

Abstract
Several lines of evidence suggest that various cofactors may be required for prion replication. PrP binds to polyanions, and RNAs were shown to promote the conversion of PrPC into PrPSc in vitro. In the present study, we investigated strain-specific differences in RNA requirement during in vitro conversion and the potential role of RNA as a strain-specifying component of infectious prions. We found that RNase treatment impairs PrPSc-converting activity of 9 murine prion strains by protein misfolding cyclic amplification (PMCA) in a strain-specific fashion. While the addition of RNA restored PMCA conversion efficiency, the effect of synthetic polynucleotides or DNA was strain dependent, showing a different promiscuity of prion strains in cofactor utilization. The biological properties of RML propagated by PMCA under RNA-depleted conditions were compared to those of brain-derived and PMCA material generated in the presence of RNA. Inoculation of RNA-depleted RML in Tga20 mice resulted in an increased incidence of a distinctive disease phenotype characterized by forelimb paresis. However, this abnormal phenotype was not conserved in wild-type mice or upon secondary transmission. Immunohistochemical and cell panel assay analyses of mouse brains did not reveal significant differences between mice injected with the different RML inocula. We conclude that replication under RNA-depleted conditions did not modify RML prion strain properties. Our study cannot, however, exclude small variations of RML properties that would explain the abnormal clinical phenotype observed. We hypothesize that RNA molecules may act as catalysts of prion replication and that variable capacities of distinct prion strains to utilize different cofactors may explain strain-specific dependency upon RNA.
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are characterized by the accumulation in the brain and sometimes in the lymphoid tissues (13, 27) of an abnormally structured form (PrPSc) of the host cellular prion protein (PrPC) (26). PrPSc is thought to be the only (5, 9, 21, 33) or the major (7, 11, 35) constituent of the infectious agent, the prion. Prions occur in the form of diverse strains exhibiting specific biological and biochemical characteristics (1, 4). Different prion strains show distinct interspecies transmission properties and, in particular, different pathogenicities for humans (20, 37). The strain phenomenon is also important from a fundamental standpoint, as strain-specific properties of infectious agents have hitherto been encoded by the nucleic acid genome of the pathogen. In the case of prions, strain-specific properties might be determined by differences in PrPSc conformation, by differences in complex glycosylation, or by a yet-to-be defined informational molecule associated with PrPSc.
The ability to convert PrP into infectious PrPSc in vitro lends support to the concept that the prion protein is the major component of prions (5, 9, 21, 33). Furthermore, it has been shown that RNA molecules facilitate in vitro amplification of infectious PrPSc (9, 15, 33). However, the exact role of RNA in the amplification process remains unknown. RNA could act as a mere catalyst of the PrP misfolding process. On the other hand, RNA may be associated with the infectious particle and contribute to prion strain characteristics. A recent study showed that the requirement of RNA for in vitro amplification of PrPSc is species dependent, with hamster-derived PrPSc being largely dependent on the presence of RNA in the protein misfolding cyclic amplification (PMCA) reaction mixture, whereas mouse-derived PrPSc does not require RNA for in vitro amplification (10). Another study showed similar RNA-dependent amplifications of six hamster prion strains (15). Other endogenous polyanions such as DNA, heparan sulfates, or lipids may come into play under conditions of RNA deficiency (10).
In the present study, we further investigated the possibility of a strain-specific dependency upon RNA during in vitro PrPSc amplification and addressed the role of RNA as a strain-specifying component of infectious prions. For this purpose, we conducted PMCA amplification of various mouse prions in RNA-depleted as well as control reaction mixtures. We studied the role of RNA in the efficiency of the amplification reaction for each of 9 strains of mouse prions. We determined RML strain characteristics by bioassay after amplification in RNA-depleted versus control PMCA reactions. We conclude that RNA dependency for in vitro conversion is prion strain specific and that RML prion strain identity is maintained after in vitro amplification under conditions of RNA depletion. We propose that RNA molecules may act as strain-specific catalysts of prion replication.
MATERIALS AND METHODS
Preparation of tissue homogenates.
Healthy mice were sacrificed by CO2 inhalation and immediately perfused with phosphate-buffered saline (PBS) plus 5 mM EDTA prior to harvesting of the brain. The perfusion was conducted by inserting a 20-gauge needle directly into the mouse heart and manually injecting an approximate volume of 30 ml of 5 mM EDTA in PBS. Terminal prion-infected mice were sacrificed by CO2 inhalation, and brains were immediately harvested. Mouse brains were either homogenized or flash frozen in liquid nitrogen. Brain homogenates (10%, wt/vol) were prepared in prechilled conversion buffer (PBS containing 150 mM NaCl, 1.0% Triton X-100, and the Complete protease inhibitor cocktail tablets containing EDTA from Roche Applied Science). Infected and normal brain homogenates were prepared manually using a glass Potter-Elvehjem homogenizer. Sample heating was avoided by keeping the homogenizer on ice throughout the process.
Dilutions of prion-infected brain homogenates (IBHs) were done in normal brain homogenates (NBHs), and they are expressed in relation to the brain; for example, a 100-fold dilution is equivalent to a 1% brain homogenate.
Preparation of PMCA substrates. (i) RNase A treatment.
Aliquots of 10% normal brain homogenates, prepared as described above, were treated with 48 μg/ml (120-U/ml final concentration) of RNase A (MP Biomedicals, Solon, OH) for 1 h at 37°C.
(ii) RNase inhibitor treatment.
Aliquots (100 μl) of 10% NBHs were treated with 5 μl of RNase Out (2,000 U/ml) (Invitrogen, Carlsbad, CA) for 1 h at 37°C.
(iii) Control substrates.
Aliquots (100 μl) of 10% NBHs were mixed with 5 μl of conversion buffer and incubated at 37°C for 1 h.
(iv) Preparation of PMCA substrates for reconstitution with exogenous RNA.
Aliquots (100 μl) of 10% NBHs were treated with 0.86 μg/ml (2 U/ml) of RNase A for 1 h at 37°C. Then, 2,000 U/ml of RNase Out was added to these preparations to inhibit RNase A activity and allow sample reconstitution with exogenous RNA molecules, added to a final concentration of 0.12 μg/μl.
(v) Preparation of PMCA substrates for reconstitution with synthetic polyanions.
Aliquots (100 μl) of 10% NBHs were treated with 8.6 μg/ml (20-U/ml final concentration) of RNase A for 1 h at 37°C. Then, synthetic poly(A) and poly(G) (Sigma, St. Louis, MO) were added to these preparations to a final concentration of 0.12 μg/μl.
(vi) Preparation of PMCA substrates for reconstitution with double-stranded DNA (dsDNA).
Aliquots (100 μl) of 10% NBHs were treated with 8.6 μg/ml (20-U/ml final concentration) of RNase A for 1 h at 37°C. Thereafter, 200- to 500-bp salmon sperm DNA fragments (Trevigen, Gaithersburg, MD) were added to these preparations to a final concentration of 0.12 μg/μl.
Prion replication by PMCA.
Aliquots of 10% brain homogenates from prion-infected mice were serially diluted into 10% normal brain homogenates pretreated as described above and loaded onto 0.2-ml PCR tubes containing zirconia/silica beads (Biospec Products Inc., Bartlesville, OK). Tubes were positioned on an adaptor placed on the plate holder of a microsonicator (model 3000; Misonix, Farmingdale, NY) and programmed to perform cycles of 30 min of incubation followed by a 20-s pulse of sonication set at potency of 9. Samples were incubated, without shaking, immersed in the water of the sonicator bath set at 37°C. The PMCA reaction was stopped after 24 h (48 cycles) by applying a 20-s sonication pulse. To avoid formation of amorphous aggregates that interfere with proteinase K (PK) digestion, this treatment was preformed prior to sample freezing.
For serial automated PMCA (saPMCA), a fraction of the amplified material specific for each strain (see below) was transferred to a new PCR tube containing fresh pretreated normal brain homogenate and beads and amplified under the conditions described above. To avoid generation of amorphous aggregates and in order to have homogenous samples, this dilution step was performed right after each PMCA round and prior to sample freezing. To monitor the amplification efficiency, 18-μl aliquots were taken prior to each round of PMCA and used as controls. To achieve similar conversion efficiencies for all prion strains, amplified RML, ME7, and 22L were diluted 10, 5, and 8 times, respectively, into NBHs to seed the next round of amplification.
PK digestion.
Samples were subjected to incubation in the presence of PK (100 μg/ml) for 60 min at 56°C with constant shaking at 450 rpm. The digestion was stopped by addition of NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA), and samples were heated at 100°C for 10 min. The presence of protease-resistant PrP was revealed by Western blotting.
Na-PTA precipitation.
Aliquots of PMCA-generated material were sodium-phosphotungstic acid (Na-PTA) precipitated as described in reference 32.
Thermolysin digestion.
Samples were subjected to incubation in the presence of 50 μg/ml thermolysin for 60 min at 70°C, with constant shaking at 450 rpm. The digestion was stopped by addition of NuPAGE LDS sample buffer (Invitrogen), and samples were heated at 100°C for 10 min. The presence of protease-sensitive PrPSc was revealed by Western blotting.
Western blotting.
Proteins in NuPAGE LDS sample buffer (Invitrogen) were fractionated by SDS-polyacrylamide gel electrophoresis, electroblotted onto a polyvinylidene difluoride (PVDF) membrane (Millipore), and probed with D18 antibody, diluted 1:30,000 from a 1-mg/ml stock, followed by a murine horseradish peroxidase (HRP)-conjugated anti-human IgG antibody at a 1:15,000 dilution (Southern Biotech, Birmingham, AL). Immunoreactive bands were visualized by incubation with West Pico (Pierce, Rockford, IL) and imaged using a BioSpectrum imaging system (UVP, Upland, CA).
RNA purification and fractionation.
RNA samples were purified from mouse brain and liver. Total RNA and large- and small-RNA preparations were purified with the mirVana kit (Ambion, Carlsbad, CA) by following the manufacturer's instructions. After total RNA purification, the samples were treated with DNase and PK to eliminate potential DNA and protein contamination that could have interfered with the result interpretation. Upon conclusion of these treatments, the samples were repurified using the same kit to remove the enzymes. Thereafter, RNA purification was either finished or continued for further RNA fractionation into large (≥200-nucleotide [nt]) or small (<200-nt) molecules according to the mirVana protocol. RNA concentration and size were evaluated by RiboGreen RNA quantitation (Molecular Probes, Eugene, OR) 1.2% denaturing agarose electrophoresis (RNA, ≥800 nt) and Novex 15% denaturing polyacrylamide Tris-borate-EDTA (TBE)-urea gels (RNA, 20 to 800 nt) (Invitrogen).
Real-time PCR (RT-PCR).
RNA samples were purified from PMCA substrates after treatment with RNase A or RNase inhibitor or after being left untreated with the mirVana kit (Ambion). Thereafter, quantification of the levels of specific microRNAs (miRNAs) (as indicated in Results), rRNA, and housekeeping RNAs was evaluated with the TaqMan nicroRNA assays or the TaqMan rRNA control reagents by following the manufacturer's instructions.
Mouse bioassay and titrations.
The use of animals was conducted according to institutional guidelines after review of the protocol by the Institutional Animal Care and Use Committee. Tga20 mice expressing wild-type PrPC at about 8 times the endogenous levels or wild-type C57BL/6 mice were used as an experimental model of scrapie. Animals were 4 to 6 weeks old at the time of inoculation. Mice anesthetized by isoflurane were injected intracerebrally in the prefrontal cortex area with 20 μl of the sample. They were euthanized by CO2 and then decapitated when they showed terminal signs of prion disease. Brains were extracted and analyzed biochemically and immunohistochemically. The right cerebral hemisphere was frozen and stored at −80°C for secondary transmission experiments and biochemical examination of PrPSc using Western blotting after PK digestion as described above. The left hemisphere was fixed in Carnoy's solution and processed as described below.
For inoculum titration, groups of 8 or 6 mice were inoculated as described above with limiting dilutions of brain-derived or PMCA-generated RML or ME7 (where indicated) in the presence or absence of RNase A and RNase inhibitor. To control the spontaneous generation of prions by PMCA, mice were inoculated with unseeded NBHs subjected to the same rounds of PMCA as seeded reactions. Additional controls included unseeded NBHs as well as brain-derived RML treated with RNase A and RNase inhibitor. The 50% lethal dose (LD50) of each inoculum was calculated according to the method of Reed and Muench (28).
Ethics statement.
The Institutional Animal Care and Use Committee (IACUC) of The Scripps Research Institute (TSRI), Scripps Florida, reviewed and approved the animal protocol entitled “Role of RNAs in Prion Replication” (animal protocol number 07-012). TSRI, Scripps Florida, maintains a centralized animal care and use program registered by the USDA, assured with the Office of Laboratory Animal Welfare (OLAW) and accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC). Housing and care of animals are consistent with the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals, the Animal Welfare Act, and other applicable state and local regulations.
Scrapie-associated fiber (SAF) purification.
One hundred-microliter PMCA- and brain-derived samples were mixed with 4 μl of Benzonase (2.5 U/μl, stock 1:10 diluted in PBS to 50 U/ml) plus 1 μl of 0.2 M MgCl2 (final concentration, 1 mM) and incubated for 30 min at 37°C. The samples were then mixed with 200 μl of buffer T7 (10% NaCl, 10% Sarkosyl, 1% SB 3-14, 2 mM Tris [pH 7.4]) and incubated for 15 min at room temperature (25°C) in a Thermomixer (Bio-Rad) set at 450 rpm. Following this incubation, the preparations were layered over 100 μl of a sucrose cushion (10% NaCl, 10% sucrose, 0.1% SB 3-14, 2 mM Tris [pH 7.4]) and spun for 2 h at maximum speed (13,200 rpm, 16,100 relative centrifugal force) on a benchtop centrifuge (Eppendorf, 5415R) set at 18°C. The pellets were resuspended by sonication in 100 μl of PBS containing 3 mg/ml of bovine serum albumin (BSA). NBHs (10%) prepared from perfused noninoculated C57BL/6 mice were used as negative controls.
Calculation of PrPSc specific infectivity.
To calculate the specific infectivity per PrPSc molecule of RML generated by PMCA in comparison with brain RML, 1:2 serial dilutions of SAF preparations obtained from PMCA- and brain-derived samples were analyzed by Western blotting (see Fig. 5A). The band intensity was quantified by densitometry using a BioSpectrum imaging system (UVP) and expressed in arbitrary pixel units. The background corresponding to the signal generated by the uninfected brain homogenate was subtracted from the signals. All gels were normalized to each other, allowing the comparison of PrP values. In our experimental setup, we defined the specific infectivity of PrPSc as the reciprocal of the pixel intensity obtained with the sample dilution corresponding to the LD50 measured by bioassay (Tables 1 and 2). Specific infectivity of PrPSc in the PMCA samples is expressed relative to that of PrPSc in the standard RML inoculum (the latter therefore has a value of 1).
Fig 5.

Analysis of RML strain characteristics amplified in RNA-depleted reactions. (A) Comparison of electrophoretic mobilities and quantitation of PrPSc levels in PMCA-generated material (round 13) prior to mouse inoculation. Scrapie-associated fibers (SAFs) were purified from PMCA samples and brain-derived RML. Serial dilutions of each sample were performed prior to Western blotting to determine the levels of PrPSc inoculated into mice. Molecular mass markers, in kilodaltons, are shown for reference. The uninfected brain homogenate (uninfected) is shown as a control of the amount of PrPC that is nonspecifically precipitated during SAF purification. (B) Difference in RML-PRNaseA electrophoretic pattern results from a higher proportion of the PrP C1 fragment. SAF preparations used for panel A were deglycosylated and analyzed by Western blotting to evaluate the levels of full-length and truncated forms of PrP. Densitometric calculations of full-length and truncated PrP, shown in the adjacent graph, demonstrate higher levels of C1 in RML-PRNaseA.
Table 1.
Survival times of Tga20 mice intracerebrally inoculated with PMCA-generated prions
| Inoculuma | Dilutionb | Incubation (dpic) | No. sick/no. inoculated |
|---|---|---|---|
| NBHRNaseA | 10−2 | >240 | 0/6 |
| NBHRNaseOut | 10−2 | >240 | 0/6 |
| NBH-P | 10−2 | >240 | 0/6 |
| RML-P | 10−1 | 77 ± 1 | 6/6 |
| 10−3 | 97 ± 2 | 6/6 | |
| RML-PRNaseOut | 10−1 | 81 ± 2 | 6/6 |
| 10−2 | 88 ± 2 | 6/6 | |
| 10−3 | 95 ± 3 | 6/6 | |
| 10−4 | 128 ± 9 | 4/8 | |
| 10−5 | >240 | 0/8 | |
| 10−6 | >240 | 0/8 | |
| RML-PRNaseA | 10−1 | 78 ± 2 | 6/6 |
| 10−2 | 92 ± 4 | 6/6 | |
| 10−3 | 98 ± 3 | 5/6 | |
| 10−4 | 157 ± 11 | 5/8 | |
| 10−5 | >240 | 0/8 | |
| RML-B | 10−4 | 77 ± 1 | 6/6 |
| 10−5 | 95 ± 2 | 6/6 | |
| 10−6 | 187 | 1/6 | |
| 10−7 | 123 | 1/8 | |
| 10−8 | 105 | 1/8 | |
| 10−9 | >240 | 0/8 | |
| RML-BRNaseA | 10−5 | 94 ± 5 | 5/6 |
| RML-BRNaseOut | 10−5 | 124 ± 20d | 6/6 |
NBHRNaseA and NBHRNaseOut, NBH treated with RNase A and RNase Out, respectively; NBH-P, NBH subjected to 18 rounds of PMCA in the absence of seed; RML-P, PMCA-generated RML; RML-PRNaseOut, PMCA-generated RML in the presence of RNase Out; RML-PRNaseA, PMCA-generated RML in the presence of RNase A; RML-B, brain-derived RML; RML-BRNaseA and RML-BRNaseOut, brain-derived RML treated with RNase A and RNase Out, respectively.
Dilutions of PMCA-generated inocula are relative to the PMCA sample. Dilutions of brain-derived inocula are relative to the brain.
dpi, days postinoculation, expressed as averages ± standard errors of the means.
The high standard error of the mean in this group is due to the presence of an outlier mouse with a prolonged survival time of 216 days postinoculation.
Table 2.
Specific infectivity of PMCA-derived PrPSc generated in the presence or absence of RNA
| Inoculum | Titer | Specific infectivity relative to RMLc |
|---|---|---|
| RML-PRNaseA | 105.8a | 3.5 |
| RML-PRNaseOut | 105.8a | 0.6 |
| RML-B | 107.5b | 1.0 |
Inoculum titer per milliliter of PMCA-generated material.
Inoculum titer per gram of brain.
Specific infectivity of PrPSc molecules present in the inoculum relative to brain-derived RML (RML-B). RML-PRNaseA, PMCA-generated RML under RNA-depleted conditions; RML-PRNaseOut, PMCA-generated RML in the presence of RNase Out.
Immunohistochemistry.
Brains were harvested immediately after euthanasia. The right cerebral hemisphere was frozen and stored at −80°C as described above. The left hemisphere was fixed in Carnoy's solution for 48 h before proceeding. Brain tissues were processed using a Leica ASP300 tissue processor (Leica, Buffalo Grove, IL) and embedded using a Leica paraffin embedding machine. Tissues were mounted in sagittal orientation and sectioned at 4 μm for immunohistochemistry or 10 μm for hematoxylin and eosin staining. For immunohistochemistry, sections were labeled with PrP primary antibody Bar233 (Cayman Chemicals, Ann Arbor, MI) according to reference 18.
Extended cell panel assay.
Cell lines were maintained and the extended cell panel assay (ECPA) was performed as described in references 3, 22, and 25.
Statistics.
The chi-square test was performed to determine whether the abnormal clinical phenotype observed in treatment groups RML-PRNaseA and RML-PRNaseOut were statistically different from each other at a 5% level.
A one-way analysis-of-variance (ANOVA) test was used to determine statistically significant differences in survival times among treatment (RML-BRNaseA and RML-BRNaseOut) and control (RML-B) groups. Additionally, if a difference was found, Scheffe's test was utilized to identify exactly which groups were different. A log rank test was also used to determine if the survival curves for each group were statistically different from each other.
SAS 9.2 (SAS, Cary, NC) was used for all calculations.
RESULTS
RNA depletion impairs in vitro prion replication in a strain-specific fashion.
To address the role of RNA molecules in prion pathogenesis, we first evaluated the effect of RNA depletion by RNase A in in vitro PrPSc amplification reactions. Aliquots of 10% normal brain homogenates (NBHs) were treated for 1 h with RNase A or RNase inhibitor (RNase Out) or left untreated, as described in Materials and Methods. The efficiency of RNA removal was evaluated by real-time quantitative PCR (RT-qPCR) using specific probes for the brain-specific miRNA miR-124a, ubiquitous miRNAs like let-7b, and the rRNA 18S (see Fig. S1 in the supplemental material). Infected brain homogenates (IBHs) prepared from C57BL/6 mice inoculated with the mouse prion strains RML, ME7, and 22L were treated with the same enzymes, added to these substrates, and amplified by serial automated PMCA (saPMCA). After initial efficient replication in all substrates, the conversion of various prion strains over several rounds was distinctively affected in the absence of RNAs (Fig. 1). While RML amplification remained unaffected for up to 9 saPMCA rounds, ME7 conversion ceased completely after 4 rounds in RNase A-treated substrates. Strain 22L exhibited an intermediate behavior, as illustrated by decreased replication efficiency after 3 rounds of saPMCA that was maintained in subsequent rounds (Fig. 1). It has been reported that PrPSc becomes sensitive to PK digestion after RNase A treatment, suggesting that RNA is complexed with PrPSc and that it confers proteinase resistance (17). To investigate whether the absence of ME7 signal in our PMCA reaction was due to an increased sensitivity to PK or to impaired replication in the absence of RNA, we evaluated the presence of PK-sensitive PrPSc (sPrPSc) in our samples. Aliquots of PMCA material generated in the presence or absence of RNA were Na-PTA precipitated and thermolysin digested according to previous reports (8). Even after this treatment, no PrPSc signal could be detected in ME7 samples generated in the absence of RNA molecules (data not shown).
Fig 1.
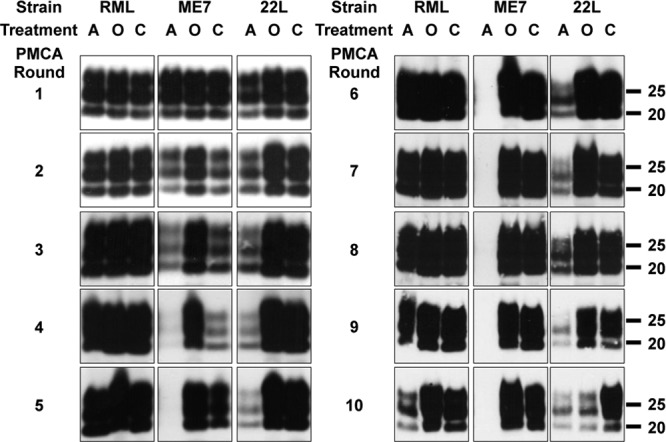
RNA depletion by RNase A impairs in vitro conversion in a strain-dependent fashion. Serial replication of mouse prion strains is shown to be affected by treatment with RNase A. Uninfected brain homogenates (substrate) (10%) treated with RNase A (A) or RNase inhibitor (RNase Out) (O) or left untreated (C) were inoculated with 10% homogenates of brains of mice infected with RML, ME7, and 22L (inocula) subjected to the same RNase treatments. These preparations were amplified by several rounds of PMCA. After each round, aliquots were PK digested and immunoblotted with the anti-PrP monoclonal antibody D18. After RNase A treatment, the ME7 PrPSc signal was significantly reduced from round 2 and disappeared after 4 rounds. Amplification of 22L prions decreased after 3 rounds in the presence of RNase A. RML was unaffected until round 9. The experiment was done 5 more times until round 3, 5, 8, or 10, with comparable results. Molecular mass markers, in kilodaltons, are indicated on the right side of the Western blots.
These findings prompted us to investigate the degree of RNA dependency for efficient in vitro amplification of several mouse prion strains. Since the three strains listed above are characterized by different conversion efficiencies under normal conditions, with RML being the most efficient, followed by 22L and ME7, we wondered whether the observed impaired amplification under RNA-depleted conditions correlated with replication efficiency. To investigate this question, we utilized several mouse-propagated prion strains characterized by different incubation times in the mouse bioassay, as well as three additional prion strains generated in vitro by saPMCA (kindly provided by J. Castilla). These samples were serially diluted into RNA-depleted or control NBHs, and aliquots of these dilutions were in vitro amplified for one PMCA round (Fig. 2A). This method highlighted the differential in vitro amplification capacities of prion strains under normal conditions (Fig. 2A, control PMCA); the extent to which the seed can be diluted while still allowing amplification to occur is indicative of the conversion efficiency of the strain under consideration. Interestingly, RNA depletion of the conversion reaction affected this parameter differently depending on the strain. These differences could also be seen between strains with comparable replication capacities, such as 139A and 766 (Fig. 2). Moreover, lower conversion efficiency was not necessarily associated with higher RNA dependency. Strains 139A, 303, and 726 exhibited lower conversion efficiency than RML and 79A, yet their in vitro conversion was less affected by RNA depletion (Fig. 2). These results indicate that RNA dependency for in vitro prion replication is strain specific.
Fig 2.
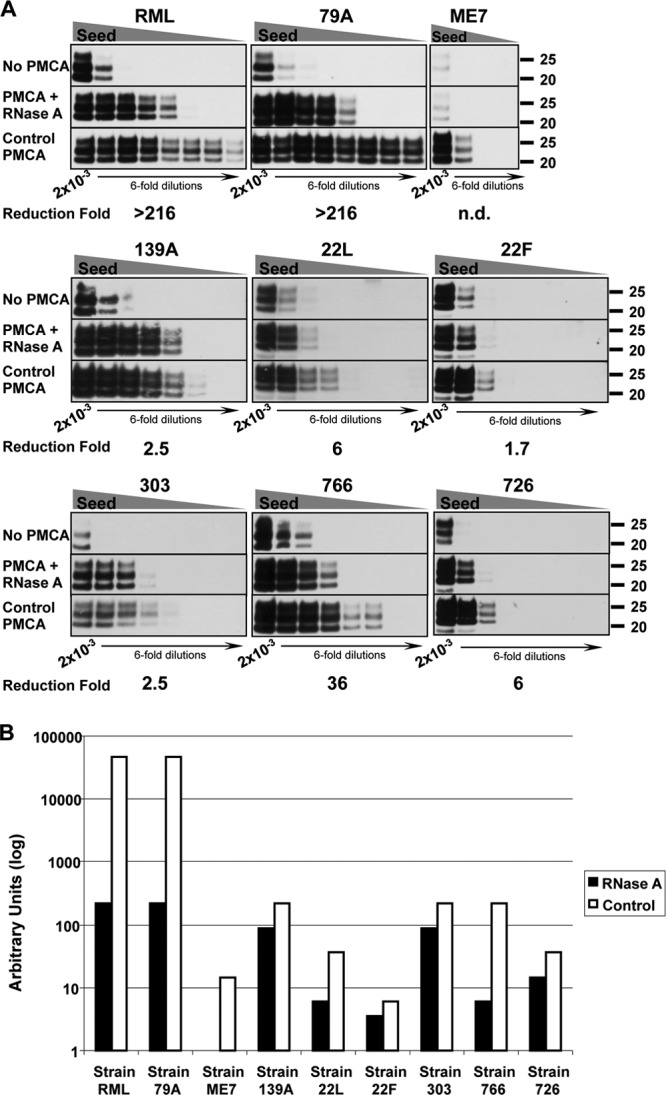
The requirement of RNA molecules for in vitro amplification is highly strain dependent. (A) Brain-derived RML, 79A, ME7, 139A, 22L, and 22F as well as three in vitro-generated prion strains—303, 766, and 726—were propagated in RNase A-treated substrates to evaluate RNA dependency. The calculated reduction of PMCA efficiency under RNA-deficient conditions compared to standard conditions is indicated under each group of Western blots. Except for 22F and 726, each strain was tested 2 to 4 times; representative blots are shown. “No PMCA” blots show the levels of PrPSc prior to PMCA conversion. PrPSc serial dilutions: 2 × 10−3, 3.3 × 10−4, 5.6 × 10−5, 9.3 × 10−6, 1.5 × 10−6, 2.6 × 10−7, 4.3 × 10−8, and 7.1 × 10−9. (B) The relative conversion efficiencies of individual prion strains in normal (open bars) and RNase A-treated brain homogenates (black bars) were calculated from the blots shown in panel A relative to the PrPSc signal observed before PMCA amplification. These results indicate that the sensitivity to RNA depletion is independent of the intrinsic conversion capacity of each strain.
Addition of exogenous RNA to RNA-depleted PMCA reactions enhances prion replication.
To demonstrate that the inhibitory effect of RNase A treatment on PMCA reactions was due to RNA depletion, we reconstituted RNA-depleted substrates with brain-extracted total RNA and evaluated the efficiency of ME7 conversion in these samples in comparison with nonreconstituted substrates. The presence of exogenous RNA enhanced ME7 amplification (Fig. 3A). Liver-derived RNA showed an efficiency similar to that of brain-derived RNA; moreover, we observed a dose-dependent reconstitution of prion conversion (Fig. 3B). While 0.01 μg/μl of total RNA was not sufficient to increase prion replication after RNase A treatment, addition of 0.06 μg/μl and 0.12 μg/μl enhanced ME7 conversion in a dose-dependent manner (Fig. 3B, parts 1, 5, and 6). We evaluated the levels of RNA in the substrates after RNase A and RNase inhibitor treatments as well as after RNA reconstitution, both before and after PMCA (see Fig. S2 in the supplemental material). Addition of 0.12 μg/μl of RNA to RNase A-treated NBHs brought RNA levels back to those found in untreated NBH as determined by RiboGreen quantitation (Fig. S2; compare lanes 3 and 9).
Fig 3.

Addition of RNA enhances prion in vitro amplification. (A) Total RNA enhances ME7 conversion efficiency. Normal brain homogenates (NBHs; 10%) were treated with RNase A or RNase Out. After treatment, RNase A was inhibited with RNase Out (parts 2 and 3) and aliquots of this mixture were either left untreated (part 2) or reconstituted by addition of brain-extracted total RNA (part 3). Serial dilutions of ME7 were performed in these substrates and amplified by PMCA. Part 1 shows the levels of ME7 PrPSc in the dilution series prior to PMCA amplification (10−3, 2.5 × 10−4, 6.3 × 10−5, and 1.6 × 10−5). The experiment was done 5 times, with similar results. (B) Dose-dependent reconstitution of ME7 conversion by liver RNA independent of RNA size. Total liver RNA was fractionated into large- and small-RNA fractions. NBHs (10%) were treated with RNase A (parts 1 to 3, 5, and 6). After treatment, RNase A was inhibited by addition of RNase inhibitor (parts 1 to 3 and 6). Increasing concentrations of total and large- and small-RNA fractions were added to RNA-depleted substrates (parts 1, 2, and 3, respectively). All substrates were seeded with various quantities of ME7 prions and amplified by PMCA (parts 1 to 3, 5, and 6). Part 4 shows the levels of ME7 PrPSc in the dilution series prior to PMCA amplification (10−4, 2.5 × 10−5, 6.3 × 10−6, and 1.6 × 10−6). Molecular mass markers, in kilodaltons, are shown for reference. For −PK lanes, undigested normal brain homogenate was used as a standard. The experiment was done twice, with similar results.
To investigate the size of RNA involved in PrPSc replication, RNA was purified and size fractionated from mouse livers. Large-RNA (≥200 nt) and small-RNA (<200 nt) samples were added at different concentrations to aliquots of serially diluted ME7 and RML on RNA-free substrates. We again observed the dose-dependent effect of RNA in PrPSc conversion. Large and small RNAs restored PrPSc amplification equally well (Fig. 3B; compare parts 2, 3, 5, and 6 and data not shown for RML). We analyzed the quality of the RNA after PMCA. A 2:1 ratio for 28S/18S rRNA has long been considered the benchmark for intact RNA. This ratio was detected in our RNase inhibitor-treated or reconstituted NBH samples prior to PMCA amplification (data not shown); however, similar to the case in a previous study (15), we observed fractionation of large RNA species into smaller fragments (<200 nt) at the end of the 48-cycle PMCA round (Fig. S2; compare odd- and even-numbered lanes).
We evaluated the capacity of synthetic RNA or DNA to reconstitute mouse prion amplification in RNA-depleted substrates. Taking advantage of the RNase A specificity for pyrimidine nucleoside linkages, heteropolymeric preparations of poly(A) or poly(G) acids were added to aliquots of RNase A-treated substrates in the absence of RNase Out. Serial dilutions of RML and ME7 prions were prepared in these substrates and aliquots of these samples amplified by PMCA. Interestingly, conversion stimulation by poly(A) and poly(G) was different between the two strains. ME7 amplification was restored to control levels by poly(A) but not poly(G) (Fig. 4A). PMCA amplification of RML, on the other hand, was reconstituted by poly(A) and poly(G) (Fig. 4A). These results suggested a higher cofactor selectivity of ME7 than of RML. In line with this hypothesis, the addition of 200- to 500-bp fragments of DNA to RNA-depleted substrates did not reconstitute ME7 conversion, whereas it did restore RML amplification (Fig. 4B).
Fig 4.
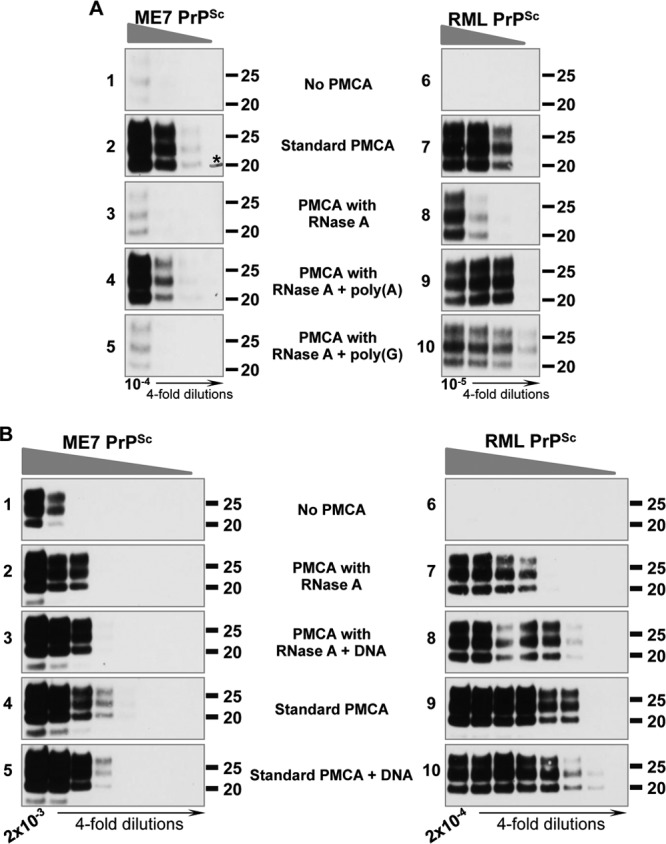
Strain-specific restoration of PMCA conversion efficiency with different polyanions. (A) RML and ME7 conversion efficiencies are restored by adding synthetic poly(A) to the PMCA reaction under RNA-depleted conditions, but only RML conversion is restored by poly(G). RNase A-treated substrates were reconstituted with the synthetic polyanions poly(A) (parts 4 and 9) and poly(G) (parts 5 and 10) and amplified by PMCA. Parts 1 and 6 show the levels of ME7 and RML PrPSc before conversion. The experiment was done 7 times for RML and 5 times for ME7, with similar results. Molecular mass markers, in kilodaltons, are shown for reference. The asterisk in part 2 indicates a hand-drawn molecular mass marker. (B) RML, but not ME7, conversion efficiency is restored by adding DNA to the PMCA reaction under RNA-depleted conditions. RNA-depleted substrates (parts 2 and 7) were reconstituted with salmon sperm DNA (parts 3 and 8). In parallel experiments, DNA was added to normal PMCA substrates (parts 5 and 10) and the amplification yield compared to that for control PMCA reactions (parts 4 and 9). These preparations were seeded with decreasing concentrations of ME7 and RML prions and amplified by PMCA. The experiment was done once with duplicate samples (one is shown) for RML and twice for ME7. ME7 and RML dilutions are shown in parts 1 and 6, respectively. Molecular mass markers, in kilodaltons, are shown for reference.
RML strain identity is maintained after propagation in RNA-depleted substrates.
The unified theory of prion propagation hypothesizes that nucleic acid molecules may confer specific characteristics on prion strains (35). Our data show that strains differ in their nucleic acid requirements for propagation in vitro. These findings prompted us to investigate whether prion amplification in RNA-depleted substrates affected their strain properties. To this end, we generated, by in vitro conversion, RML and ME7 preparations devoid of RNA and brain-derived PrPSc molecules (RML-PRNaseA and ME7-PRNaseA). We performed rounds of saPMCA until a 10−15 dilution of the infected brain homogenate (IBH) was achieved according to previous calculations (5). Due to differences in replication efficiency between strains, dilution factors of 10 and 5 had to be utilized for RML and ME7, respectively, to obtain comparable amplification between rounds (Fig. 1), requiring 13 and 18 rounds of saPMCA for RML and ME7, respectively, to achieve a 10−15 dilution of the IBH. As controls, these strains were serially propagated in untreated (RML-P and ME7-P) and RNase Out-treated (RML-PRNaseOut and ME7-PRNaseOut) brain homogenates used as substrates. Similar to the experiment depicted in Fig. 1, amplification of ME7 in the absence of RNA was lost after 5 rounds of saPMCA, while RML amplification was unchanged for up to 8 rounds. Despite a decrease in conversion efficiency in RNA-depleted substrates, RML conversion was maintained until round 13 of saPMCA. Propagation of RML and ME7 in control substrates was efficiently maintained over 13 and 18 rounds, respectively (data not shown).
PrPSc levels of RML prions serially amplified in vitro were evaluated by Western blot analysis and compared to brain-derived RML preparations (Fig. 5A). Intriguingly, the PrPSc glycosylation pattern in RML-PRNaseA samples was more similar to that observed in brain homogenate samples than RML-P and RML-PRNaseOut samples. We performed PrP deglycosylation experiments to analyze the proportion of truncated PrP species C1 (generated from PrPC in the absence of prion infection) and C2 (generated from PrPSc during prion infection) (6, 38). The C1 level was significantly higher in RML-PRNaseA than in RML-PRNaseOut and RML-P samples, whereas brain-derived RML exhibited a high level of C2 (Fig. 5B). The higher level of C1 or C2 in both preparations is likely to account for the similar pattern observed in the complex glycosylated samples in Fig. 5A. Higher C2 levels in the infected brain homogenate than in PMCA samples is likely due to the activity of endogenous PrPSc-cleaving enzymes, as described previously (14, 38), whereas protease inhibitors present in the PMCA reactions prevent this activity. At this time, we can only speculate about the reasons underlying the difference in the proportions of full-length PrP to C1 observed between RML-PRNaseA and the other PMCA samples. Absence of RNA binding to PrP might lead to a loss of steric protection or to a conformational change rendering PrP slightly more sensitive to the limited proteolytic cleavage that occurs despite the presence of protease inhibitors. To investigate if the absence of RNA during RML conversion affected its strain-specific characteristics, 1/10 dilutions of RML-PRNaseA were prepared and titrated by intracerebral inoculation into Tga20 mice. Incubation periods were not statistically different from those resulting from titration of control samples performed in parallel (Fig. 6 and Table 1). As expected, none of the NBH samples treated with either RNase A or RNase Out caused prion disease in inoculated mice. Likewise, NBHs subjected to 18 rounds of saPMCA in the absence of prion seeds were not infectious to Tga20 mice, ruling out the spontaneous generation of PrPSc under these conditions (Fig. 6 and Table 1). We did not detect infectivity in any of the ME7-PRNaseA samples, showing that the absence of RNA abolished both in vitro PrPSc conversion and amplification of infectious ME7 prions (Fig. 6). Treatment of brain-derived RML with RNase A or RNase Out for 1 h at 37°C prior to mouse inoculation had no effect on the disease incubation and clinical manifestation (Table 1; compare RML-B 10−5 with RML-BRNaseA and RML-BRNaseOut 10−5; P = 0.17 as determined by one-way ANOVA). However, an atypical phenotype characterized by forelimb paresis (see movies S1 and S2 in the supplemental material) was observed in groups RML-PRNaseA (12 out of 22 mice) and RML-PRNaseOut (2 out of 22 mice). The higher incidence of this phenotype in the RML-PRNaseA group was statistically significant (P = 0.0012 as determined by chi-square). The RML-PRNaseA inoculum produced incubation times in Tga20 comparable to those of RML-PRNaseOut and RML-P inocula (Fig. 5A and Table 1). We measured specific infectivity of PrPSc in samples generated with and without RNAs. Our estimations indicated that PrPSc in RML-PRNaseA samples was associated with 3.5 times more infectivity than that in control samples (Table 2); however, this difference cannot be considered significant given the ±1 log degree of accuracy of animal bioassays.
Fig 6.

Curves representing the survival times resulting from the inoculation of PMCA material generated in the presence or absence of RNA for Tga20 mice. One mouse in each of groups NBHRNaseA and NBH-P died of intercurrent disease. Due to overlapping curves, some data points cannot be observed in the graph. NBHRNaseA, NBH treated with RNase A; NBHRNaseOut, NBH treated with RNase Out; NBH-P, NBH subjected to 18 rounds of PMCA; RML-PRNaseA, RML amplified by PMCA in the presence of RNase A; RML-PRNaseOut, RML amplified by PMCA in the presence of RNase Out; RML-P, RML amplified by PMCA under standard conditions; RML-B, brain-derived RML; ME7-PRNaseA, ME7 amplified by PMCA in the presence of RNase A.
To further test the possibility of the emergence of a new RML-derived prion strain, we performed intracerebral inoculations of PMCA material amplified in the presence (RML-P) or absence (RML-PRNaseA) of RNA into wild-type C57BL/6 mice. We evaluated the infectivity of RML-PRNaseA by inoculating the same amount of PrPSc in those and in PMCA control samples (RML-P) and comparing disease incubation times. We found clinical endpoints at 159 ± 1 days after RML-P inoculation and 154 ± 2 days after RML-PRNaseA injection, and we found classical RML clinical signs in 100% of the mice (Table 3), suggesting that RML-PRNaseA and RML-P are the same prion strain. Secondary transmissions in C57BL/6 or Tga20 mice of RML-PRNaseA propagated in C57BL/6 and Tga20 mice, respectively, were characterized by typical clinical signs in all mice and incubation periods of 141 ± 4 and 66 ± 2 days, respectively, very similar to the 144 ± 5.5 and 59 days induced by brain-derived RML (Table 4) (18).
Table 3.
Survival times of C57BL/6 mice intracerebrally inoculated with RML-P and RML-PRNaseAa
| Inoculum | Incubation (dpib) | No. sick/no. inoculated |
|---|---|---|
| RML-P | 159 ± 1 | 6/6 |
| RML-PRNaseA | 154 ± 2 | 6/6 |
Densitometric calculations of the Western blots shown in Fig. 5A indicated that PMCA-generated RML (RML-P) contains 3 times more PrPSc than PMCA-generated RML under RNA-depleted conditions (RML-PRNaseA); therefore, to inject equal amounts of PrPSc, we inoculated a 10−1 dilution of RML-PRNaseA and a 3 × 10−2 dilution of RML-P.
dpi, days postinoculation, expressed as averages ± standard errors of the means.
Table 4.
Survival times after secondary transmission of RML-PRNaseA in C57BL/6 and Tga20 micea
| Mouse strain | Inoculum/origin | Incubation (dpib) | No. sick/no. inoculated |
|---|---|---|---|
| C57BL/6 | RML-PRNaseA/C57BL/6 | 141 ± 4 | 6/6 |
| Tga20 | RML-PRNaseA/Tga20 | 66 ± 2 | 6/6 |
Brain homogenates (1%) of C57BL/6 and Tga20 mice infected with PMCA-generated RML under RNA-depleted conditions (RML-PRNaseA) were inoculated intracerebrally into C57BL/6 and Tga20 mice, respectively.
dpi, days postinoculation, expressed as averages ± standard errors of the means.
To investigate if the forelimb paresis induced by RML-PRNaseA inoculation reflected a change in neurotropism, we conducted a blind immunohistochemical analysis of Tga20 and C57BL/6 mouse brains inoculated with RML-PRNaseA as well as RML-P. Analysis of sections of the hippocampus, cortex, thalamus, cerebellum, medulla, and pons from these animals did not reveal any differences in vacuolation patterns (Fig. 7A and B). We also failed to detect differences in PrPSc accumulation patterns, suggesting that RML was not modified by replication in the absence of RNA (Fig. 7C). In addition, we examined RML properties in vitro in an extended cell panel assay (ECPA) (3, 22, 25). This assay examines the capacity of a given prion strain to infect different cell types in culture and its susceptibility to different pharmacological inhibitors of prion replication. These responses have been shown to vary from one strain to another and can even be used to distinguish between “substrains” of prions. PMCA-generated material was either ultracentrifuged (100,000 × g, 1 h, 4°C) or diluted 2,000-fold prior to cell inoculation to reduce Triton X-100 levels contained in these preparations that are toxic to cultured cells. Unfortunately, these procedures yielded preparations with very low titers of infectivity that hindered us from drawing any conclusions (data not shown). Alternatively, we used Tga20 mouse brain homogenates infected with RML-PRNaseA, RML-PRNaseOut, and original RML. All RML-related inocula showed similar properties in the ECPA. ME7 and 22L properties in the ECPA are shown for reference (Fig. 8).
Fig 7.

Comparison of the histopathological features in the brains of mice inoculated with RML prions amplified in vitro in the presence or absence of RNAs. (A and B) Hematoxylin and eosin staining. The profiles of spongiform degeneration are similar in hematoxylin-eosin-stained sagittal sections prepared from brains of C57BL/6 mice inoculated with equivalent amounts of RML PrPSc generated by PMCA in the absence (A) and presence (B) of RNAs. Scale bars, 100 μm except for the sagittal brain section, where the bar is 300 μm. (C) PrPSc staining. The PrPSc accumulation pattern in brains of Tga20 mice inoculated with RML PrPSc generated by PMCA in the absence (left) or presence (right) of RNA was visualized by staining with the anti-PrP monoclonal antibody Bar233 as described in Materials and Methods. Scale bar, 100 μm.
Fig 8.
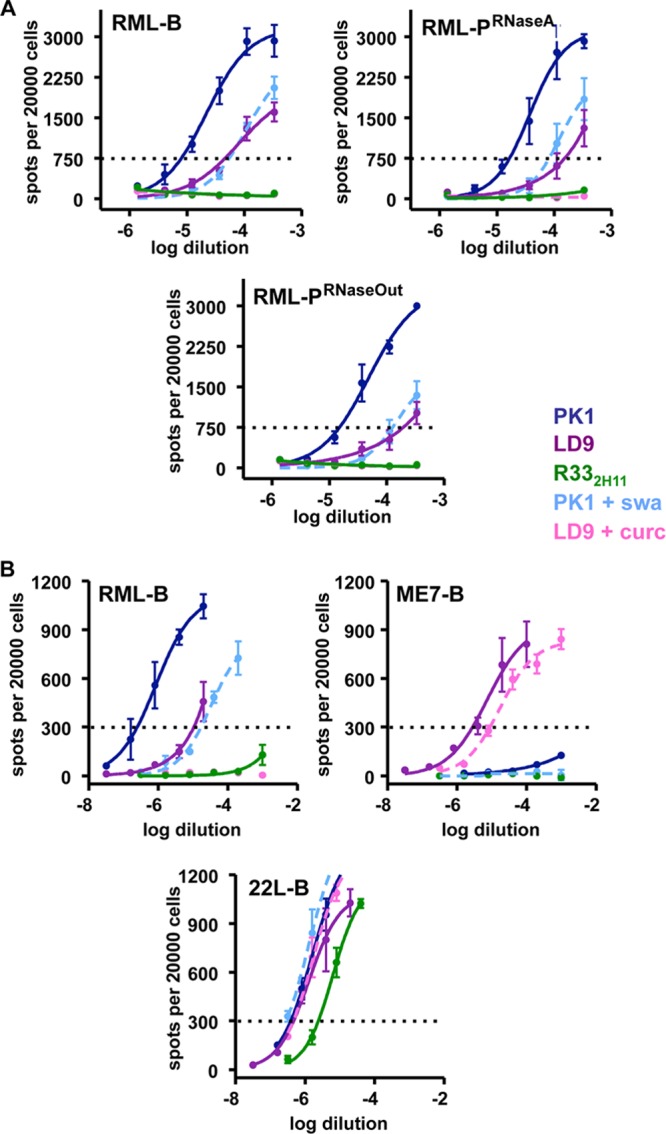
Extended cell panel assay (ECPA). (A) PMCA-generated material was passaged into Tga20 mice and used to infect PK1 (blue), LD9 (purple), and R332H11 (green) cell lines subjected to several pharmacological treatments affecting prion replication in a strain-specific fashion. Amplification of RML in the absence of RNA did not change strain properties in the ECPA. (B) ECPA profiles of brain-derived RML (RML-B) ME7 (ME7-B), and 22L (22L-B) strains are shown in comparison. Panels A and B represent two different experiments. Prion inhibitors included curcumin (cur; pink dashed line) and swainsonine (swa; light blue dashed line). The horizontal dotted line indicates the number of positive cells (“spots”) used to calculate the index of response of each cell line to a particular prion strain and pharmacological treatment (shown in Table S1 in the supplemental material).
DISCUSSION
In the present study, we showed that RNA enhances in vitro amplification of several mouse prions strains: RML, 79A, ME7, 139A, 22L, 22F, 303, 766, and 726 (the last three murine prion strains were generated by PMCA, a generous gift of J. Castilla). Therefore, the effect of RNA as a cofactor for PMCA is not restricted to hamster prions. Deleault et al. previously reported an absence of effect of RNase treatment on the amplification of RML and 22L scrapie strains (10). This difference might be due to the fact that we used an 80-times-higher dose of RNase A and that the PMCA reaction was performed in the presence of beads, a condition that was shown to enhance in vitro conversion in general as well as the RNA stimulating effect on in vitro amplification by a mechanism that has yet to be determined (15).
However, the extent of reduction of amplification efficacy by RNA depletion varied from one strain to another. The effect of RNA depletion on conversion efficiency could be best appreciated by the serial seed dilution approach. Amplification efficiency was assessed by the number of 6-fold dilutions of the inoculum that could be performed while still allowing in vitro conversion. Even two RML-derived strains, 79A and 139A, exhibited a 2-log difference in their responses to RNA depletion. Importantly, the sensitivity to RNA depletion was independent of the intrinsic, and strain-specific, in vitro conversion capacity. For example, RML and 79A, two strains with high in vitro conversion efficiency, were more RNA sensitive than the less PMCA-efficient strains 139A, 303, 726, and 22F. Two strains with similar conversion efficiencies, namely, 766 and 139A, exhibited different responses to RNA depletion. The effect of RNA depletion on ME7 could not be quantitatively assessed due to the low conversion efficiency of this strain. Similar to previous findings with hamster prions (10, 15), we could not find a mouse prion strain that is completely unresponsive to RNA depletion. Reconstitution of the reaction mixtures with brain or liver RNA restored in vitro amplification. We did not observe any difference between the effects of large- and small-RNA populations. Another strain-specific phenomenon was the capacity to utilize polyanions other than endogenous RNA for replication. RML amplification in RNA-depleted substrates was restored by the addition of poly(A), poly(G), or DNA, whereas ME7 amplification could be restored by poly(A) but not poly(G) or DNA. A previous study showed that infectious PrPSc can be generated in vitro from recombinant PrP amplified in the presence of poly(A) and lipids (34). Our results demonstrate a strain-specific ability to utilize a variety of cofactors. It is logical to stipulate that this phenomenon underlies the strain-specific effect of RNA depletion, with the more promiscuous strains being less affected than those strictly dependent on RNA or certain RNA structures for PrPC-to-PrPSc conversion.
The role of cofactors in prion strain determination, although hypothesized a long time ago (35), has never been specifically addressed. We exploited the fact that RML could amplify in vitro in the absence or quasiabsence of RNA to determine if RNA confers strain-specific properties. We amplified ME7 and RML over 18 and 13 rounds, respectively, to a level where no initial inoculum was present in the sample anymore and inoculated Tga20 as well as wild-type mice with the amplification product. We cannot exclude the presence of undetectable amounts of RNA in the PMCA reactions, but these would be out of proportion with the amounts of newly amplified PrPSc. ME7 PMCA samples from RNA-depleted PMCA conditions were devoid of detectable PrPSc and infectivity, showing that loss of PrPSc was accompanied by a loss of infectivity. In vitro-amplified RML under standard and RNA-depleted conditions exhibited similar incubation periods and histopathological features in either Tga20 or C57BL/6 mice. The similarity between the two types of prions was confirmed using ECPA, which allows prion strain discrimination in cultured cells by exploiting the differential capacity of prion strains to replicate in different cell types and pharmacological conditions (3, 22, 25). Interestingly, we observed an unusual clinical symptom (forelimb paresis) with a statistically higher incidence in the group of mice inoculated with RML amplified under RNA-depleted conditions than in the standard PMCA group or the group that received RML brain homogenate. This observation might be an epiphenomenon or might indicate a slight modification of the RML strain characteristics too small to be considered a strain change and be detected by the tools used in this study. Recent studies show that prion strains are not homogenous but are populated by substrains of prions detectable in vitro. A shift from the main substrain to another may account for a transient change of phenotype of a given strain under particular replication conditions (36). Since our PMCA-generated material could not be directly evaluated in the ECPA, we cannot rule out the possibility that a substrain of RML had been selected in vitro under RNA-depleted conditions and reverted to the original strain upon transmission to mice (23). Nonetheless, our results strongly suggest that RNA is not essential to maintain strain-specific characteristics of RML prions.
Our study suggests that RNA molecules behave like catalysts of in vitro amplification of prions, the extent of the catalytic effect being strain dependent, and that RNA molecules are not required to specify strain-specific information. Purified PrPSc amplified with recombinant PrP in the absence of addition of mammalian cofactor leads to the formation of infectivity at a relatively low level, as judged by some incomplete transmission rates and variability of survival times (19), which is in good agreement with the concept that RNAs constitute prion replication catalysts. As poly(A) (this study and reference 34) and, for one of two tested strains, poly(G) can reconstitute PMCA efficacy, it will be of high interest to evaluate strain characteristics of prions amplified under conditions of such synthetic catalysts.
The alternative hypothesis to that stipulating that a PrP-associated cofactor is strain defining is that strain characteristics are supported by differences in PrPSc conformation. Direct experimental evidence of strain-specific differences in PrPSc conformation has been provided (29, 30). Different PrPSc conformations are likely to result in variable PrPC seeding efficiencies, leading to different PrPSc amplification kinetics in vitro and replication efficiencies in vivo, and variable degrees of reliance on catalytic activity. Some PrPSc conformations may require highly specific and efficient RNA catalysts to overcome the energy barrier to PrPC conversion, while other PrPSc conformations might be less demanding in the type of polyanionic catalyst required, meaning that “weaker” catalysts might be sufficient for them to trigger PrPC-to-PrPSc conversion.
The question arises as to the cellular source of RNA able to catalyze PrPC-to-PrPSc conversion. RNA molecules, in particular noncoding RNAs, are present in the extracellular environment and body fluids either associated with proteins (90%) or within multivesicular bodies like exosomes (12, 40). Extracellular RNA may interact with PrP present in the outer leaflet of the plasma membrane and catalyze the conversion of PrPC into PrPSc either in cholesterol-rich microdomains (16, 31, 39) or along the endosomal recycling pathway (2, 24), two sites hypothesized to sustain prion replication.
Supplementary Material
ACKNOWLEDGMENTS
We thank Charles Weissmann for his insight and advice on the project and for critical reading of the manuscript. We thank Joaquín Castilla for the gift of the PMCA-generated murine prion strains and for bringing to our attention the use of beads for PMCA reactions. We thank Nicole Salès for help with histology and immunohistochemistry. We gratefully acknowledge the expert help of the Animal Resource Center personnel at Scripps Florida. We are grateful to Larisa Cervenakova for her comments on the manuscript.
Footnotes
Published ahead of print 18 July 2012
Supplemental material may be found at http://jvi.asm.org/.
REFERENCES
- 1. Bessen RA, Marsh RF. 1992. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66:2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borchelt DR, Taraboulos A, Prusiner SB. 1992. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267:16188–16199 [PubMed] [Google Scholar]
- 3. Browning S, et al. 2011. Abrogation of complex glycosylation by swainsonine results in strain- and cell-specific inhibition of prion replication. J. Biol. Chem. 286:40962–40973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruce ME. 1993. Scrapie strain variation and mutation. Br. Med. Bull. 49:822–838 [DOI] [PubMed] [Google Scholar]
- 5. Castilla J, Saa P, Hetz C, Soto C. 2005. In vitro generation of infectious scrapie prions. Cell 121:195–206 [DOI] [PubMed] [Google Scholar]
- 6. Chen SG, et al. 1995. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 270:19173–19180 [DOI] [PubMed] [Google Scholar]
- 7. Couzin J. 2004. Biomedicine. An end to the prion debate? Don't count on it. Science 305:589. [DOI] [PubMed] [Google Scholar]
- 8. Cronier S, et al. 2008. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem. J. 416:297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deleault NR, Harris BT, Rees JR, Supattapone S. 2007. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104:9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dickinson AG, Outram GW, Taylor DM, Foster JD. 1989. Further evidence that scrapie agent has an independent genome, p 446–459 In Court LA, Dormont D, Brown P, Kingsbury DT. (ed), Unconventional virus diseases of the central nervous system (Paris 2–6 December 1986). CEA Diffusion, Fontenay-aux Roses, France [Google Scholar]
- 12. Dinger ME, Mercer TR, Mattick JS. 2008. RNAs as extracellular signaling molecules. J. Mol. Endocrinol. 40:151–159 [DOI] [PubMed] [Google Scholar]
- 13. Doi S, et al. 1988. Western blot detection of scrapie-associated fibril protein in tissues outside the central nervous system from preclinical scrapie-infected mice. J. Gen. Virol. 69:955–960 [DOI] [PubMed] [Google Scholar]
- 14. Dron M, et al. 2010. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J. Biol. Chem. 285:10252–10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez-Montalban N, Makarava N, Savtchenko R, Baskakov IV. 2011. Relationship between conformational stability and amplification efficiency of prions. Biochemistry 50:7933–7940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goold R, et al. 2011. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeong BH, et al. 2009. Reduction of prion infectivity and levels of scrapie prion protein by lithium aluminum hydride: implications for RNA in prion diseases. J. Neuropathol. Exp. Neurol. 68:870–879 [DOI] [PubMed] [Google Scholar]
- 18. Karapetyan YE, et al. 2009. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One 4:e5730 doi:10.1371/journal.pone.0005730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim JI, et al. 2010. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285:14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lasmézas CI, et al. 2001. Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt-Jakob disease: implications for human health. Proc. Natl. Acad. Sci. U. S. A. 98:4142–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Legname G, et al. 2004. Synthetic mammalian prions. Science 305:673–676 [DOI] [PubMed] [Google Scholar]
- 22. Mahal SP, et al. 2007. Prion strain discrimination in cell culture: the cell panel assay. Proc. Natl. Acad. Sci. U. S. A. 104:20908–20913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahal SP, Browning S, Li J, Suponitsky-Kroyter I, Weissmann C. 2010. Transfer of a prion strain to different hosts leads to emergence of strain variants. Proc. Natl. Acad. Sci. U. S. A. 107:22653–22658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marijanovic Z, Caputo A, Campana V, Zurzolo C. 2009. Identification of an intracellular site of prion conversion. PLoS Pathog. 5:e1000426 doi:10.1371/journal.ppat.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oelschlegel AM, Fallahi M, Ortiz-Umpierre S, Weissmann C. 2012. The extended cell panel assay characterizes the relationship of prion strains RML, 79A, and 139A and reveals conversion of 139A to 79A-like prions in cell culture. J. Virol. 86:5297–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan KM, et al. 1993. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U. S. A. 90:10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144 [DOI] [PubMed] [Google Scholar]
- 28. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–496 [Google Scholar]
- 29. Safar J, et al. 1998. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4:1157–1165 [DOI] [PubMed] [Google Scholar]
- 30. Smirnovas V, et al. 2011. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18:504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taraboulos A, et al. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129:121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wadsworth JD, et al. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180 [DOI] [PubMed] [Google Scholar]
- 33. Wang F, Wang X, Yuan CG, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang F, et al. 2012. Genetic informational RNA is not required for recombinant prion infectivity. J. Virol. 86:1874–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weissmann C. 1991. A ‘unified theory’ of prion propagation. Nature 352:679–683 [DOI] [PubMed] [Google Scholar]
- 36. Weissmann C, Li J, Mahal SP, Browning S. 2011. Prions on the move. EMBO Rep. 12:1109–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Will RG, et al. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347:921–925 [DOI] [PubMed] [Google Scholar]
- 38. Yadavalli R, et al. 2004. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J. Biol. Chem. 279:21948–21956 [DOI] [PubMed] [Google Scholar]
- 39. Ying YS, Anderson RG, Rothberg KG. 1992. Each caveola contains multiple glycosyl-phosphatidylinositol-anchored membrane proteins. Cold Spring Harbor Symp. Quant. Biol. 57:593–604 [DOI] [PubMed] [Google Scholar]
- 40. Zhu H, Fan GC. 2011. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am. J. Cardiovasc. Dis. 1:138–149 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.