the course of an HIV-1 infection, the macrophage activation status changes from M1 during the early phase of the disease to M2a during the chronic phase of the infection and, finally, to deactivated macrophages at later stages (14). Macrophages might therefore change in terms of their susceptibilities to HIV-1 infection during the course of the infection, and this could be reflected in their capacities to become reservoirs for the virus. Variations in HIV-1 endocytosis should therefore be considered when designing antiretroviral strategies.
ACKNOWLEDGMENTS
We thank Odette Simard, Caroline Côté, and Marc-André Roy for technical help.
This study was supported by an emerging team grant in HIV pathogenesis to M.J.T. from the Canadian Institutes of Health Research (CIHR) (grant HET-85519). L.-A.G. is the recipient of CIHR and Fonds de la Recherche en Santé du Québec doctoral awards. M.J.T. is the recipient of the Canada Research Chair in Human Immuno-Retrovirology (tier 1 level).
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Baldwin CL, Jiang X, Fernandes DM. 1993. Macrophage control of Brucella abortus: influence of cytokines and iron. Trends Microbiol. 1:99–104 [DOI] [PubMed] [Google Scholar]
- 2. Bosch B, et al. 2008. A clathrin-dynamin-dependent endocytic pathway for the uptake of HIV-1 by direct T cell-T cell transmission. Antiviral Res. 80:185–193 [DOI] [PubMed] [Google Scholar]
- 3. Bounou S, Leclerc JE, Tremblay MJ. 2002. Presence of host ICAM-1 in laboratory and clinical strains of human immunodeficiency virus type 1 increases virus infectivity and CD4(+)-T-cell depletion in human lymphoid tissue, a major site of replication in vivo. J. Virol. 76:1004–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bourinbaiar AS, Phillips DM. 1991. Transmission of human immunodeficiency virus from monocytes to epithelia. J. Acquir. Immune Defic. Syndr. 4:56–63 [PubMed] [Google Scholar]
- 5. Bowman EJ, Siebers A, Altendorf K. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U. S. A. 85:7972–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter GC, Bernstone L, Baskaran D, James W. 2011. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology. 409:234–250 [DOI] [PubMed] [Google Scholar]
- 7. Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. 2009. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 182:6237–6246 [DOI] [PubMed] [Google Scholar]
- 8. Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944 [DOI] [PubMed] [Google Scholar]
- 9. Daecke J, Fackler OT, Dittmar MT, Krausslich HG. 2005. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 79:1581–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de la Vega M, et al. 2011. Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology 8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dornadula G, Zhang H, Shetty S, Pomerantz RJ. 1999. HIV-1 virions produced from replicating peripheral blood lymphocytes are more infectious than those from nonproliferating macrophages due to higher levels of intravirion reverse transcripts: implications for pathogenesis and transmission. Virology 253:10–16 [DOI] [PubMed] [Google Scholar]
- 12. Fernandez-Boyanapalli R, et al. 2010. Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFN-gamma in a nitric oxide-dependent manner. J. Immunol. 185:4030–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gordon S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3:23–35 [DOI] [PubMed] [Google Scholar]
- 14. Herbein G, Varin A. 2010. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janas AM, Dong C, Wang JH, Wu L. 2008. Productive infection of human immunodeficiency virus type 1 in dendritic cells requires fusion-mediated viral entry. Virology 375:442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lifson JD, Reyes GR, McGrath MS, Stein BS, Engleman EG. 1986. AIDS retrovirus induced cytopathology: giant cell formation and involvement of CD4 antigen. Science 232:1123–1127 [DOI] [PubMed] [Google Scholar]
- 17. Mackaness GB. 1964. The immunological basis of acquired cellular resistance. J. Exp. Med. 120:105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantovani A, et al. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25:677–686 [DOI] [PubMed] [Google Scholar]
- 19. Maréchal V, Clavel F, Heard JM, Schwartz O. 1998. Cytosolic Gag p24 as an index of productive entry of human immunodeficiency virus type 1. J. Virol. 72:2208–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maréchal V, et al. 2001. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 75:11166–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McClure MO, Marsh M, Weiss RA. 1988. Human immunodeficiency virus infection of CD4-bearing cells occurs by a pH-independent mechanism. EMBO J. 7:513–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Montaner LJ, et al. 1999. Type 1 and type 2 cytokine regulation of macrophage endocytosis: differential activation by IL-4/IL-13 as opposed to IFN-gamma or IL-10. J. Immunol. 162:4606–4613 [PubMed] [Google Scholar]
- 24. Mosser DM. 2003. The many faces of macrophage activation. J. Leukoc. Biol. 73:209–212 [DOI] [PubMed] [Google Scholar]
- 25. Ouellet M, Roy J, Barbeau B, Geleziunas R, Tremblay MJ. 2003. NF-kappaB induction by bisperoxovanadium compounds requires CD45, p36(LAT), PKC, and IKK activity and exhibits kinetics of activation comparable to those of TCR/CD28 coengagement. Biochemistry 42:8260–8271 [DOI] [PubMed] [Google Scholar]
- 26. Pai RK, Askew D, Boom WH, Harding CV. 2002. Regulation of class II MHC expression in APCs: roles of types I, III, and IV class II transactivator. J. Immunol. 169:1326–1333 [DOI] [PubMed] [Google Scholar]
- 27. Pauza CD, Price TM. 1988. Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J. Cell Biol. 107:959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Permanyer M, Ballana E, Este JA. 2010. Endocytosis of HIV: anything goes. Trends Microbiol. 18:543–551 [DOI] [PubMed] [Google Scholar]
- 29. Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72:2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaeffer E, Soros VB, Greene WC. 2004. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J. Virol. 78:1375–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schuitemaker H, et al. 1992. Proliferation-dependent HIV-1 infection of monocytes occurs during differentiation into macrophages. J. Clin. Invest. 89:1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaughnessy LM, Swanson JA. 2007. The role of the activated macrophage in clearing Listeria monocytogenes infection. Front. Biosci. 12:2683–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith AE, Helenius A. 2004. How viruses enter animal cells. Science 304:237–242 [DOI] [PubMed] [Google Scholar]
- 34. Souza KL, Elsner M, Mathias PC, Lenzen S, Tiedge M. 2004. Cytokines activate genes of the endocytotic pathway in insulin-producing RINm5F cells. Diabetologia 47:1292–1302 [DOI] [PubMed] [Google Scholar]
- 35. Stein BS, et al. 1987. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 49:659–668 [DOI] [PubMed] [Google Scholar]
- 36. Vidricaire G, Imbeault M, Tremblay MJ. 2004. Endocytic host cell machinery plays a dominant role in intracellular trafficking of incoming human immunodeficiency virus type 1 in human placental trophoblasts. J. Virol. 78:11904–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vidricaire G, Tremblay MJ. 2007. A clathrin, caveolae, and dynamin-independent endocytic pathway requiring free membrane cholesterol drives HIV-1 internalization and infection in polarized trophoblastic cells. J. Mol. Biol. 368:1267–1283 [DOI] [PubMed] [Google Scholar]
- 38. Wang J, Roderiquez G, Oravecz T, Norcross MA. 1998. Cytokine regulation of human immunodeficiency virus type 1 entry and replication in human monocytes/macrophages through modulation of CCR5 expression. J. Virol. 72:7642–7647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei X, et al. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu L, KewalRamani VN. 2006. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat. Rev. Immunol. 6:859–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao B, et al. 2006. Constitutive receptor-independent low density lipoprotein uptake and cholesterol accumulation by macrophages differentiated from human monocytes with macrophage-colony-stimulating factor (M-CSF). J. Biol. Chem. 281:15757–15762 [DOI] [PubMed] [Google Scholar]