

Abstract
Antistaphylococcal beta-lactams enhance daptomycin activity and have been used successfully in combination for refractory methicillin-resistant Staphylococcus aureus (MRSA) infections. Ceftaroline possesses MRSA activity, but it is unknown if it improves the daptomycin potency comparably to other beta-lactams. We report a complex patient case of endocarditis who was treated with daptomycin in combination with ceftaroline, which resulted in clearance of a daptomycin-nonsusceptible strain. An in vitro pharmacokinetic/pharmacodynamic model of renal failure was used to simulate the development of daptomycin resistance and evaluate the microbiologic effects of daptomycin plus ceftaroline treatment. Combination therapy with daptomycin and ceftaroline restored daptomycin sensitivity in vivo and resulted in clearance of persistent blood cultures. Daptomycin susceptibility in vitro was increased in the presence of either ceftaroline or oxacillin. Daptomycin at 6 mg/kg of body weight every 48 h was bactericidal in the model but resulted in regrowth and daptomycin resistance (MIC, 2 to 4 μg/ml) with continued monotherapy. The addition of ceftaroline at 200 mg every 12 h after the emergence of daptomycin resistance enhanced bacterial killing. Importantly, daptomycin plus ceftaroline as the initial combination therapy produced rapid and sustained bactericidal activity and prevented daptomycin resistance. Both in vivo- and in vitro-derived daptomycin resistance resulted in bacteria with more fluid cell membranes. After ceftaroline was added in the model, fluidity was restored to the level of the initial in vivo isolate. Daptomycin-resistant isolates required high daptomycin exposures (at least 10 mg/kg) to optimize cell membrane damage with daptomycin alone. Ceftaroline combined with daptomycin was effective in eliminating daptomycin-resistant MRSA, and these results further justify the potential use of daptomycin plus beta-lactam therapy for these refractory infections.
INTRODUCTION
Daptomycin is increasingly used in the treatment of complex methicillin-resistant Staphylococcus aureus (MRSA) infections. Reduced susceptibility to daptomycin in S. aureus (MIC, >1 μg/ml) has been attributed to a number of genetic mutations, most notably in the mprF locus, that correlate with a phenotype characterized by altered cell walls, surface charge, and membrane function (18, 21). These characteristics have often been associated with concomitant vancomycin-intermediate susceptibility in S. aureus (VISA), with vancomycin MICs of ≥4 μg/ml, but this cross-resistance is not absolute (22).
A number of recent studies have described novel interactions between beta-lactam antibiotics and daptomycin in MRSA strains (7, 11, 31). As first described by Yang et al., reduced daptomycin susceptibility correlates with a simultaneous increase in oxacillin susceptibility, even though the beta-lactam resistance element mecA remains intact (31). In vitro and in vivo static and dynamic assessments of daptomycin in combination with antistaphylococcal beta-lactams demonstrate rapid antimicrobial synergy by enhanced daptomycin binding to the cell membrane (7, 26). Via a similar mechanism, beta-lactam antibiotics enhance the killing activities of cationic host defense peptides (26). The potential therapeutic implications for this combination are compelling, especially in cases with a high risk of treatment failure and when treatment options are limited.
We present a recent case of a woman admitted to the intensive care unit from an outside hospital who had MRSA bacteremia and septic shock. The patient's past medical history was significant for end-stage renal disease with thrice-weekly hemodialysis, diabetes mellitus type II, and morbid obesity. The patient had failed 11 total days of daptomycin treatment at 6 mg/kg of body weight every 48 h at the outside hospital, as well as in our hospital, and had 12 consecutive total positive blood cultures. Due to access issues, intravenous catheters could not be removed. As shown in the antibiotic timeline in Fig. 1, the failing daptomycin therapy was supplemented by the addition of ceftaroline after the development of reduced daptomycin susceptibility, persistent positive cultures, and clinical sepsis. A transesophageal echocardiogram (TEE) on hospital day 11 demonstrated a 23-mm mass in the right atrium. On hospital day 18, the patient complained of right wrist pain; subsequent aspiration of this tissue revealed MRSA. A repeat TEE on hospital day 63 (day 54 of combination therapy) showed the atrial mass was smaller (11 mm by 10 mm) and no longer mobile. Combination therapy was followed by clearance of blood cultures within 4 days and slow improvement in symptoms, but other medical complications resulted in hospital discharge to home hospice.
Fig 1.

Course of therapy timeline until definitive clearance of bacteremia in a patient with prosthetic valve endocarditis treated with daptomycin alone and then combined with ceftaroline. Following the collection of the timeline data presented here, the patient was hospitalized for an additional 48 days, received combination therapy throughout, and all remaining blood cultures were negative.
In this study, we further investigate the microbiologic effects of the antimicrobial treatment noted in this case by utilizing an in vitro pharmacokinetic/pharmacodynamic (PK/PD) model of daptomycin alone and in combination with ceftaroline to (i) treat a daptomycin-nonsusceptible strain that develops while on therapy and (ii) attempt to prevent the emergence of this phenotype with combination therapy used as the initial regimen.
MATERIALS AND METHODS
Bacterial isolates.
All four isolates for this study were obtained from the woman with native valve endocarditis described above. The timeline for these isolates and corresponding antibiotic therapies is presented in see Table 1 (below) and Fig. 1. The initial blood isolate in this study, W3148, was obtained on presentation of bacteremia at our institution, while the final blood isolate (W3248) was obtained after 2 weeks of consecutive daily positive blood cultures. The infection seeded to the subject's wrist, and two resulting isolates were obtained from this tissue 3 and 10 days (W3278 and W3348, respectively) after the final positive blood culture.
Table 1.
Antibiotic susceptibilities in clinical isolates from the study case
| Isolate | Description | Day of DAP CPT combination therapy | MIC (μg/ml)a |
||||||
|---|---|---|---|---|---|---|---|---|---|
| DAPb | VANb | CPTc | DAP/VAN with CPT 0.5 μg/mlb | OXAc | DAP/VAN with OXA 128 μg/mld | DAP/VAN with OXA 0.5 μg/mld | |||
| W3148 | Initial blood culture | −6 | 0.75–1.0 | 2.0 | 1.0 | 0.06/0.5 | 256 | 0.25/0.75 | 0.38/1.5 |
| W3248 | Final blood culture | 4 | 2.0 | 3.0 | 1.0 | 0.06/0.5 | 256 | 0.06/1.0 | 0.25/1.5 |
| W3278 | Initial seeded wrist culture | 7 | 0.13 | 1.5 | 1.0 | 0.06/0.5 | 256 | 0.06/1.5 | 0.13/1.5 |
| W3348 | Final seeded wrist culture | 14 | 0.19 | 1.5 | 1.0 | 0.06/0.5 | 256 | 0.13/0.75 | 0.13/1 |
Abbreviations: DAP, daptomycin; VAN, vancomycin; CPT, ceftaroline; OXA, oxacillin.
Determined by Etest.
Determined by broth microdilution.
Values represent DAP and VAN Etest results after incorporation of CPT or OXA into the MHA medium.
Antibiotics and media.
Daptomycin was provided by the manufacturer (Cubist Pharmaceuticals, Lexington MA). Ceftaroline powder for in vitro analysis was kindly provided by Gary Doern with support of the manufacturer (Forest Laboratories, Inc., New York, NY), and oxacillin was purchased from Sigma-Aldrich, St. Louis, MO. Mueller-Hinton II broth (BD, Difco, Sparks, MD) supplemented with 50 μg/ml calcium (as CaCl2) and 12.5 μg/ml magnesium (as MgCl2) was used for all in vitro PK/PD analyses (MHB50). For any assays using oxacillin, this medium was further supplemented with 2% sodium chloride.
Susceptibility.
MICs of daptomycin and vancomycin were determined in duplicate by Etest and broth microdilution, while ceftaroline and oxacillin susceptibilities were determined by broth microdilution (6). All samples were incubated at 35°C for 24 h. Daptomycin MICs were then determined in the presence of ceftaroline or oxacillin by placing a daptomycin Etest disc on Mueller-Hinton agar containing ceftaroline (0.5× MIC, at 0.5 μg/ml) or oxacillin (0.5 μg/ml or 0.5× MIC at 128 μg/ml) for each isolate (7).
PK/PD model.
A previously described in vitro PK/PD model was used for simulating one-compartment antibiotic exposures of daptomycin and ceftaroline (1). This dynamic model allows for analysis of the pharmacodynamic effects of antibiotics under various simulated clearance conditions in humans (1, 25). All models were performed in duplicate experiments. The initial isolate, W3148, was grown overnight in 25 ml MHB50 to a 4.0 McFarland standard and added into the model (250 ml, total) to obtain a starting inoculum of 1 × 108 CFU/ml. Two monotherapy antibiotic regimens with free (f) drug concentrations were evaluated, using end-stage renal disease doses (9, 23) consistent with the doses used for the clinical subject and modeled over a 7-day (168-h) duration: (i) daptomycin at 6 mg/kg every 48 h (targeted free maximum free drug concentration [fCmax], 8.0 μg/ml; half-life, 18.9 h) and (ii) ceftaroline at 200 mg every 12 h (targeted fCmax, 7.0 μg/ml; half-life, 6.2 h).
PK/PD modeling of combination therapy of antibiotics with two different elimination rates was performed according to the methods described by Blaser et al. (4). Briefly, the total elimination rate was set to that of the antibiotic with the faster clearance (ceftaroline). A supplemental dosing chamber was designed to continuously add antibiotic into the reaction (infection) chamber for the antibiotic that is normally cleared at the lower rate (daptomycin). Antibiotic doses were delivered into the supplement and reaction chambers for the slower-clearing antibiotic (daptomycin) to account for its elimination by the more rapid total rate. Combination therapy was simulated by two separate PD models: (iii) ceftaroline combined with daptomycin upon the first detection of daptomycin resistance with daptomycin therapy alone, and (iv) daptomycin plus ceftaroline initiated on day 1. In this fashion, we were able replicate antibiotic treatment from the clinical subject as well as determine the individual effects of these antibiotics.
Pharmacokinetic concentrations of daptomycin in test samples were evaluated by high-performance liquid chromatography (HPLC). The HPLC apparatus consisted of a Shimadzu controller SIL10A autosampler, LC10AD isocratic pump (Columbia, MO), and Gilson UV detector at 220 nm (Middleton, WI). The mobile phase was pumped through a Kinetix C18 column (75 by 4.6 μm column; sub-2μm for breakthrough; Phenomenex, Torrance, CA) at a rate of 0.75 ml/min and consisted of 0.05 M formic acid adjusted to pH 3.0 and 35% acetonitrile. Aliquots (75 μl) of the standard and samples were injected directly without extraction or protein precipitation. Ceftaroline concentrations were evaluated by bioassay with Bacillus subtilis ATCC 6633 as the indicator organism and using standard drug concentrations of 1.0, 2.5, 5.0, and 10 μg/ml. The method for this assay has been described in detail elsewhere (28). The pharmacokinetic parameters were estimated and calculated using NONMEM modeling.
In vitro nonsusceptibility screening.
Susceptibility to daptomycin was evaluated at the start of each model and subsequently every 24 h during the simulation (29). Samples of 50 μl from the model were spot plated onto Mueller-Hinton agar plates containing 3 μg/ml daptomycin (3× the starting MIC). Plates were then examined for growth, and CFU/ml were quantified after 48 h of incubation at 35°C. Both Etest and broth microdilution methods were used to determine the MICs related to any resulting growth on screening plates.
Membrane integrity.
Staphylococcal membrane permeability after antibiotic exposure was assayed with the LIVE/DEAD BacLight kit (Invitrogen, Madison, WI) (3). In vivo- and in vitro-derived isolates were grown in MHB50 and resuspended to an optical density at 600 nm (OD600) of 0.25 in 5 mM HEPES buffer plus 50 μg/ml Ca2+. Daptomycin exposures were derived from the known Cmax values for each dose, as follows: 6 mg/kg (8 μg/ml), 8 mg/kg (10 μg/ml), 10 mg/kg (12 μg/ml), and 12 mg/kg (16 μg/ml). Suspensions were incubated for 3 h at room temperature, brought to reagent concentrations of 5 μM SYTO-9 and 30 μM propidium iodide, and incubated for an additional 15 min at room temperature in the dark. Fluorescence was measured in a Molecular Devices SpectraMax M2e spectrofluorometer.
Membrane fluidity.
Staphylococcal membrane fluidity was determined for in vivo- and in vitro-derived strains as previously described (2, 5). Overnight bacterial cultures were inoculated into fresh MHB50 medium and grown to an OD600 of 0.2 to 0.5. Bacteria were pelleted, washed with normal saline, resuspended to an OD600 of 0.3 in normal saline plus 10 μM 1,6-diphenyl-1,3,5-hexatriene (DPH), and incubated at 37°C for 1 h. Aliquots were transferred to preheated cuvettes, and fluorescence was measured in an ISS Koala spectrofluorometer with a temperature-controlled cuvette holder maintained at 37°C. The probe was excited with vertically polarized light (λex, 360 nm), and emission intensity was detected in both parallel and perpendicular planes (λem, 426 nm). Results were corrected by subtracting data from an unlabeled control reaction. The polarization index (pI) is the sensitivity ratio of the instrument for detecting DPH emission and was determined using the following formula: pI = [IV − IH(IHV/IHH)]/[IV + IH(IHV/IHH)],where I is the fluorescence intensity and V and H indicate vertical and horizontal orientation instrument values, respectively (2). Data represent averages ± standard deviations of 3 experiments performed on separate days. A lower fluorescence polarization value indicates a higher degree of membrane integrity.
spa typing and multilocus sequence typing.
The spa typing was performed following the method of Koreen et al. (15). The spa type identification was done using the Ridom StaphType (version 2.1.1) software (Ridom GmbH, Wurzburg, Germany). The multilocus sequence typing (MLST) was done as described by Enright et al. (10).
Statistical analysis.
Comparisons were conducted by using Student's t test. For multiple comparisons within each measurement, reported P values are Holm adjusted.
RESULTS
In vitro antibiotic susceptibility.
The antibiotic susceptibility results of the study isolates are shown in Table 1. These strains belong to spa type t002 and MLST sequence type 5. The initial isolate, W3148, was susceptible to daptomycin, vancomycin, and ceftaroline, while the final blood culture was nonsusceptible to both daptomycin and vancomycin by Etest but remained susceptible to ceftaroline. The daptomycin MIC for this strain was 1 mg/liter (susceptible), and the vancomycin MIC was 4 mg/liter by broth microdilution. The secondary cultures obtained from the wrist were susceptible to each of these antibiotics by both Etest and broth microdilution. The daptomycin and vancomycin MICs in the blood culture isolates were lower (4- to 32-fold reductions) in the presence of both ceftaroline and oxacillin at 0.5× the respective MICs. A greater reduction in daptomycin MICs occurred in the presence of these antibiotics, compared to the change in vancomycin MICs. At an equivalent concentration of 0.5 μg/ml, ceftaroline reduced daptomycin MICs up to 32-fold, compared to up to an 8-fold reduction with oxacillin.
Simulated antibiotic exposures in the PK/PD model.
An in vitro pharmacodynamic model of renal failure (creatinine clearance, <30 ml/min) was utilized to simulate the in vivo antibiotic regimen of the patient. Daptomycin at 6 mg/kg every 48 h was modeled until the emergence of daptomycin nonsusceptibility, defined as an absolute rise in the MIC to a level of 2 μg/ml, and then ceftaroline at 200 mg every 12 h was added in combination for the remainder of the 7-day duration. Alternative regimens were also modeled to compare these effects to single antibiotic therapy or combination antibiotic therapy for the entire duration. The pharmacokinetic results of daptomycin and ceftaroline from the PK/PD model are displayed in Fig. 2 and were consistent with the range of values for the representative doses in patients with renal failure: for daptomycin, ƒCmax, 7.6 ± 1.1 μg/ml; ƒCmin, 0.8 ± 0.3 μg/ml; t½, 18.5 h; area under the concentration-time curve for the free drug from 0 to 24 h (ƒAUC0–24), 110.8 μg/ml · h; for ceftaroline, ƒCmax, 8.2 ± 0.2 μg/ml; ƒCmin, 1.1 ± 0.3 μg/ml; t½, 4.3 h; 98% of the time above the MIC, and 36% of the time above 4× the MIC.
Fig 2.
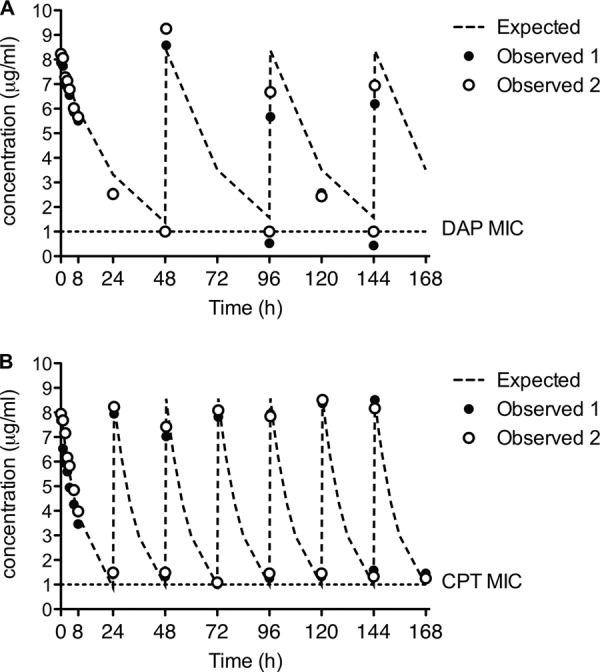
Pharmacokinetic concentration-time profiles of expected and observed concentrations in the one-compartment in vitro pharmacodynamic model over the 7-day simulation. (A) Daptomycin; (B) ceftaroline.
Daptomycin alone at 6 mg/kg every 48 h was bactericidal in the model within the first 8 h of simulated therapy (mean maximum kill, 5.3 ± 0.3 log CFU/ml). As demonstrated in Fig. 3A, bacterial regrowth was detected after 24 h of therapy and increased for the entire 7-day duration with this dose. Daptomycin resistance after the 6-mg/kg dose, indicated by a MICs of 2 to 4 μg/ml by Etest and 2 μg/ml by broth microdilution, was detected at 72 h of therapy and was the predominant organism phenotype at the end of the model simulation (Fig. 4). Following these results, we replicated daptomycin monotherapy for 3 days and then added ceftaroline into the model to simulate combination therapy in the patient. The results in Fig. 3B demonstrate that the effects of daptomycin alone with this strain for up to 72 h were reproducible, and combining ceftaroline with daptomycin after the emergence of daptomycin resistance resulted in enhanced bacterial killing. This did not result in bactericidal activity (2.4 ± 0.8 log kill), but the burden of organisms at the end of therapy with this regimen at 7 days was significantly lower than with daptomycin therapy alone (mean, 5.7 ± 0.5 versus 8.9 ± 0.2 log CFU/ml, respectively; P < 0.001). Daptomycin resistance occurred in each model of daptomycin monotherapy but varied in the initiation and degree of formation up to 72 h (Fig. 4). Notably, within 72 h after the introduction of ceftaroline combination therapy in the model, daptomycin resistance could no longer be detected. Isolates recovered during the last 24 h of daptomycin plus ceftaroline therapy had MICs of ≤0.75 μg/ml by Etest and ≤0.5 μg/ml by broth microdilution, which are equivalent to or less than the starting MIC of 1 μg/ml for the wild-type strain.
Fig 3.

Antibiotic activity in the in vitro PK/PD model of daptomycin alone and in combination with ceftaroline in the initial daptomycin-susceptible clinical isolate W3148. (A) Single antibiotic exposure. black circles, growth control; black squares, daptomycin at 6 mg/kg every 48 h; white circles, ceftaroline at 200 mg every 12 h. (B) Combination antibiotic exposure results. Black circles, growth control; black triangles, DAP treatment every 48 h with CPT added on day 3; white diamonds, initial therapy with DAP every 48 h combined with CPT on day 1.
Fig 4.
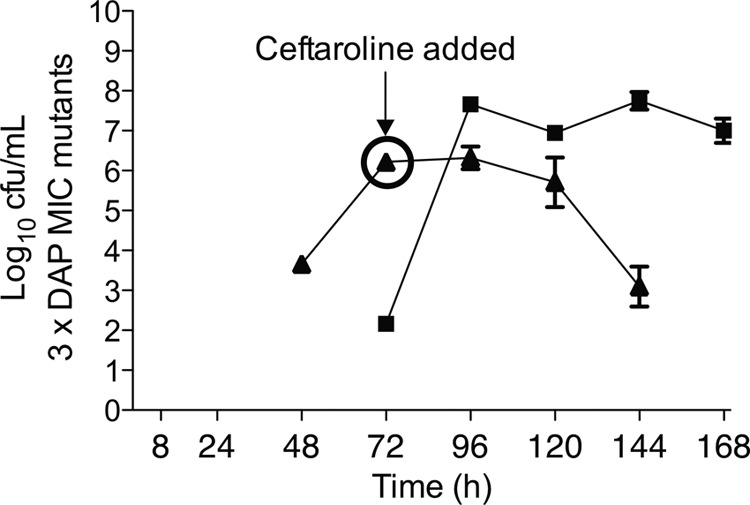
Emergence of daptomycin (DAP) resistance in the pharmacodynamic model detected after screening plates containing daptomycin at 3 μg/ml. Daptomycin resistance occurred in each model of daptomycin monotherapy, but it varied in the initiation and degree of formation up to 72 h. The addition of ceftaroline (CPT) at 72 h to the DAP regimen eliminated DAP resistance in model isolates. Black squares, DAP at 6 mg/kg every 48 h; black triangles, DAP at 6 mg/kg every 48 h with CPT added on day 3.
Ceftaroline in combination with daptomycin was also simulated as initial therapy in the PK/PD model (Fig. 3B). At least a 6.0-log decrease in inoculum was observed within the first 4 h of therapy, and this effect was maintained throughout the 7-day duration, with less than a 2-log regrowth by the end of therapy. These effects were greater than those observed with any other regimen tested. This initial combination therapy approach also inhibited the emergence of daptomycin resistance.
Membrane integrity with daptomycin.
The effects of daptomycin concentrations on membrane integrity of the in vivo-derived strains are shown in Table 2. As expected, the wild-type daptomycin-susceptible parent strain exhibited a high degree of membrane damage when exposed to daptomycin conditions equivalent to peak concentrations with 6-mg/kg doses. No additional membrane damage was apparent with higher daptomycin exposures. Conversely, membrane integrity was relatively preserved with the 6-mg/kg and 8-mg/kg exposures in the resistant in vivo mutant. Higher-dose exposures of 10 mg/kg and 12 mg/kg produced membrane damage similar to that of the susceptible parent strain following lower daptomycin exposures. The isolates obtained from the wrist during daptomycin plus ceftaroline clinical treatment displayed similar membrane integrity profiles to the initial parent strain.
Table 2.
Daptomycin cell membrane effects in isolates from the patient
| Isolate | % of intact cell membranes after daptomycin exposure ata: |
Membrane fluidity (pI)b | |||
|---|---|---|---|---|---|
| 6 mg/kg | 8 mg/kg | 10 mg/kg | 12 mg/kg | ||
| W3148 | 22.4 ± 4.2 | 25.7 ± 1.4 | 17.9 ± 1.7 | 18.3 ± 3.2 | 0.294 ± 0.018 |
| W3248 | 43.0 ± 10.0 | 29.7 ± 1.4 | 16.9 ± 1.9 | 17.1 ± 3.1 | 0.288 ± 0.016 |
| W3278 | 22.1 ± 4.2 | 20.6 ± 0.5 | 20.2 ± 1.3 | 18.5 ± 0.7 | 0.284 ± 0.020 |
| W3348 | 32.7 ± 5.7 | 20.5 ± 1.4 | 19.5 ± 1.0 | 18.3 ± 0.5 | 0.287 ± 0.020 |
Values are means ± standard deviations. A lower percentage of intact cell membranes indicates greater daptomycin activity.
A lower pI value indicates a greater degree of membrane fluidity.
Membrane fluidity.
Daptomycin nonsusceptibility has been linked to changes in membrane fluidity (18). It is suggested that cationic peptides, including daptomycin and host peptides, exert maximal activity in the presence of an optimal or stable cell membrane order (30). A more fluid cell membrane may inhibit insertion of cationic peptides into the target cell membrane and has been associated with reduced cationic peptide susceptibility (17).
In comparing membrane fluidity between the daptomycin-susceptible parent strain and the in vivo-derived resistant mutant, the development of daptomycin resistance resulted in more-fluid cell membranes, although this difference was not statistically significant. In vivo, daptomycin susceptibility was restored in isolates W3278 and W3348 following ceftaroline combination therapy in the patient, but the enhanced fluidity persisted (Table 2).
Significant increases in membrane fluidity occurred in daptomycin-resistant isolates derived from the in vitro PK/PD model. The greatest increase in fluidity occurred soon after the emergence of daptomycin resistance (MIC, 3 μg/ml) at 96 h (pI, 0.281 ± 0.014; P < 0.05), but this change was not sustained in the final resistant isolate at 168 h (MIC, 4 μg/ml) from this regimen (pI, 0.286 ± 0.021; P > 0.05). Membrane fluidity at the end of ceftaroline combination exposure in the model appeared to return to level comparable to the initial isolate (pI, 0.289 ± 0.039).
DISCUSSION
This study presents a novel clinical case of microbiologically successful daptomycin and ceftaroline combination therapy following the emergence of daptomycin resistance. The noted clinical effects of combination in this case included sterilization of blood cultures following persistently positive daily cultures for 2 weeks with daptomycin monotherapy, reduction in the size of the cardiac vegetative infection source during combination therapy, and daptomycin resensitization by two susceptibility methods in genetically identical isolates that seeded to the patient's wrist during the endovascular infection. To our knowledge, this is the first reported case of improvement and restoration of daptomycin sensitivity in vivo with concomitant beta-lactam exposure.
In vitro pharmacodynamic modeling revealed microbiological findings similar to this case, including improved antibacterial activity after the addition of ceftaroline and the restoration of daptomycin susceptibility with combination therapy. Importantly, combination modeling of both antibiotics on the first day of simulated therapy displayed rapid and sustained bactericidal activity and prevented the emergence of daptomycin resistance. Although this study was limited to one clinical case and subsequent evaluation of a single clinical isolate in the model, we and others have successfully treated similar cases of complicated MRSA infections with this combination that were refractory and/or resistant to primary daptomycin therapy (G. Sakoulas, personal communication). The potent antimicrobial activity with an initial daptomycin-plus-ceftaroline combination treatment may be useful in complex MRSA infections, including isolates with daptomycin MICs approaching the susceptibility breakpoint of 1 μg/ml.
Ceftaroline is a new cephalosporin antibiotic with activity against a diverse range of Gram-negative and Gram-positive organisms, including MRSA and VISA (27). Ceftaroline activity against MRSA is maintained through its affinity for the penicillin-binding protein 2a, which is responsible for resistance to other antistaphylcooccal beta-lactams (27). Although ceftaroline alone is active against daptomycin-nonsusceptible strains in animal and in vitro models (13, 28), it is unknown if its added exposure improves daptomycin efficacy against these strains analogous to that found with other beta-lactam antibiotics (7, 11). Single-drug therapy with ceftaroline in the PK/PD model was bacteriostatic against the parent strain in our study over the 7-day duration, but antibiotic activity and susceptibility were further enhanced when ceftaroline was used in combination with daptomycin. We highlight that the ceftaroline dose used in this case and in simulation are based upon the regimen for its approved indications. More frequent dosing every 8 h has been successfully used as salvage therapy for bacteremia and endocarditis (12), and this may be desirable for complicated infections due to MRSA to optimize the pharmacodynamic parameter of time above the MIC (8).
A recent patient investigation of daptomycin in combination with antistaphylcoccal beta-lactams for complicated MRSA infections demonstrated important microbiologic findings. Daptomycin susceptibility was restored upon in vitro exposure to nafcillin in a daptomycin-resistant strain that emerged while on therapy (7). Similar effects were noted in our study with both oxacillin and ceftaroline. Based upon susceptibility testing, a 32-fold reduction in the daptomycin MIC was observed with the in vivo daptomycin-resistant strain when tested in the presence of ceftaroline. Interestingly, ceftaroline overall had a 2- to 6-fold larger effect on lowering daptomycin MICs than did an equivalent concentration of oxacillin. We anticipate future studies comparing the effects of ceftaroline and oxacillin or nafcillin on daptomycin activity in pharmacodynamic models.
Enhanced daptomycin membrane binding and reduction in the net positive membrane surface charge are known to occur in the presence of beta-lactams (7, 26). Restoration of daptomycin susceptibility in our in vitro study correlated with a change in cell membrane fluidity near that of the parent isolate and may provide another mechanistic explanation for the phenotypic change in susceptibility. Prior investigations have shown a link between cell wall degradation and more rigid cell membranes (30). These findings support the hypothesis that cell wall damaging effects of ceftaroline in MRSA may lead to a compensatory more rigid cell membrane and thus restoration of daptomycin susceptibility. Although the changes in membrane fluidity were less apparent in our study, minor changes in cell membrane fluidity can have profound impacts on cell function and antibiotic susceptibility (14, 19).
Daptomycin exhibits concentration-dependent activity, and several in vitro and animal models have reported improved bactericidal activity with simulated doses ranging from 8 to 12 mg/kg, compared to 4 to 6 mg/kg daily (20, 24). High-dose therapies in these models particularly improve the efficacy of daptomycin against nonsusceptible strains compared to conventional doses of 6 mg/kg daily (20, 24). In a recent noncomparative and retrospective study of higher-dose daptomycin therapy for cases refractory to vancomycin treatment, doses of at least 8 mg/kg were both safe and effective for the treatment of complicated infections (16).
Although the daptomycin dose in this present case was increased to 10 mg/kg, this occurred only after the clearance of blood cultures with 6-mg/kg doses when combined with ceftaroline. We did not assess this high-dose regimen against the initial blood isolate in the PK/PD model, but a membrane damage assay revealed that daptomycin concentrations did have notable effects on cell membrane integrity. As expected, the daptomycin-resistant isolate W3248 retained greater cell membrane integrity at exposures analogous to Cmax for the 6-mg/kg and 8-mg/kg doses, indicating that daptomycin activity at these doses is reduced. Membrane damage was comparable between susceptible and resistant strains at higher-dose exposures of 10 mg/kg and 12 mg/kg. It would be interesting to discern if ceftaroline further enhances daptomycin activity at these higher doses in the in vitro model against strains with reduced daptomycin susceptibilities.
The patient's pharmacokinetic profile in this case was a challenge for direct pharmacodynamic modeling in vitro. One major limitation was to simulate daptomycin distributions during hemodialysis. During the two interdialytic periods with thrice-weekly hemodialysis, daptomycin is administered every 48 h for 2 doses and then every 72 h for one dose. For these reasons and due to the difficulty in mimicking 4-hour hemodialysis sessions, a severe renal dysfunction model (creatinine clearance of <30 ml/min) with every 48-hour dosing was used as a surrogate. Also, ceftaroline pharmacokinetic concentrations in the combination model with daptomycin could not be evaluated due to interactions with the microbioassay organism, but this is a well-described method to appropriately simulate model combination therapies in vitro (4). Nonetheless, the microbiologic findings from the model correspond well with those noted during patient treatment. It is likely that similar activity relationships with daptomycin and ceftaroline occur under normal clearance conditions.
Although these results are promising, additional studies are needed to determine the ideal beta-lactam agent and doses to use in combination with daptomycin for refractory MRSA infections with reduced daptomycin susceptibility.
Footnotes
Published ahead of print 6 August 2012
REFERENCES
- 1. Akins RL, Rybak MJ. 2000. In vitro activities of daptomycin, arbekacin, vancomycin, and gentamicin alone and/or in combination against glycopeptide intermediate-resistant Staphylococcus aureus in an infection model. Antimicrob. Agents Chemother. 44:1925–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayer AS, et al. 2000. In vitro resistance of Staphylococcus aureus to thrombin-induced platelet microbicidal protein is associated with alterations in cytoplasmic membrane fluidity. Infect. Immun. 68:3548–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belley A, et al. 2009. Oritavancin kills stationary-phase and biofilm Staphylococcus aureus cells in vitro. Antimicrob. Agents Chemother. 53:918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blaser J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J. Antimicrob. Chemother. 15(Suppl A):125–130 [DOI] [PubMed] [Google Scholar]
- 5. Chamberlain NR, et al. 1991. Correlation of carotenoid production, decreased membrane fluidity, and resistance to oleic acid killing in Staphylococcus aureus 18Z. Infect. Immun. 59:4332–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clinical and Laboratory Standards Institute 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 11th ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 7. Dhand A, et al. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin. Infect. Dis. 53:158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drusano GL. 2010. Pharmacodynamics of ceftaroline fosamil for complicated skin and skin structure infection: rationale for improved anti-methicillin-resistant Staphylococcus aureus activity. J. Antimicrob. Chemother. 65(Suppl 4):iv33–iv9 [DOI] [PubMed] [Google Scholar]
- 9. Dvorchik B, et al. 2004. Population pharmacokinetics of daptomycin. Antimicrob. Agents Chemother. 48:2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enright MC, et al. 2002. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. U. S. A. 99:7687–7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Entenza JM, Giddey M, Vouillamoz J, Moreillon P. 2010. In vitro prevention of the emergence of daptomycin resistance in Staphylococcus aureus and enterococci following combination with amoxicillin/clavulanic acid or ampicillin. Int. J. Antimicrob. Agents 35:451–456 [DOI] [PubMed] [Google Scholar]
- 12. Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS., II 2012. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J. Antimicrob. Chemother. 67:1267–1270 [DOI] [PubMed] [Google Scholar]
- 13. Jacqueline C, et al. 2011. Comparison of ceftaroline fosamil, daptomycin and tigecycline in an experimental rabbit endocarditis model caused by methicillin-susceptible, methicillin-resistant and glycopeptide-intermediate Staphylococcus aureus. J. Antimicrob. Chemother. 66:863–866 [DOI] [PubMed] [Google Scholar]
- 14. Jones T, et al. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother. 52:269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koreen L, et al. 2004. spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J. Clin. Microbiol. 42:792–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kullar R, et al. 2011. High-dose daptomycin for treatment of complicated gram-positive infections: a large, multicenter, retrospective study. Pharmacotherapy 31:527–536 [DOI] [PubMed] [Google Scholar]
- 17. Mishra NN, et al. 2011. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob. Agents Chemother. 55:526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mishra NN, et al. 2009. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:2312–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukhopadhyay K, et al. 2007. In vitro susceptibility of Staphylococcus aureus to thrombin-induced platelet microbicidal protein-1 (tPMP-1) is influenced by cell membrane phospholipid composition and asymmetry. Microbiology 153:1187–1197 [DOI] [PubMed] [Google Scholar]
- 20. Murillo O, et al. 2009. Efficacy of high doses of daptomycin versus alternative therapies against experimental foreign-body infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:4252–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patel D, et al. 2011. Mechanisms of in-vitro-selected daptomycin-non-susceptibility in Staphylococcus aureus. Int. J. Antimicrob. Agents 38:442–446 [DOI] [PubMed] [Google Scholar]
- 22. Peleg AY, et al. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316 doi:10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riccobene T, Jakate A, Rank D, Thye D. 2009. An open-label, pharmacokinetic, safety, and tolerability study of single-dose intravenous ceftaroline in subjects with end-stage renal disdaese on intermittent haemodialysis, abstr P1455. Abstr. 19th Eur. Congr. Clin. Microbiol. Infect. Dis., European Society of Clinical Microbiology and Infectious Diseases, Basel, Switzerland [Google Scholar]
- 24. Rose WE, et al. 2008. Daptomycin activity against Staphylococcus aureus following vancomycin exposure in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob. Agents Chemother. 52:831–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rose WE, Rybak MJ, Kaatz GW. 2007. Evaluation of daptomycin treatment of Staphylococcus aureus bacterial endocarditis: an in vitro and in vivo simulation using historical and current dosing strategies. J. Antimicrob. Chemother. 60:334–340 [DOI] [PubMed] [Google Scholar]
- 26. Sakoulas G, et al. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 56:838–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saravolatz LD, Stein GE, Johnson LB. 2011. Ceftaroline: a novel cephalosporin with activity against methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 52:1156–1163 [DOI] [PubMed] [Google Scholar]
- 28. Steed M, Vidaillac C, Rybak MJ. 2011. Evaluation of ceftaroline activity versus daptomycin (DAP) against DAP-nonsusceptible methicillin-resistant Staphylococcus aureus strains in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 55:3522–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steed ME, Vidaillac C, Rybak MJ. 2012. Evaluation of telavancin activity versus daptomycin and vancomycin against daptomycin-nonsusceptible Staphylococcus aureus in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 56:955–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong YQ, Mukhopadhyay K, Yeaman MR, Adler-Moore J, Bayer AS. 2005. Functional interrelationships between cell membrane and cell wall in antimicrobial peptide-mediated killing of Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3114–3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang SJ, et al. 2010. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”). Antimicrob. Agents Chemother. 54:3161–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]