

Abstract
MicroRNAs (miRNAs) have been implicated in the pathogenesis and progression of brain tumors. miR-21 is one of the most highly overexpressed miRNAs in glioblastoma multiforme (GBM), and its level of expression correlates with the tumor grade. Programmed cell death 4 (PDCD4) is a well-known miR-21 target and is frequently downregulated in glioblastomas in accordance with increased miR-21 expression. Downregulation of miR-21 or overexpression of PDCD4 can inhibit metastasis. Here, we investigate the role of heterogeneous nuclear ribonucleoprotein C1/C2 (hnRNPC) in the metastatic potential of the glioblastoma cell line T98G. hnRNPC bound directly to primary miR-21 (pri-miR-21) and promoted miR-21 expression in T98G cells. Silencing of hnRNPC lowered miR-21 levels, in turn increasing the expression of PDCD4, suppressing Akt and p70S6K activation, and inhibiting migratory and invasive activities. Silencing of hnRNPC reduced cell proliferation and enhanced etoposide-induced apoptosis. In support of a role for hnRNPC in the invasiveness of GBM, highly aggressive U87MG cells showed higher hnRNPC expression levels and hnRNPC abundance in tissue arrays and also showed elevated levels as a function of brain tumor grade. Taken together, our data indicate that hnRNPC controls the aggressiveness of GBM cells through the regulation of PDCD4, underscoring the potential usefulness of hnRNPC as a prognostic and therapeutic marker of GBM.
INTRODUCTION
Glioblastoma multiforme (GBM) is the most aggressive type of primary brain tumor, associated with high mortality and morbidity. Gliomas are classified into four grades: grades I and II are less aggressive astrocytomas, and grades III and IV are gliomas, malignant tumors with high proliferation rates. Despite advanced therapeutics for GBM, including surgery, radiation, and chemotherapy, the prognosis of GBM patients has not improved (31).
MicroRNAs (miRNAs) are short RNAs of about 22 nucleotides in length and play critical roles in various cellular processes, including proliferation, differentiation, tumorigenesis, and metastasis. They typically function as negative posttranscriptional regulators of gene expression by binding to target mRNAs (often at their 3′ untranslated regions [3′UTRs]) and triggering target mRNA cleavage and translational suppression (7). Based on bioinformatic analysis, 30% of all human genes may be regulated by miRNAs (4). A number of miRNAs have been closely associated with different aspects of tumorigenesis, including metastasis, raising the possibility that miRNAs could serve as diagnostic and prognostic markers (12, 17). Among the cancer-associated miRNAs, miR-21 is frequently overexpressed in diverse types of cancer, including breast cancer (21), liver cancer (43), pancreatic cancer (38), and glioblastoma (5), and plays a critical role in proliferation, differentiation, and apoptosis. Recently, it was reported that downregulation of miR-21 inhibits tumorigenicity (15), invasion (1, 20, 49), and metastasis (49). Aberrant expression of miR-21 contributes to the malignancy of cancer cells through the regulation of apoptosis- and metastasis-related genes, and it increases drug resistance, a major cause of cancer recurrence (30). A number of miR-21 target transcripts have been reported, including those that encode programmed cell death 4 (PDCD4), PTEN, TPM1, maspin, SPRY, and RECK.
PDCD4 was first identified as a protein that was upregulated during apoptosis and suppressed tumorigenesis (10, 26). Loss of PDCD4 expression was closely associated with the progression and prognosis of various tumors, including glioblastomas (16) and cancers of the lung, ovary, and kidney (6, 30, 45). Through its MA-3 domain, PDCD4 interacts with the translation factors eIF4A and sIF4G and functions as a translational suppressor, potently influencing protein expression patterns. In addition, high expression of PDCD4 inhibits AP-1 activity by suppressing Jun N-terminal protein kinase (JNK)-mediated c-Jun phosphorylation (2). Conversely, silencing of PDCD4 induces the expression of p21 and other p53 target genes and inhibits UV-induced apoptosis (3). Diminished expression of PDCD4 is further implicated in the metastatic potential of tumor cells by increasing the levels of urokinase-type plasminogen activator receptor (uPAR), which degrades extracellular matrix proteins (29). PDCD4 expression is regulated via two main mechanisms: miR-21 binds the PDCD4 3′UTR and suppresses PDCD4 translation, and PDCD4 is phosphorylated by Akt and p70S6K at serine 67 and 457 (35), leading to ubiquitinylation of PDCD4 via the ubiquitin ligase β-TRCP and degradation by the proteasome (13).
Heterogeneous nuclear ribonucleoprotein C1/C2 (hnRNPC) is a ubiquitously expressed RNA-binding protein (RBP) that consists of two isoforms, hnRNPC1 and hnRNPC2, with hnRNPC1 being the more abundant form and hnRNPC2 being the larger splice variant, with an additional 13 amino acids (9). hnRNPC influences various aspects of mRNA metabolism, including pre-mRNA processing, mRNA transport, and stabilization (8, 36, 39). hnRNPC can also enhance translation via internal ribosome entry sites (IRESs) (24) and promote amyloid precursor protein (APP) translation by competing with the RBP fragile X mental retardation protein (FMRP) for recruitment of APP mRNA to processing (P) bodies (28).
Here, we investigated the role of hnRNPC in the progression of GBM. We found that hnRNPC was capable of binding to primary miR-21 (pri-miR-21) and induced miR-21 expression; the ensuing inhibition of Akt and p70S6K pathways led to increased PDCD4 expression in the glioblastoma cell line T98G. We further discovered that hnRNPC silencing enhanced apoptosis of T98G cells, particularly after treatment with etoposide. Importantly, the hnRNPC levels correlated with the metastatic activity of T98G and hnRNPC was overexpressed in the highly invasive cell line U87MG (U87R4) and in high-grade (grades III and IV) gliomas.
MATERIALS AND METHODS
Reagents and antibodies.
Actinomycin D, cycloheximide, and MG-132 were from Sigma-Aldrich and etoposide was from Calbiochem. Antibodies recognizing PDCD4 and hnRNPC1C2 were from Santa Cruz Biotechnology; the antibody recognizing Flag was from Sigma-Aldrich; the antibodies recognizing cleaved PARP, PTEN, p-Akt, p-p70, and p-p6 were from Cell Signaling; and the antibody recognizing the loading control protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Abcam.
Cell culture and transfection.
T98G, U87MG, U87L4, and U87R4 cells were maintained at 37°C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (GIBCO-BRL) and 1% antibiotic-antimycotic solution (GIBCO-BRL); U87L4 and U87R4 cells were kindly supplied by Do-Hyun Nam (Samsung Medical Center, Seoul, Republic of Korea). For transfection, cells were plated at a density of 2 × 105 cells/dish and transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Small interfering RNAs (siRNAs) directed to hnRNPC and PDCD4 were from Santa Cruz Biotechnology, and control siRNA was synthesized by Genolution (Seoul, Republic of Korea). Precursor miR-21 (pre-miR-21) and antisense miR-21 (anti-miR-21) were from Ambion.
RNP analysis: RNP IP and biotin pulldown experiment.
To check the interaction between pri-miR-21 and hnRNPC, RNP immunoprecipitation (IP) was used as previously described (23). Briefly, transfected cells were lysed with polysome lysis buffer, and lysates were divided into two parts that were incubated with protein A-Sepharose beads coated with 10 μg each of mouse IgG or mouse hnRNPC antibody for 1 h at 4°C. The beads were then washed three times with NT2 buffer (23), the RNA was isolated after incubation with 20 U RNase-free DNase I (15 min, 30°C) and 0.5 mg/ml proteinase K (15 min, 55°C) to eliminate contaminating DNA and to degrade protein, respectively. For biotin pull down analysis, cDNA from T98G was used as the template for synthesizing biotinylated transcripts spanning pri-miR-21. Each forward primer contained the T7 RNA polymerase promoter sequence as described previously (23). Whole-cell lysates prepared from T98G cells were incubated with purified biotinylated transcripts, and the complex of hnRNPC-biotin RNA was isolated by binding with streptavidin-coupled Dynabeads (Invitrogen). The level of hnRNPC in IP materials was determined by Western blot analysis.
Western blot and quantitative real-time PCR.
For Western blot analysis, cells were rinsed with ice-cold phosphate-buffered saline (PBS) and lysed with radioimmunoprecipitation (RIPA) buffer containing protease inhibitors and phosphatase inhibitors (Roche). Cell debris was removed by centrifugation (12,000 × g for 10 min at 4°C), and the protein concentration of the supernatant was quantified using Bradford reagent (Sigma-Aldrich). Proteins were fractionated in SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were incubated with primary antibodies overnight, washed with Tris-buffered saline (TBS), incubated with appropriate secondary antibodies, and visualized using an enhanced chemiluminescence (ECL) solution (Amersham). For reverse transcription (RT) and quantitative real-time PCR (qPCR), total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's procedure and used for the synthesis of cDNA using SuperScript III reverse transcriptase (Invitrogen). mRNA expression levels were quantified by RT-qPCR (ABI Prism 7600) using power SYBR green PCR master mix (Applied Biosystems, Foster City, CA). Primer sequences are shown in Fig. S1 in the supplemental material.
Migration and invasion assay.
Twenty-four hours after transfection, the migratory activity was determined by assessing the ability of the cells to cross the pores of migration chambers in the presence of chemoattractants (QCM colorimetric cell migration assay kit; Millipore). Briefly, transfected cells were cultured in serum-free medium overnight, harvested, and resuspended in serum-free medium at a concentration of 1 × 106 cells/ml. The cell suspension in serum-free medium (0.3 ml = 3 × 105 cells) was added into the upper chambers of the 24-well Falcon cell culture transwell plates (8-μm pore size), and serum-containing medium (0.6 ml) was added into the lower chambers to stimulate cell migration. After incubation at 37°C overnight, cells on the surface of the polycarbonate membrane (nonmigrating cells) were removed by scraping with a cotton swab, and migrated cells were stained for 20 min. The stained cells were eluted with extraction buffer, and then the optical density (at 560 nm) was quantified. For the invasion assay, we used Matrigel transwell plates (BD Biocoat Matrigel invasion chamber). Transfected cells were suspended in culture medium, and 5 × 104 cells (0.5 ml) were added to the upper chamber. By adding 0.7 ml of DMEM containing 10% fetal bovine serum into the lower compartment, cells were stimulated to invade. Cells were subsequently fixed with 100% methanol and stained with 0.1% crystal violet. Following several washes with water, the membranes were removed and mounted on a microscope slide. The average number of invading cells on the lower surface of the membranes was determined under a microscope by scoring several fields.
Immunohistochemical analysis.
For tissue microarray (TMA), we employed TMA slides (NGL481) containing 24 cases in duplicate with various grades and stages of brain tumors (Pantomics, Richmond, CA). The slides were subjected to heat-induced epitope retrieval, incubation with mouse monoclonal anti-hnRNPC antibody overnight, and subsequent incubation with the corresponding anti-mouse secondary antibody. Stained slides were imaged using a ScanScope CS system (Aperio, Vista, CA), and regions of interest (ROI) were selected for each slide using Image J-based macros. The levels of hnRNPC expression were determined using the LSAB+ system (Dako, Carpinteria, CA). A count of all pixels above an arbitrary threshold was calculated in order to exclude background staining and to establish a mean threshold of staining.
RESULTS
hnRNPC is able to bind to pri-miR-21 and regulates miR-21 expression.
We surveyed RNA-binding proteins (RBPs) that might regulate miR-21 expression by employing an RNP IP assay using several anti-RBP antibodies. Briefly, RNP complexes present in T98G cell lysates were immunoprecipitated using RBP antibodies that recognized various RBPs or the corresponding control IgG. The levels of primary miR-21 (pri-miR-21) in the IP materials were determined by RT-qPCR using four different sets of primers (see Fig. S1 in the supplemental material); since each set of primers showed similar results, we represent here the results using primer set 2. pri-miR-21 is RNA transcribed from within an intron of host gene VMP1 (vacuole membrane protein 1, also termed transmembrane protein 49 [TMEM49]) (14). Among several RBPs tested, pri-miR-21 was selectively enriched in hnRNPC IP (Fig. 1A; also see Fig. S2A in the supplemental material), and therefore, we chose hnRNPC for further study. The specificity of the interaction between pri-miR-21 and hnRNPC was further assessed by Western blot analysis of hnRNPC in hnRNPC IP samples and by comparing the levels of other pri-miR transcripts in the same IP complexes (see Fig. S2B to D). Since hnRNPC associated with pri-miR-21, we first studied the effect of hnRNPC silencing on miR-21 expression. As shown in Fig. 1B, the levels of pri-miR-21, pre-miR-21, and mature miR-21 decreased after hnRNPC silencing. In order to check whether silencing of hnRNPC influences general miRNA biogenesis, we assessed the levels of miRNAs in hnRNPC-silenced T98G cells by using a QuantiMir miRNA array kit (see Fig. S3 in the supplemental material). The levels of several miRNAs were affected by hnRNPC silencing; however, miR-21 showed the most significant change. These results suggested that hnRNPC probably regulated miR-21 expression by binding directly to pri-miR-21. To map the region of pri-miR-21 to which hnRNPC binds, we synthesized biotinylated transcripts representing six fragments of pri-miR-21 (see Fig. S4 in the supplemental material) and used them for biotin pull-down analysis. Briefly, each of six fragments (each ∼600 bp) was incubated with cell lysates, whereupon the level of hnRNPC was determined by Western blot analysis (Fig. 1C). The strongest interaction was seen with fragment C, which contained 3 AU-rich elements (AREs), distinct RNA sequences involved in altering mRNA stability and translation. To check if Drosha is involved in hnRNPC-mediated miR-21 regulation, protein-protein interaction between Drosha and hnRNPC was assessed by coimmunoprecipitation (co-IP) analysis. As shown in Fig. S5 in the supplemental material, there was no direct interaction between Drosha and hnRNPC, indicating that hnRNPC-mediated regulation of miR-21 did not result from interference with Drosha function.
Fig 1.

hnRNPC binds to pri-miR-21 and regulates miR-21 expression in T98G cells. (A) Lysates from T98G cells were immunoprecipitated with mouse IgG or mouse hnRNPC antibody. pri-miR-21 levels in IP were determined by RT-qPCR. GAPDH mRNA level in IP was used as normalization control. Data are means and standard deviations from three different experiments. (B) T98G cells were transfected with control (CTRL) or hnRNPC siRNA. At 48 h after transfection, levels of pri-miR-21, pre-miR-21, and mature miR-21 were determined by RT-qPCR. Data are means and standard deviations from three different experiments. (C) Six fragments of pri-miR-21 were synthesized in vitro and incubated with T98G cell lysates. RNP complexes of biotinylated transcripts and hnRNPC were detected using Western blot analysis. pri-miR-21, primary miR-21; pre-miR-21, precursor miR-21.
Silencing of hnRNPC increases the expression of PDCD4 and PTEN.
The expression of miR-21 is an important prognostic marker of GBM patients and correlates closely with the GBM grade. The experiments described above revealed that pri-miR-21 was a target of hnRNPC and that silencing of hnRNPC decreased the levels of pri-miR-21, pre-miR-21, and mature miR-21 (Fig. 1B). To check the effect of hnRNPC on two miR-21 targets, the mRNAs encoding PDCD4 and PTEN, T98G cells were transfected with control or hnRNPC siRNA and, 48 h later, protein and RNA were analyzed. Silencing of hnRNPC significantly increased the PDCD4 and PTEN mRNA and protein levels (Fig. 2A and B). To study whether this effect was seen in other human cell lines, two glioma cells (U87MG and A172) and cervical carcinoma HeLa cells were studied similarly. As shown in Fig. S6A in the supplemental material, in each case hnRNPC silencing increased the expression of PDCD4. Next, we tested the effect of miR-21 on the expression of PDCD4 and PTEN in T98G cells. Transfection of T98G cells with antisense miR-21 (anti-miR-21) increased the expression of PDCD4 and PTEN, and conversely, precursor miR-21 (pre-miR-21)-transfected cells showed decreased expression of both proteins (Fig. 2C). It was also found that increased expression of PDCD4 by hnRNPC silencing was reversed by overexpression of miR-21 (see Fig. S6B), and the stability of PDCD4 and PTEN mRNA was not influenced by hnRNPC silencing (see Fig. S7 in the supplemental material). From these results, we concluded that silencing of hnRNPC increased the expression of PDCD4 and PTEN at least in part by diminishing the expression of miR-21, a negative regulator of PDCD4 and PTEN.
Fig 2.

Silencing of hnRNPC increases expression of PDCD4 and PTEN. (A, B) T98G cells were transfected with siRNAs shown, and 48 h later, the expression of PDCD4 and PTEN was determined by Western blot analysis (A) and the levels of PDCD4 and PTEN mRNAs by RT-qPCR analysis (B). Data are means and standard deviations from three different experiments. (C) T98G cells were transfected with control (CTRL) siRNA, anti-miR-21, or pre-miR-21, and 48 h later, whole-cell lysates were prepared and the levels of hnRNPC, PDCD4, and PTEN were determined by Western blot analysis. anti-miR-21, antisense miR-21.
Silencing of hnRNPC increases PDCD4 protein stability by inhibiting the Akt and p70S6K pathways.
In addition to miR-21-mediated regulation, PDCD4 expression is influenced by its phosphorylation status. Akt and p70S6K phosphorylate PDCD4 at serine 457 and serine 67, respectively, and phosphorylated PDCD4 is a substrate of ubiquitinylation by β-TRCP ubiquitin ligase, causing its degradation via the proteasome or its translocation to the nucleus (13, 35). To investigate in detail the mechanism whereby hnRNPC silencing increased PDCD4 expression, we checked whether hnRNPC silencing inhibited the Akt and p70S6K pathways. T98G cells were transfected with control or hnRNPC siRNAs, and 48 h later, whole-cell lysates were prepared and the levels of p-Akt and p-p70S6K were determined by Western blot analysis. As shown in Fig. 3A, silencing of hnRNPC diminished the phosphorylated forms of Akt and p70S6K, indicating that hnRNPC affects the Akt and p70S6K activation pathways. To investigate the role of Akt in PDCD4 stability, we tested the effect of inhibiting phosphatidylinositol 3-kinase (PI3K) using wortmannin; as shown in Fig. 3B, T98G cells treated with wortmannin showed increased expression of PDCD4, as reported previously. Since the Akt and p70S6K pathways were reported to influence PDCD4 protein stability, we measured the stability of PDCD4 protein after hnRNPC silencing. T98G cells were transfected as described above and treated with cycloheximide (10 μg/ml) to block protein synthesis. At the times indicated in Fig. 3C, cells were harvested, whole-cell lysates prepared, and the levels of PDCD4 and GAPDH determined by Western blot analysis. While control siRNA-transfected cells showed dramatic decreases in PDCD4 levels by 40 min of incubation in cycloheximide, PDCD4 abundance in hnRNPC-silenced cells remained elevated for a longer time period (up to 60 min) (Fig. 3C). Given these results, we propose that silencing of hnRNPC increased PDCD4 expression in two ways: one by lowering miR-21 expression, and the other by suppressing the Akt and p70S6K pathways.
Fig 3.

Silencing of hnRNPC increases the stability of PDCD4 protein by inhibiting the Akt and p70S6K pathways. (A) T98G cells were transfected with control (CTRL) or hnRNPC siRNAs; 48 h later, whole-cell lysates were prepared and the levels of hnRNPC, PDCD4, phosphorylated Akt (p-Akt), Akt, p-p70S6K, and p-S6 were determined by Western blot analysis. (B) T98G cells were treated with various concentrations of PI3K inhibitor wortmannin; 24 h later, whole-cell lysates were prepared and the levels of PDCD4 and p-Akt were determined by Western blot analysis. (C) To check whether hnRNPC influenced the protein stability of PDCD4, T98G cells were transfected with control (CTRL) or hnRNPC siRNAs; 24 h later, cells were transferred into 6-well plates, treated with cycloheximide (CHX; 10 μg/ml), and harvested at the indicated times. The levels of PDCD4 and GAPDH were determined by Western blot analysis. Data are means and standard deviations from three different experiments.
Silencing of hnRNPC inhibits proliferation of T98G cells and enhances etoposide-induced apoptosis.
Although many miRNAs are downregulated in tumor cells, miR-21 is usually overexpressed and promotes the proliferation of cancer cells (40). To investigate the effect of hnRNPC silencing on the proliferation of T98G cells, control or hnRNPC siRNAs were transfected into them; 48 h later, counting of viable cells using trypan blue revealed that hnRNPC silencing inhibited the proliferation of T98G cells (Fig. 4A). Since the reduction of miR-21 expression induces apoptosis in glioma cells by activating caspase-3 and -9 (48), we checked the effect of hnRNPC silencing on apoptosis. T98G cells were transfected with control or hnRNPC siRNA, and 24 h later, they were treated with dimethyl sulfoxide (final 0.1%) or etoposide (50 μM, which is the 50% inhibitory concentration [IC50] of etoposide) (see Fig. S8A in the supplemental material) for 16 h, whereupon whole-cell lysates were prepared and apoptosis was determined by measuring the levels of cleaved poly(ADP-ribose) polymerase (PARP). As shown in Fig. 4B and also in Fig. S8B and C, hnRNPC silencing induced apoptosis and enhanced etoposide-induced apoptosis. In summary, hnRNPC influenced the apoptosis of T98G cells by modulating miR-21 and PDCD4 levels, in keeping with previous evidence that downregulation of miR-21 decreased proliferation and induced apoptosis of glioblastoma cells (5).
Fig 4.
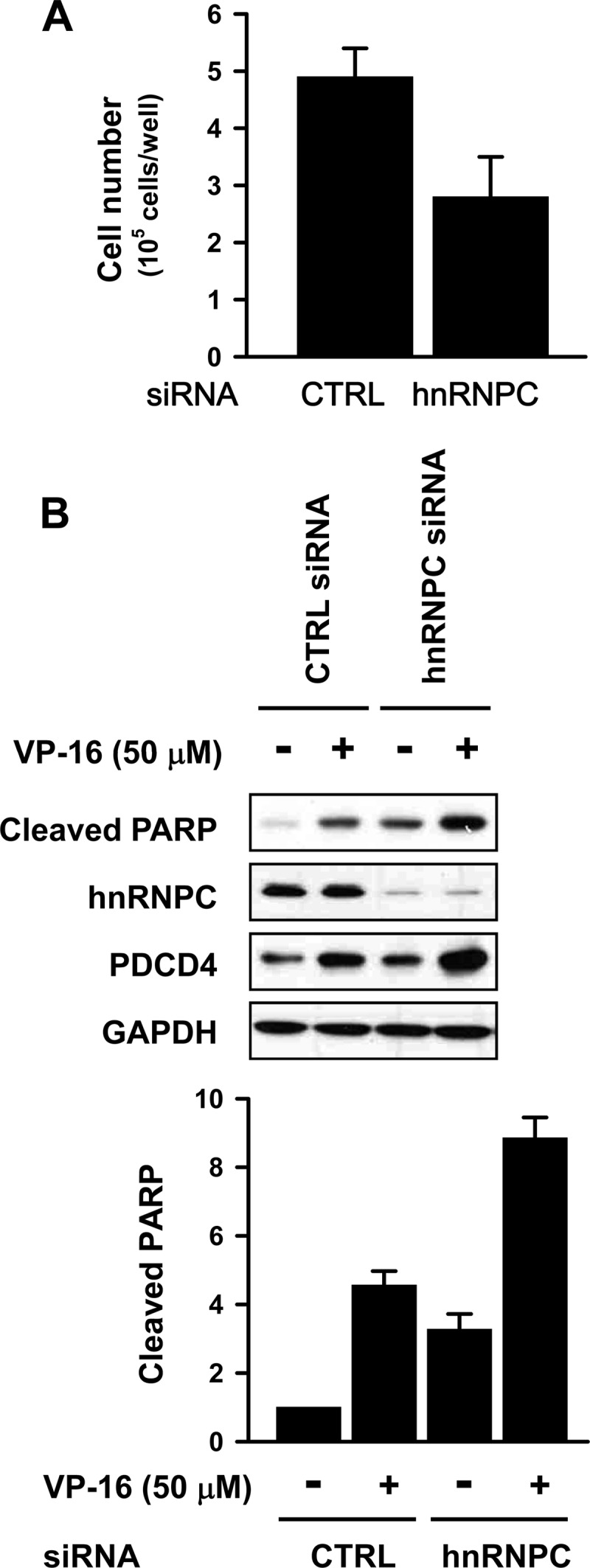
Silencing of hnRNPC inhibits proliferation and enhances etoposide-induced apoptosis. (A) T98G cells were transfected with control (CTRL) or hnRNPC siRNA; 24 h later, cells were counted and equal cell numbers were placed into 12-well plates. After an additional 48 h of incubation, cells were counted in the presence of trypan blue. Data are means and standard deviations from three different experiments. (B) T98G cells were transfected with control (CTRL) or hnRNPC siRNAs, and 24 h later, cells were transferred into 6-well plates and treated with etoposide (VP-16; 50 μM) for 16 h. Apoptosis was assessed by Western blot analysis of the levels of cleaved PARP. Data are means and standard deviations from three different experiments.
Silencing of hnRNPC suppresses the metastatic potential of T98G cells.
PDCD4 was reported to inhibit migration and invasion by regulating urokinase receptor (uPAR) and by suppressing the Akt pathway (29, 44), while miR-21 modulates the invasion of hepatocellular carcinoma, colorectal cancer, and breast cancer (1, 11, 20, 49). We investigated the effect of hnRNPC silencing on the migration and invasion of T98G cells. T98G cells were transfected with control or hnRNPC siRNA, and 24 h later, cells were counted and equal cell numbers were added into transwell and Matrigel transwell plates to study migration and invasion, respectively. Silencing of hnRNPC suppressed the migratory and invasive activity of T98G cells (Fig. 5A and B), further supporting the notion that hnRNPC can regulate the metastatic potentials of T98G cells, including migration and invasion, by regulating PDCD4 expression, as reported earlier (37).
Fig 5.
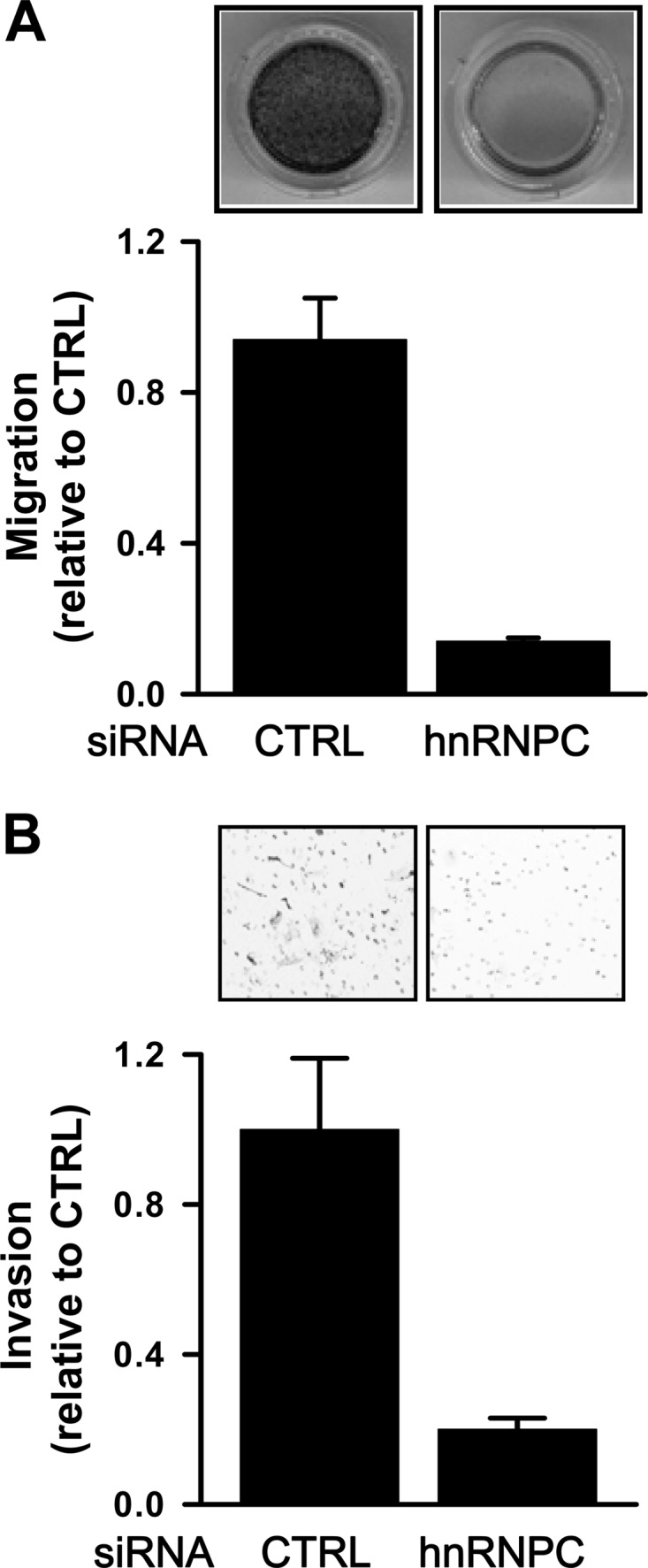
Silencing of hnRNPC suppresses the metastatic potential of T98G cells. (A) T98G cells were transfected with control (CTRL) or hnRNPC siRNAs; 24 h later, cells were counted and equal numbers of cells were added into transwell plates (24 wells, 8-μm pore size). After nonmigrated cells were completely removed, migrated cells were stained and photographed. The inhibitory effect of hnRNPC silencing on migration was determined by measuring signal absorbance at 560 nm. Data are means and standard deviations from three different experiments. (B) At 24 h after T98G cells were transfected as described above and equal numbers of cells added into Matrigel transwell plates, invading cells were fixed with 100% methanol, stained with 0.1% crystal violet, and counted in several fields using a microscope. Data are means and standard deviations from three different experiments.
hnRNPC is elevated in highly invasive (U87R4) U87MG cells compared with its levels in noninvasive (U87L4) U87MG cells.
A recent report describes the generation of highly aggressive U87MG cells through serial intracranial transplantation associated with the expression of frizzled 4 (FZD4), which regulates the stemness and invasiveness of migrating glioma cells (22). To investigate the role of hnRNPC in the invasiveness of glioma cells, we used U87R4 cells (migrating), which show higher levels of invasiveness than U87L4 cells (nonmigrating). Since the miR-21 target PTEN negatively regulates the PI3K/Akt pathway and U87MG is a PTEN-deficient glioma, we examined whether PDCD4 expression was increased by hnRNPC silencing in U87MG cells. First, we assessed the effect of hnRNPC silencing on PDCD4 expression in PTEN-deficient U87MG cells. Forty-eight hours after transfection of U87MG cells with control or hnRNPC siRNA, PDCD4 expression was found to be elevated (Fig. 6A), indicating that hnRNPC regulated PDCD4 expression in a PTEN-independent manner. To investigate the role of hnRNPC in the invasive activity of glioblastomas, we compared the abundance of hnRNPC, PDCD4, and activated Akt between nonmigrating U87L4 and migrating U87R4 cells. Besides showing higher expression of hnRNPC, U87R4 cells showed decreased expression of PDCD4, lower levels of activated Akt (Fig. 6B), and increased miR-21 levels (Fig. 6C) compared to the levels in U87L4 cells. It also showed higher invasiveness (Fig. 6D), as reported previously (22). Furthermore, silencing of hnRNPC increased PDCD4 expression and inhibited Akt activation (Fig. 6E). Moreover, the invasive activity of U87R4 cells was suppressed by hnRNPC silencing, indicating that the elevated hnRNPC expression contributes to the invasiveness of U87MG glioblastoma cells (Fig. 6F). In sum, glioblastoma cells showing high invasiveness expressed lower levels of PDCD4 and correspondingly higher levels of hnRNPC, revealing that hnRNPC may play a key role in the metastatic potential of glioma cells.
Fig 6.

hnRNPC is overexpressed in highly invasive U87MG (U87R4) cells. (A) PTEN-deficient glioma cell line U87MG was transfected with control (CTRL) or hnRNPC siRNAs, and 48 h later, the levels of hnRNPC and PDCD4 were determined by Western blot analysis. (B) To compare the expression of hnRNPC and PDCD4 in U87L4 cells relative to their levels in U87R4 cells, whole-cell lysates were prepared and the levels of hnRNPC, PDCD4, p-Akt, Akt2, and GAPDH expression were determined by Western blot analysis. (C) The levels of miR-21 in U87L4 and U87R4 cells were determined by RT-qPCR using the TaqMan microRNA assay kit. (D) To isolate highly invasive U87R4 cells, U87MG cells were subjected to serial intracranial injection. After 4 cycles of selection, U87L4 (nonmigrating) and U87R4 (migrating) cells were isolated. Equal cell numbers of each cell population were added into a Biocoat Matrigel invasion chamber; 24 h later, invaded cells were stained and photographed under a microscope. The inhibitory activity of hnRNPC was determined by counting the average number of invaded cells from 9 different fields. Data are means and standard deviations from three different experiments. (E) U87R4 (migrating) cells were transfected with control (CTRL) or hnRNPC siRNAs, and 48 h later, the levels of hnRNPC, PDCD4, p-Akt, Akt2, and GAPDH expression were determined by Western blot analysis. (F) U87R4 cells were transfected as described above, and invasive activity was determined by counting numbers of invaded cells from 9 different fields.
hnRNPC expression level correlates with brain tumor grade.
miR-21 is widely overexpressed in many solid tumors, particularly in glioblastoma tissue and in cultured glioblastoma cell lines, where it functions as an antiapoptotic factor (18). PDCD4 mRNA and protein are frequently lost in human glioma tissue, and loss of PDCD4 appeared to contribute to the development of glioma (16). To evaluate whether the expression of hnRNPC correlates with the glioblastoma grade, we used a tissue microarray (TMA) that included 24 duplicates of brain tumor samples of various grades and stages. hnRNPC expression levels increased with brain tumor grade, as shown in Fig. 7 (also see Fig. S9 in the supplemental material), lending support to the idea that hnRNPC expression might be clinically significant and may represent a novel marker of prognosis for GBM patients.
Fig 7.

hnRNPC expression level correlates with brain tumor grade. To investigate hnRNPC expression levels in brain tumors, we studied tissue microarrays containing 24 samples of brain tumors of different grades and stages in duplicates. The levels of hnRNPC expression were then assessed by immunostaining and statically analyzed. AVE, average.
Taken together, hnRNPC is closely implicated in the metastatic potential of glioblastomas through regulation of miR-21 and Akt activation. The proposed action of hnRNPC is summarized in Fig. 8.
Fig 8.
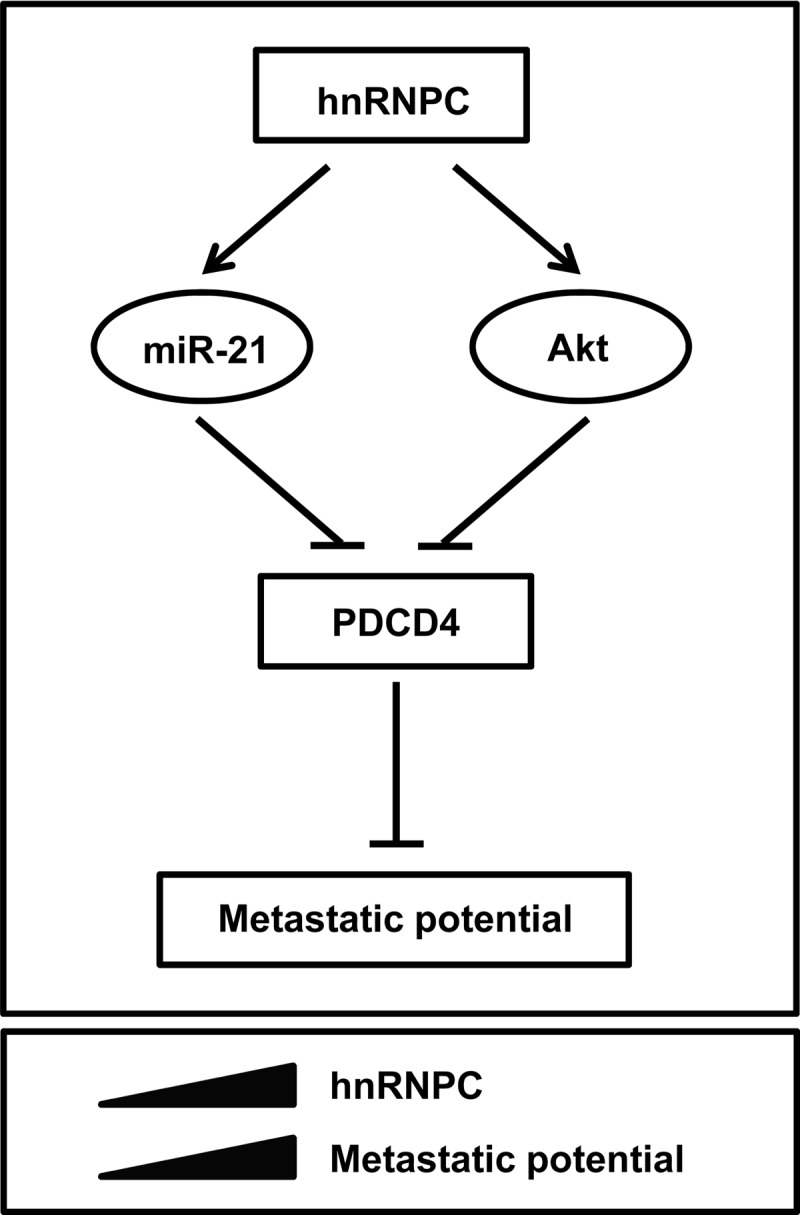
Schematic of proposed influence of hnRNPC on metastatic traits of glioblastoma. See the text for details.
DISCUSSION
GBM is the most aggressive human brain tumor, and despite therapeutic advances, GBM remains an incurable disease. GBM is difficult to eradicate in part because it has high metastatic activity, including migration and invasion. Metastasis is a complex, still poorly understood process whereby cancer cells disseminate. Here, we found that hnRNPC promoted the expression of miR-21, one of the most highly overexpressed miRNAs in GBM, and correspondingly inhibited the expression of PDCD4, a well-known target of miR-21.
RNA-binding proteins (RBPs) recognize specific sequences in target mRNAs (32) and control many aspects of RNA metabolism, including splicing, transport, localization, stability, and translation. The function of RBPs is determined through their expression levels, nucleocytoplasmic translocation, and posttranslation modifications, including phosphorylation. In addition to their role in mRNA metabolism, RBPs play critical roles in miRNA function by regulating the biogenesis, localization, and degradation of miRNAs (42). KH-type splicing regulatory protein (KSRP or KHSRP) is known to induce decay of mRNA containing AU-rich elements (ARE) and was recently reported to regulate the biogenesis of several miRNAs by serving as a component of microprocessor complex. KSRP promotes the biogenesis of a subset of miRNAs containing a GGG triplet motif in their terminal loop by enhancing the Drosha-mediated microprocessing activity (41). The kinase ATM (ataxia telangiectasia mutated) is activated by DNA damage and phosphorylates KSRP, increasing the accessibility of KSRP to pri-miRNA and thus enhancing miRNA biogenesis (47). Like KSRP, hnRNPA1 binds to a conserved loop region of miRNA and facilitates the cleavage by Drosha (34). However, in the case of let-7 miRNA biogenesis, hnRNPA1 negatively regulates the biogenesis of let-7a by interfering with KSRP binding to the terminal loop of pri-let-7a, which enhances let-7 biogenesis (33). In this study, we show evidence that hnRNPC regulates miR-21 expression, probably by associating with pri-miR-21. Further study is needed to elucidate the precise mechanism whereby hnRNPC silencing decreases pri-/pre-miR-21 and mature miR-21. Since hnRNPC is known to regulate RNA splicing and mRNA stability, these functions are possibly required for hnRNPC-associated miR-21 biogenesis.
So far, many miR-21 target transcripts have been reported (25), including the mRNAs that encode PDCD4, PTEN, TPM1, SPRY2, TIMP3, RECK, and APAF1. miR-21 plays a crucial role in tumor progression, particularly metastasis, and functions as a strong antiapoptotic factor, promoting a metastatic phenotype in diverse solid tumors, including GBM. It is also closely associated with the clinical stage and prognosis of cancer patients. Accordingly, GBMs often display lower levels of the miR-21 target PDCD4, a protein capable of influencing the expression of different cancer-related molecules, like p21 (3, 19), mitogen-activated protein kinase kinase kinase kinase 1 (MAP4K1) (46), JNK (46), and carbonic anhydrase II (27). PDCD4 also suppresses cell invasion and intravasation by inhibiting the expression of matrix metalloproteinases (MMPs) and uPAR (29, 46). Here, we found that silencing of hnRNPC increased PDCD4 expression, possibly in two parallel ways: one by downregulating miR-21, and the other by suppressing the Akt and p70S6K pathways. Understanding the detailed mechanisms whereby hnRNPC regulates Akt and p70S6K pathways independently of miR-21 awaits further study. The results showing that hnRNPC-regulated PDCD4 expression influenced the proliferation and apoptosis of T98G cells were consistent with previous reports that overexpression of PDCD4 decreased proliferation and increased apoptosis (18).
Since the metastatic activity of GBM is a main cause of cancer recurrence, it is important to understand the precise mechanism of GBM metastasis. We found here that silencing of hnRNPC diminished the migratory and invasive activity in T98G cells without altering proliferation. We also found that highly aggressive U87MG cells (U87R4) showed higher hnRNPC abundance and correspondingly lower PDCD4 levels than nonmigrating U87MG cells (U87L4). Finally, tissue microarray (TMA) analysis confirmed that hnRNPC was overexpressed in highly aggressive brain tumors (grades III and IV). We propose that hnRNPC controls the metastatic potential of GBM and may be used as a prognostic and therapeutic marker in GBM.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from the Basic Science Research Program, National Research Foundation of Korea by the Ministry of Education, Science, and Technology (2011-009329 to H. H. Kim) and the Korea Health Care Technology R&D project, Ministry of Health and Welfare Affairs, Republic of Korea (A092255). M.G. was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, United States.
Footnotes
Published ahead of print 20 August 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Asangani IA, et al. 2008. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27:2128–2136 [DOI] [PubMed] [Google Scholar]
- 2. Bitomsky N, Bohm M, Klempnauer KH. 2004. Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene 23:7484–7493 [DOI] [PubMed] [Google Scholar]
- 3. Bitomsky N, Wethkamp N, Marikkannu R, Klempnauer KH. 2008. siRNA-mediated knockdown of Pdcd4 expression causes upregulation of p21(Waf1/Cip1) expression. Oncogene 27:4820–4829 [DOI] [PubMed] [Google Scholar]
- 4. Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan JA, Krichevsky AM, Kosik KS. 2005. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65:6029–6033 [DOI] [PubMed] [Google Scholar]
- 6. Chen Y, et al. 2003. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J. Pathol. 200:640–646 [DOI] [PubMed] [Google Scholar]
- 7. Chien CH, et al. 2011. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 39:9345–9356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi YD, Grabowski PJ, Sharp PA, Dreyfuss G. 1986. Heterogeneous nuclear ribonucleoproteins: role in RNA splicing. Science 231:1534–1539 [DOI] [PubMed] [Google Scholar]
- 9. Christian KJ, Lang MA, Raffalli-Mathieu F. 2008. Interaction of heterogeneous nuclear ribonucleoprotein C1/C2 with a novel cis-regulatory element within p53 mRNA as a response to cytostatic drug treatment. Mol. Pharmacol. 73:1558–1567 [DOI] [PubMed] [Google Scholar]
- 10. Cmarik JL, et al. 1999. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc. Natl. Acad. Sci. U. S. A. 96:14037–14042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly EC, Van Doorslaer K, Rogler LE, Rogler CE. 2010. Overexpression of miR-21 promotes an in vitro metastatic phenotype by targeting the tumor suppressor RHOB. Mol. Cancer Res. 8:691–700 [DOI] [PubMed] [Google Scholar]
- 12. Deng S, Calin GA, Croce CM, Coukos G, Zhang L. 2008. Mechanisms of microRNA deregulation in human cancer. Cell Cycle 7:2643–2646 [DOI] [PubMed] [Google Scholar]
- 13. Dorrello NV, et al. 2006. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314:467–471 [DOI] [PubMed] [Google Scholar]
- 14. Fujita S, et al. 2008. miR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 378:492–504 [DOI] [PubMed] [Google Scholar]
- 15. Gao F, et al. 2009. PDCD4 gene silencing in gliomas is associated with 5′CpG island methylation and unfavourable prognosis. J. Cell. Mol. Med. 13:4257–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao F, et al. 2007. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol. Rep. 17:123–128 [PubMed] [Google Scholar]
- 17. Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. 2006. MicroRNA expression and function in cancer. Trends Mol. Med. 12:580–587 [DOI] [PubMed] [Google Scholar]
- 18. Gaur AB, Holbeck SL, Colburn NH, Israel MA. 2011. Downregulation of Pdcd4 by mir-21 facilitates glioblastoma proliferation in vivo. Neuro Oncol. 13:580–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goke R, et al. 2004. Programmed cell death protein 4 (PDCD4) acts as a tumor suppressor in neuroendocrine tumor cells. Ann. N. Y. Acad. Sci. 1014:220–221 [DOI] [PubMed] [Google Scholar]
- 20. Huang TH, et al. 2009. Up-regulation of miR-21 by HER2/neu signaling promotes cell invasion. J. Biol. Chem. 284:18515–18524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iorio MV, et al. 2005. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 65:7065–7070 [DOI] [PubMed] [Google Scholar]
- 22. Jin X, et al. 2011. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 71:3066–3075 [DOI] [PubMed] [Google Scholar]
- 23. Kim HH, et al. 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 23:1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim JH, et al. 2003. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol. Cell. Biol. 23:708–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krichevsky AM, Gabriely G. 2009. miR-21: a small multi-faceted RNA. J. Cell. Mol. Med. 13:39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lankat-Buttgereit B, Goke R. 2009. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol. Cell 101:309–317 [DOI] [PubMed] [Google Scholar]
- 27. Lankat-Buttgereit B, et al. 2004. Pdcd4 inhibits growth of tumor cells by suppression of carbonic anhydrase type II. Mol. Cell. Endocrinol. 214:149–153 [DOI] [PubMed] [Google Scholar]
- 28. Lee EK, et al. 2010. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 17:732–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leupold JH, et al. 2007. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 26:4550–4562 [DOI] [PubMed] [Google Scholar]
- 30. Li Y, et al. 2009. MicroRNA-21 targets LRRFIP1 and contributes to VM-26 resistance in glioblastoma multiforme. Brain Res. 1286:13–18 [DOI] [PubMed] [Google Scholar]
- 31. Louis DN. 2006. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 1:97–117 [DOI] [PubMed] [Google Scholar]
- 32. Lunde BM, Moore C, Varani G. 2007. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell. Biol. 8:479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michlewski G, Caceres JF. 2010. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 17:1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Michlewski G, Guil S, Semple CA, Caceres JF. 2008. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol. Cell 32:383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palamarchuk A, et al. 2005. Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res. 65:11282–11286 [DOI] [PubMed] [Google Scholar]
- 36. Pinol-Roma S, Dreyfuss G. 1993. hnRNP proteins: localization and transport between the nucleus and the cytoplasm. Trends Cell Biol. 3:151–155 [DOI] [PubMed] [Google Scholar]
- 37. Reis PP, et al. 2010. Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR-21 in oral squamous cell carcinoma. Mol. Cancer 9:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roldo C, et al. 2006. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 24:4677–4684 [DOI] [PubMed] [Google Scholar]
- 39. Shetty S. 2005. Regulation of urokinase receptor mRNA stability by hnRNP C in lung epithelial cells. Mol. Cell. Biochem. 272:107–118 [DOI] [PubMed] [Google Scholar]
- 40. Si ML, et al. 2007. miR-21-mediated tumor growth. Oncogene 26:2799–2803 [DOI] [PubMed] [Google Scholar]
- 41. Trabucchi M, et al. 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459:1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Kouwenhove M, Kedde M, Agami R. 2011. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 11:644–656 [DOI] [PubMed] [Google Scholar]
- 43. Volinia S, et al. 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A. 103:2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wei N, Liu SS, Chan KK, Ngan HY. 2012. Tumour suppressive function and modulation of programmed cell death 4 (PDCD4) in ovarian cancer. PLoS One 7:e30311 doi:10.1371/journal.pone.0030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei NA, et al. 2009. Loss of programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Mol. Cancer 8:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang HS, et al. 2006. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol. Cell. Biol 26:1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang X, Wan G, Berger FG, He X, Lu X. 2011. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell 41:371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou X, et al. 2010. Reduction of miR-21 induces glioma cell apoptosis via activating caspase 9 and 3. Oncol. Rep. 24:195–201 [DOI] [PubMed] [Google Scholar]
- 49. Zhu S, et al. 2008. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 18:350–359 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.