

Abstract
Three hepatitis C virus (HCV) inhibitors, asunaprevir (ASV; BMS-650032), daclatasvir (DCV; BMS-790052), and BMS-791325, each targeting a different nonstructural protein of the virus (NS3, NS5A, and NS5B, respectively), have independently demonstrated encouraging preclinical profiles and are currently undergoing clinical evaluation. Since drug-resistant variants have rapidly developed in response to monotherapy with almost all direct-acting antiviral agents (DAAs) for HCV, the need for combination therapies to effectively eradicate the virus from infected patients is clear. These studies demonstrated the additive-synergistic effects on replicon inhibition and clearance of combining NS3 protease or NS5B RNA polymerase inhibitors with the first-in-class, NS5A replication complex inhibitor daclatasvir (DCV) and reveal new resistance pathways for combinations of two small-molecule inhibitors that differ from those that develop during monotherapy. The results suggest that under a specific selective pressure, a balance must be reached in the fitness costs of substitutions in one target gene when substitutions are also present in another target gene. Further synergies and additional novel resistance substitutions were observed during triple-combination treatment relative to dual-drug therapy, indicating that, in combination, HCV inhibitors can exert cross-target influences on resistance development. Enhanced synergies in replicon inhibition and a reduced frequency of resistance together lend strong support to the utility of combinations of DAAs for the treatment of HCV, and the identification of altered resistance profiles during combination treatment provides useful information for monitoring resistance in the clinic.
INTRODUCTION
Hepatitis C virus (HCV) is a positive-stranded RNA virus in the Flaviviridae family of enveloped virions which affects an estimated 170 million people worldwide and is the major cause of chronic hepatitis. Currently, approximately 50% of patients infected with genotype 1 (gt 1), the most prevalent form of the virus, fail to achieve a sustained reduction in viral load with therapy employing pegylated alpha interferon (IFN-α) plus ribavirin (alfa/RBV) (52, 54, 56). A substantial fraction (20%) of chronically infected patients develop serious progressive liver disease, including cirrhosis or hepatocellular carcinoma. alfa/RBV treatment is associated with a high incidence (>30%) of adverse effects, some of which are of sufficient severity to cause patients to discontinue therapy (56). Despite the recent approval of two new direct-acting antiviral agents (DAAs), boceprevir and telaprevir, for use in combination with alfa/RBV (18, 47), their use may be limited by poor efficacy in some patient populations, inconvenient 3-times-daily dosing of the DAA, and association with side effects, including anemia, rash, and gastrointestinal effects, in addition to the well-documented spectrum of adverse effects associated with alfa/RBV. Although addition of these DAAs to the standard of care for HCV represents a significant improvement in patient therapy, there is still an unmet medical need for new agents and more-tolerable treatment regimens for newly diagnosed patients and those failing current therapies.
The 9.6-kb HCV genome encodes a polyprotein of about 3,000 amino acids via translation of a single, uninterrupted open reading frame. The polyprotein is cleaved co- and posttranslationally in infected cells by cellular and virus-encoded proteases to produce a multicomponent replication complex (8, 33). The serine protease encoded by the N-terminal region of NS3 is thought to be responsible for all downstream cis and trans proteolytic cleavages (9, 17). NS5A possesses no known enzymatic activity, but exists in different states of phosphorylation, and influences multiple functions at various stages of the viral replication cycle (41, 58). It has been shown to interact with an extensive array of host proteins and to play a role in IFN resistance (37, 40). NS5B is the RNA-dependent RNA polymerase responsible for replication of HCV RNA (1, 4).
The essential roles of nonstructural proteins NS3 to NS5 in viral replication render each an attractive target for antiviral intervention (2). Clinical proof of concept has been achieved for a number of DAAs targeting some of these proteins, including the serine protease activity of NS3 (11, 16, 25, 31, 32, 45, 53) and the RNA-dependent RNA polymerase activity of NS5B (20, 26, 46; H. Tatum et al., poster 1163, presented at the 47th European Association for the Study of the Liver [EASL] Congress, Barcelona, Spain, 18 to 22 April 2011). More recently, daclatasvir (DCV) (Table 1) was the first NS5A replication complex inhibitor to show proof of concept in the clinic, demonstrating in early clinical testing the potential for this class of inhibitor to become a valuable component of an all-oral treatment regimen for HCV (15).
Table 1.
In vitro antiviral profiles of BMS HCV inhibitorsa
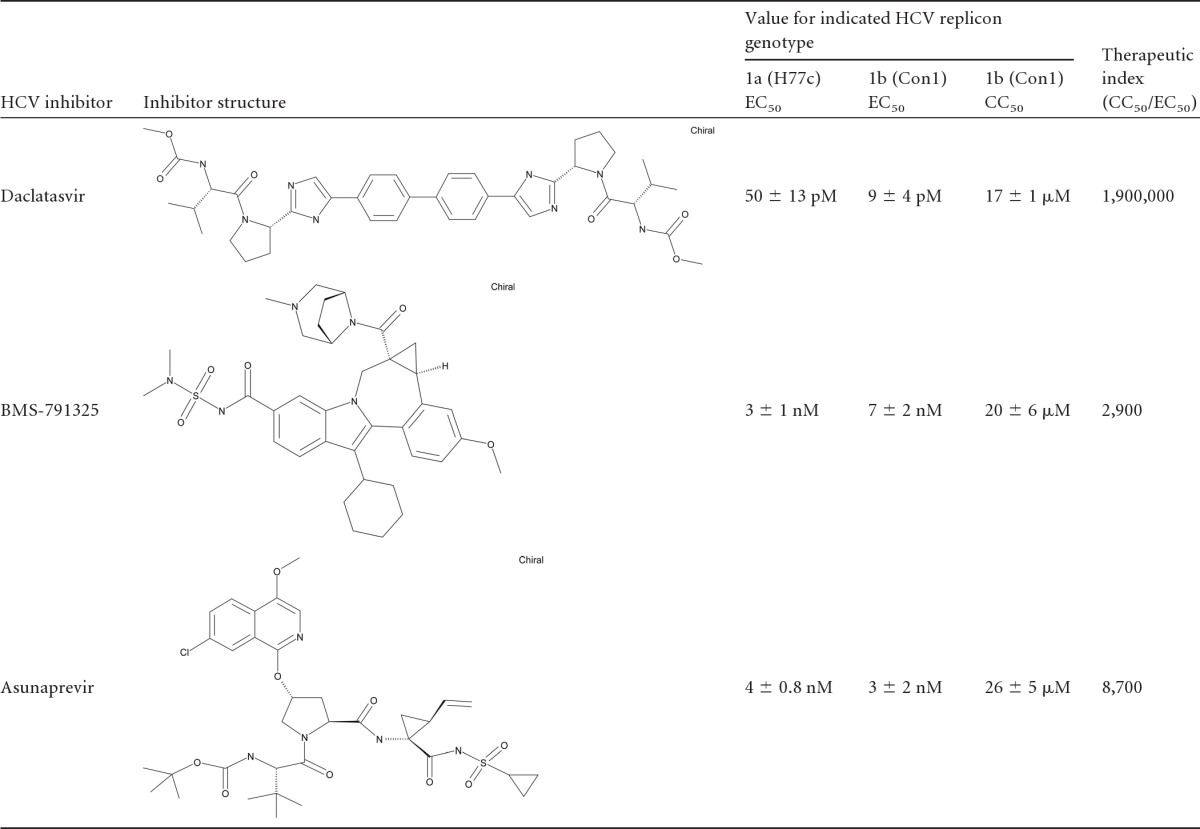
Data shown represent results of ≥3 independent tests ± standard deviations.
The high turnover rate and error-prone nature of the HCV RNA polymerase contribute to the production of potentially resistant viral quasispecies. In practice, resistance has emerged to all small-molecule inhibitors of HCV tested as monotherapy except some nucleoside-nucleotide NS5B inhibitors. Resistance mutations have been identified both in vitro and in vivo upon treatment with nearly all inhibitors of HCV serine protease, NS5A, or allosteric RNA polymerase inhibitors advanced to date (3, 19, 22, 23, 24, 30, 36, 38, 51, 52, 57, 60, 62), with good correlation observed between resistance emergence in the replicon system and in vivo. Recent literature indicates that treatment with combinations of non-cross-resistant inhibitors not only improves antiviral activity during treatment but also suppresses the posttreatment viral rebound often associated with monotherapy (19, 21, 26). To achieve a sustained viral response (SVR), it would be essential to use combination therapies similar to those that have recently been explored in replicon studies (5, 10, 29), animal models (44), and patient studies (14, 51) as a viable approach to improving the efficacy, tolerability, and compliance issues associated with current therapies. For this report, the effects of a combination approach to HCV therapy have been studied in the HCV replicon system using two- and three-drug combinations that include an NS5A replication complex inhibitor (DCV), the NS3 protease inhibitor asunaprevir (ASV), and the nonnucleoside NS5B RNA polymerase inhibitor BMS-791325.
MATERIALS AND METHODS
Cell lines.
Bovine viral diarrhea virus (BVDV) and HCV replicon cell lines were previously described (27, 43) and were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 2 mM l-glutamine, 10% fetal bovine serum (FBS), and penicillin-streptomycin, with or without 0.3 to 0.5 mg/ml Geneticin (G418).
Efficiency of replicon clearance from cultured cells.
HCV replicon cells (6 × 104 per well in 6-well plates) and BVDV replicon cells (4 × 104 per well) were treated with various 50% effective concentrations (EC50s) of inhibitors in cell growth media for 1 week. After 7 days, media containing inhibitor were removed and cells were maintained in growth media containing 0.5 mg/ml G418. Media were changed twice weekly for a period of ∼4 weeks. Plates were washed, and colonies were counted after cells were stained with 0.2% crystal violet. All conditions were tested in duplicate and repeated in separate experiments.
Selection of populations with reduced susceptibility to HCV inhibitors.
HCV replicon cells were plated at a density of 6 × 104 per 60-mm-diameter plate and maintained in growth media with 0.3 mg/ml G418 and various concentrations of inhibitor(s). BVDV replicon cells, plated at 2 × 104 per 60-mm-diameter plate, were maintained in growth media containing 0.5 mg/ml G418 and treated in parallel with the same inhibitors. Fresh media containing compound were added every 3 to 5 days for a total of 4 weeks, after which plates were washed and cells were stained with 0.2% crystal violet or further selected for testing. All concentrations were tested in duplicate, and selections were repeated in separate experiments. gt 1b cultures undergoing triple-combination treatment required gradual (1.5- to 2-fold) ramping of drug concentrations, starting with the 5×-selected population, allowing 4 weeks incubation for each escalation in concentration in order to select high-level resistance.
HCV replicon luciferase and FRET assays.
To evaluate compound efficacy, HCV replicon cells were incubated in 96-well plates in the presence of compound for 3 days. For replicons containing a luciferase reporter gene, Renilla luciferase activity was then assayed using a Renilla Luciferase Assay System or DualGlo Luciferase Assay System (Promega Corporation, Madison, WI), according to the manufacturer's directions. Plates were read on a TopCount NXT Microplate Scintillation and Luminescence Counter (Packard Instrument Company, Meriden, CT). For replicons lacking a reporter gene, a fluorescence resonance energy transfer (FRET) assay monitored NS3 protease activity as a measure of HCV replicon levels (43). The 50% effective concentration (EC50) was calculated using the four-parameter logistic formula Y = A + {(B − A)/[1 + (C/x)D]}, where A and B denote minimal and maximal percent inhibition, respectively, C represents the EC50, D represents the hill slope, and x represents the compound concentration.
Cell-based inhibitor combination assays.
For combination studies, each inhibitor was tested at 11 concentrations. The compounds were tested as monotherapies and in combinations at various concentration ratios. Cells were exposed to compounds for 3 days, and the amount of HCV inhibition was then determined using the Dual-Glo luciferase assay as described above. The potential cytotoxicities of these combined agents were also analyzed in parallel by alamarBlue staining. The 50% cytotoxic concentration (CC50) values were calculated using the four-parameter logistic formula described above.
The degree of antagonism, additivity, or synergy was determined from combination dose-response curves which were fit to assess the antiviral effects of the drug treatment combinations. The concentration ratios were analyzed using the method of Chou (6). All estimates were computed using SAS Proc NLIN biostatistical software and a four-parameter logistic. Combination indices were tested for departure from additivity using isobologram methods. Asymptotic confidence intervals were also calculated for each of the combination indices. These intervals are used to test for departure from additivity by comparing the bounds to a value of 1—a lower bound of the interval greater than 1 indicates antagonism, an upper bound of less than 1 indicates synergism, and a value of 1 contained in the interval indicates additivity.
Identification of mutations selected in resistant populations.
RNA was isolated from populations of resistant cells using either TRIzol or an RNeasy 96 kit (Qiagen Inc., Valencia, CA) in accordance with the manufacturer's directions. First-strand cDNA synthesis was performed using 1 to 3 μg of total RNA and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) primed with gene-specific oligonucleotide primers. PCR was performed using the cDNA and pairs of primers flanking the genes of interest (NS3 to NS5B). PCR products were sequenced, and mutations were identified relative to vehicle-treated populations. PCR products were purified and cloned using TOPO PCR cloning methods (Invitrogen, Carlsbad, CA). DNA was sequenced from ∼100 clones to establish the frequency and coincidence of mutations in a population.
To generate mutant replicons, point mutations were generated with a QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions, and mutations were confirmed by sequencing.
Transient replication assays.
Replicon clones were linearized with Sca1 and transcribed in vitro using an Ambion T7 MegaScript kit (Ambion, Austin, TX) or a T7 RiboMAX Express Large-Scale RNA Production System (Promega, Madison, WI) according to manufacturer's directions. Transcribed RNA (3 to 5 μg) was transfected into cured Huh-7 cells (∼2 × 106 in 60-mm-diameter dishes) with DMRIE-C reagent (Invitrogen Corporation, Carlsbad, CA) following the manufacturer's protocol. After 4 to 6 h, transfected cells were transferred to 96-well assay plates (104 cells/well) and incubated in the presence of inhibitors for 72 h. Renilla luciferase assays were performed as described above.
RESULTS
Inhibition of replicon with combinations of HCV inhibitors.
The HCV replicon system is widely acknowledged as a predictive tool for the development of DAAs that target HCV RNA replication (2). In the gt 1 HCV replicon system, DCV, ASV, and BMS-791325 exhibit picomolar to low-nanomolar potency (Table 1). The virology profile and clinical effects of DCV (15, 42, 45), ASV (37, 38), and BMS-791325, a nonnucleoside allosteric inhibitor of NS5B that binds a site on the surface of the thumb domain of the RNA-dependent RNA polymerase (Tatum et al., poster 1163; J. F. Kadow et al., presented at the American Chemical Society Spring 2012 National Meeting and Exposition, San Diego, CA, 25 to 29 March, 2012; Gentles et al., unpublished data), have been reported. These inhibitors were used to investigate the spectrum of events resulting from simultaneously targeting multiple essential proteins of HCV in vitro. Of particular interest was determining the effect of the NS5A replication complex inhibitor DCV in combination with other DAAs.
We used the method of Chou (6) to evaluate the additive, antagonistic, or synergistic effects of combination therapies on HCV inhibition in a 3-day replicon assay. Table 2 shows the results of testing DCV and other inhibitors as monotherapies or in combination in HCV replicon experiments. The CC50s of these combined agents, analyzed in parallel by alamarBlue staining, were greater than the highest tested inhibitor concentration. The effects of DCV in combination with ASV or BMS-791325 indicate mixed additivity and/or synergy over a range of molar ratios of inhibitors. When the NS5A, NS5B, and NS3 inhibitors were tested in a 3-drug combination, additive effects were observed at all effective doses. Taken as a whole, the results from multiple experiments demonstrate that combinations of these inhibitors display mixed additivity and/or synergy at the 50%, 75%, and 90% effective dose levels of drug. Importantly, no antagonistic effects were observed with any of these combinations and no increase in cytotoxicity was observed.
Table 2.
Combination of daclatasvir with asunaprevir or BMS-791325
| Expt | Ratio of indicated inhibitor to daclatasvir in paired combinations | Overall result(s) for indicated combination |
||
|---|---|---|---|---|
| Asunaprevir + daclatasvir | BMS-791325 + daclatasvir | Asunaprevir + BMS-791325 + daclatasvir (1:1:1) | ||
| 1 | 1:1 | Synergy/additivity | Synergy | Additivity |
| 2.5:1 | Synergy | Additivity | ||
| 1:2.5 | Synergy | Synergy | ||
| 2 | 1:1 | Additivity | Synergy/additivity | Additivity |
| 2.5:1 | Synergy/additivity | Additivity | ||
| 1:2.5 | Synergy/additivity | Synergy | ||
| 3 | 1:1 | Synergy | Synergy/additivity | Additivity |
| 2.5:1 | Additivity | Synergy/additivity | ||
| 1:2.5 | Synergy | Synergy | ||
Resistance selection with combinations of HCV inhibitors.
Upon prolonged exposure of replicon cells to HCV inhibitors, colonies develop as resistant variants are selected. Figure 1 shows the relative densities of resistant colonies under conditions in which DCV was combined with either ASV (panel a) or BMS-791325 (panel b) in cultures examined after continuous 4-week DAA exposure in the presence of G418. In multiple experiments, populations of resistant colonies were markedly reduced when cells were treated with a combination of DCV and ASV (Fig. 1a) or of DCV and BMS-791325 (Fig. 1b) compared with resistant populations resulting from treatment with similar EC50 multiples of the DAAs alone. BVDV replicon cells treated in parallel showed no reduction in colony formation, suggesting a specific anti-HCV effect (data not shown). Interestingly, fewer resistant colonies were observed with combinations composed of 5× the EC50 of DCV with any concentration of NS5B or NS3 inhibitor than were seen with alternate combinations pairing 5× the EC50 of either the NS5B or NS3 inhibitor with any concentration of DCV (Fig. 1a and b). For example, although both plates were exposed to a total of 35× the EC50 of DAA, the plate combining 5× DCV with 30× BMS-791325 developed far fewer resistant colonies than the plate combining 30× DCV with 5× BMS-791325 (Fig. 1b). A similar phenomenon was observed for DCV in combinations with ASV (Fig. 1a).
Fig 1.

Combination treatment reduces the emergence of resistant colonies. gt 1b HCV replicon cells were incubated for 4 weeks with BMS-790052 (DCV), BMS-791325, or BMS-650032 (ASV) as monotherapy and dual therapy (a and b) and triple therapy (c) at 5×, 10×, and 30× EC50. Colonies were visualized by crystal violet staining. Data shown are representative of the results of three independent experiments.
Triple-DAA treatment of HCV replicon cells with inhibitors at 10× and 30× their respective EC50s resulted in complete clearance of HCV replicon (Fig. 1c) while having no effect on BVDV replicon cells (not shown). In the presence of 5× the EC50 of the three DAAs (total, 15 EC50 multiples), fewer colonies were observed than after treatment with a 30× concentration of any single DAA or many pairs of DAAs. Similarly, in the gt 1a replicon, combinations of two or three DAAs more effectively reduced resistant colony formation than any single DAA at similar EC50 multiples. The overall suppressive effect in gt 1a was slightly reduced compared to that in gt 1b (not shown).
Replicon clearance studies were also performed with the DAAs in the absence of G418 selection to evaluate the ability of various inhibitor combinations to eradicate HCV replicon RNA from the cells. Similar to the resistance studies, replicon clearance occurred more efficiently in gt 1b than gt 1a. In both gt 1a and gt 1b, low EC50 multiples of DCV effected a much greater reduction in resistant-colony formation than the same multiple of the other DAAs and a greater-than-additive effect on replicon clearance was observed using combinations of DAAs. Overall clearance with the DAA dual combinations in gt 1a was reduced 2- to 4-fold compared to gt 1b at 3× and 9× the EC50; the difference was greater (6- to 17-fold) at 27× the EC50.
Genotypic and phenotypic analysis of resistant variants.
Resistant variants identified from selection with each DAA inhibitor class used in these studies have been previously described (12, 13, 27, 29, 30, 38, 45, 52, 61). The major gt 1b-resistant substitutions are at residues 168 for NS3, 31 and 93 for NS5A, and 495 for NS5B, while the major gt 1a-resistant substitutions are at residues 155 for NS3, 28, 30, 31, and 93 for NS5A and 495 for NS5B. To study the impact of different DAA combinations on the emergence of resistance, and in an attempt to use these HCV-specific inhibitors as a tool to study potential interactions of viral proteins, this report emphasizes only the genotypic and phenotypic analysis of unique or less frequently observed substitutions identified from dual- and triple-DAA selections. Resistant variants selected with one, two, and three DAAs in HCV 1a and 1b replicons were analyzed by both population and clonal sequencing to determine the percentage and linkage of mutations within and between genes in the population. Substitutions observed with a frequency of ≥10% were engineered into wild-type (WT) replicon to evaluate the contribution of the altered residue to resistance and impact on replication fitness in transient replication assays.
Analysis of NS5A and NS3 dually resistant populations.
Similar to previous reports, the major gt 1b resistance substitutions selected by DCV and ASV were L31M and Y93H in NS5A (12, 27) and D168V in NS3 (38, 45) (Table 3), respectively. For the dual selection performed in parallel, the most striking substitution was N77S in NS3. The N77S variant in gt 1b under dual selection conditions with boceprevir and a 2-C-methyl-adenosine NS5B inhibitor has been previously reported (5). In that case, N77S exhibited no phenotype, as it conferred no resistance to boceprevir and did not affect replication efficiency. However, in this case, N77S conferred a low level (3- to 9-fold) of resistance to ASV by itself and, in combination with R155Q, which was itself associated with 8- to 9-fold ASV resistance, demonstrated an amplified level of resistance of 93- to 113-fold in the dually substituted N77S-R155Q variant. In NS5A, R30Q/H linked with L31M emerged in the dual selection. R30H, which has not been reported before, conferred minimal (2- to 8-fold) resistance to DCV but displayed significant (150- to 330-fold) resistance in combination with L31M. When linkage of N77S-R155Q in NS3 occurred with multiple substitutions in NS5A (R30Q-L31M-Y93H), very high levels of resistance to both DCV (31,000- to 37,000-fold) and ASV (80- to 210-fold) were observed. The observation of the N77S substitution in NS3 only during dual selection was unexpected and indicates the influence of the NS5A inhibitor on the emergence of NS3 resistance in vitro. In gt 1a replicon cells, R155K was selected by ASV, whereas a D168G substitution was observed also during dual selection with ASV and DCV, although substitutions at R155 and D168 in NS3 were not linked (Table 4). Selection with DCV yielded two predominant NS5A substitutions, Q30H and K68R, while in the dual selection, an additional NS5A resistance substitution, M28T, was also observed. The novel arginine substitution at residue 68 of NS5A did not confer any resistance but appeared to increase the relative fitness of the 1a variant (1 to 4 times the efficiency of the WT), whereas replication of the Q30H-K68R mutant was greatly impaired. A dramatic improvement was observed with the dually selected D168G/Q30H-K68R clone, which replicated at 30% to 70% of the level of the WT whereas each of the NS3 and NS5A mutants alone had extremely impaired replication efficiency (4% to 9% the level of the WT), suggesting an NS3-NS5A interaction that restored replicase activity. Interestingly, combination of the Q30H-K68R substitutions in NS5A with D168G in NS3 reduced the level of NS5A resistance.
Table 3.
Characterization of gt 1b variants identified in dual selections using transient replication assays
| Selection condition | Varianta |
Fold resistanceb |
Replication efficiency relative to WT level | ||||
|---|---|---|---|---|---|---|---|
| NS3 | NS5A | NS5B | Asunaprevir (ASV) | Daclatasvir (DCV) | 325c | ||
| 5× or 30× ASV | D168V | — | — | >100 | 0.9 | 2 | 0.3–0.5 |
| 5× DCV | — | L31M | — | 1 | 2–5 | 1 | 0.5–1.5 |
| 30× DCV | — | Y93H | — | 1 | 10–33 | 1 | 0.1–0.3 |
| — | L31M-Y93H | — | NDd | 4,200e | ND | 0.4 | |
| 5× or 20× 325 | — | — | P495A | 1 | 1 | 9–16 | 0.3–0.8 |
| 20× 325 | — | — | P495S | 1 | 1 | 40–86 | 0.01–0.1 |
| 5× ASV/DCV | N77S | R30Q-L31M | — | 5–11 | 6–16 | 1 | 0.5–1.2 |
| 30× ASV/DCV | N77S-R155Q | R30Q-L31M-Y93H | — | 80–210 | 31,000–37,000 | 1 | 0.2–0.5 |
| N77S-R155Q | R30H-L31M | — | 62–170 | 150–330 | 1 | 0.3–0.7 | |
| 5× DCV/325 | — | R30Q-L31M | P495A | 1 | 12–22 | 9–20 | 0.4–0.8 |
| 20× DCV/325 | — | R30Q-L31F | P495S | 1 | 46–62 | 111–128 | 0.1 |
| Site-directed mutant | N77S | — | — | 3–9 | 1 | 1 | 0.2–0.5 |
| Site-directed mutant | R155Q | — | — | 8–9 | 1 | 1 | 0.01–0.02 |
| Site-directed mutant | N77S-R155Q | — | — | 93–113 | 1 | 1 | 0.02–0.1 |
| Site-directed mutant | — | R30Q | — | 1 | 2 | 1 | 0.7–1.1 |
| Site-directed mutant | — | R30H | — | 1 | 2–8 | 1 | 0.1–0.2 |
| Site-directed mutant | — | R30Q-L31M | — | 1 | 4–16 | 1 | 0.9–1.2 |
| Site-directed mutant | — | R30Q-L31F | — | 2 | 57–85 | 1 | 1.0–1.2 |
| Site-directed mutant | — | R30Q-L31M-Y93H | — | 1 | 25,000–51,000 | 1 | 0.7–1.4 |
Replicons containing specific substitution(s) identified from selections were tested for resistance and replication fitness. Major selected variant constructs are in bold font. Site-directed mutants were not selected but were generated as controls in the WT backbone to assess impact on phenotype. For each selection condition, each row represents a unique variant that includes any linked mutations. —, WT sequence.
Fold resistance was determined by dividing the mutant HCV replicon EC50 by the WT replicon EC50. WT EC50s were 2.0 ± 0.6, 0.002 ± 0.001, and 8.3 ± 2.2 nM for ASV, DCV, and BMS-791325, respectively. Results represent the ranges of values determined from two or three independent experiments.
325, BMS-791325.
ND, not determined.
Value was taken from the work of Fridell et al. (12).
Table 4.
Characterization of gt 1a variants identified in the dual selections using transient replication assays
| Selection condition | Varianta |
Fold resistanceb |
Replication efficiency relative to WT level | ||||
|---|---|---|---|---|---|---|---|
| NS3 | NS5A | NS5B | Asunaprevir (ASV) | Daclatasvir (DCV) | 325c | ||
| 10× or 30× ASV | R155K | — | — | 17–48 | 1 | 1 | 0.3–0.5 |
| 10× or 30× DCV | — | Q30H-K68R | — | 1 | 1,280–2,000 | 3 | 0.04–0.07 |
| 10× or 30× 325 | — | — | A421V | 1 | 1 | 1–3 | 0.3–0.9 |
| — | — | P495L | 2 | 1 | 88–100 | 0.07–0.1 | |
| 30× DCV/ASV | R155K | Q30H-K68R | — | 46–68 | 667–700 | 1 | 0.5–0.8 |
| R155K | M28T-K68R | — | 34–61 | 1,125–13,333 | 1 | 0.4–1 | |
| D168G | Q30H-K68R | — | 18–30 | 111–333 | 1 | 0.3–0.7 | |
| 30× DCV/325 | — | Q30H-K68R | A421V | 1 | 1000 | 2 | 0.8–1 |
| — | Q30H-K68R | P495L | 2 | 1600 | 61–84 | 0.03–0.07 | |
| — | Q30H-K68R | L392I | 2 | 2200 | 6–16 | 0.06–0.19 | |
| Site-directed mutant | D168G | — | — | 8–16 | 1 | 1 | 0.07–0.09 |
| Site-directed mutant | — | M28T | — | 1 | 750–820 | 1 | 0.2–0.4 |
| Site-directed mutant | — | Q30H | — | 1 | 1111–1967 | 1 | 0.4–0.6 |
| Site-directed mutant | — | K68R | — | 1 | 1 | 1 | 1–4.6 |
| Site-directed mutant | — | — | L392I | 1 | 1 | 5–7 | 0.1–0.4 |
Replicons containing specific substitution(s) identified from selections were tested for resistance and replication fitness. Major selected variant constructs are in bold font. Site-directed mutants were not selected but were generated as controls in the WT backbone to assess impact on phenotype. For each selection condition, each row represents a unique variant that includes any linked mutations. —, WT sequence.
Fold resistance was determined by dividing the mutant HCV replicon EC50 by the WT replicon EC50. WT EC50s were 0.7 ± 0.3, 0.006 ± 0.002, and 2.2 ± 0.9 nM for ASV, DCV, and BMS-791325, respectively. Results represent the range of values determined from two or three independent experiments.
325, BMS-791325.
Analysis of NS5A-NS5B dually resistant populations.
gt 1b replicon cells treated with DCV and BMS-791325 alone or in combination yielded resistant populations with substitutions at known positions in NS5A (R30, L31, Y93) (12) and NS5B (P495) (J. A. Lemm et al., unpublished data) (Table 3). For dual selection with NS5A and NS5B inhibitors, L31M and L31F were linked with R30Q (Table 3) whereas only L31M was selected by DCV alone. Phenotypic analysis of R30Q-L31F, with or without NS5B substitution P495S, revealed much greater (46- to 85-fold) resistance to DCV than the single L31F (5-fold) (12) and R30Q (Table 3) substitutions. While the NS5A R30Q-L31F variant replicated as efficiently as the WT, linkage with the NS5B P495S substitution greatly impaired replication (Table 3).
In gt 1a, selection with BMS-791325 yielded substitutions of A421V and P495L in NS5B whereas, as stated earlier, DCV selection generated the Q30H-K68R variant in NS5A (Table 4). In dual selections, an additional L392I substitution in NS5B was also observed. Clonal analysis of the dual selections revealed linkage of Q30H-K68R in NS5A with each of the single NS5B substitutions (L392I, A421V, P495L), as no two NS5B changes were found in the same clone. The A421V NS5B substitution conferred no resistance to BMS-791325; however, it did increase the replication efficiency of the Q30H-K68R NS5A variant 10- to 15-fold (Table 4). The L392I substitution in NS5B has not been reported for gt 1a, although it was previously shown in gt 1b to confer low-level resistance (15- to 20-fold) to an indole-N-acetamide nonnucleoside inhibitor (50). In gt 1a, L392I conferred low-level (5- to 16-fold) resistance to BMS-791325 while the P495L variant gave the expected high-level (60- to 100-fold) resistance.
Triple selection with NS5A, NS5B, and NS3 inhibitors.
gt 1b cells treated with a fixed concentration greater than 5× the EC50 of DCV, ASV, and BMS-791325 together were unable to survive. However, resistant variants emerged when cells were subjected to serial passage with gradually increasing concentrations of the three inhibitors. Populations treated in this way with up to 15× the EC50 selection consisted of an interesting mix of previously seen and new variants. Compared to the dual selections, a different set of NS3 substitutions, Q41R with Q80R and Q80R with R155Q, were selected during triple-DAA treatment. Presumed to be adaptive or compensatory changes, substitutions of glutamine residues at positions 41 and 80 in NS3 are well documented, and Q80R was selected with very low frequency in a gt 1b replicon upon treatment with ASV (38). ASV resistance to Q41R or Q80R was low but measurable (4- to 5-fold) and roughly additive (7- to 12-fold) under conditions in which both substitutions were present (Table 5). The combination of Q80R with R155Q conferred 30- to 100-fold-greater resistance to ASV, which was 4× to 10× higher than was observed with each NS3 mutation alone. Despite differences in the NS3 sites affected by the triple-DAA regimen, the sites of resistance in NS5A and NS5B during treatment with a triple-drug combination were the same as those targeted with dual-drug selection regimens: R30, L31, and Y93 in NS5A and P495 in NS5B (Table 5). Replication efficiency for these reconstituted triply resistant variants was significantly impaired.
Table 5.
Characterization of gt 1b variants identified in the triple selection using transient replication assays
| Selection condition | Varianta |
Fold resistanceb |
Replication efficiency relative to WT level | ||||
|---|---|---|---|---|---|---|---|
| NS3 | NS5A | NS5B | Asunaprevir (ASV) | Daclatasvir (DCV) | BMS-791325 | ||
| 10× or 15× triplec | Q80R-R155Q | R30Q-L31M | P495A | 30–93 | 5–15 | 17–22 | 0.02–0.1 |
| Q80R-R155Q | R30Q-L31M-Y93H | P495A | 86–99 | >500 | 8–22 | 0.01 | |
| Site-directed mutant | Q41R | — | — | 4 | 1 | 1 | 1.0–1.4 |
| Site-directed mutant | Q80R | — | — | 4–5 | 1 | 1 | 0.5–1.0 |
| Site-directed mutant | Q41R-Q80R | — | — | 7–12 | 1 | 1 | 1.0–1.2 |
| Site-directed mutant | Q80R-R155Q | — | — | 42–73 | 1 | 1 | 0.5–1.0 |
Replicons containing a specific substitution(s) identified from selections were tested for resistance and replication fitness. Major selected variant constructs are in bold font. Site-directed mutants were not selected but were generated as controls in the WT backbone to assess impact on phenotype. For each selection condition, each row represents a unique variant including any linked mutations. —, WT sequence.
Fold resistance was determined by dividing the mutant HCV replicon EC50 by the WT replicon EC50. WT EC50s were 2.0 ± 0.6, 0.002 ± 0.001, and 8.3 ± 2.2 nM for ASV, DCV, and BMS-791325, respectively. Results represent the range of values determined from two or three independent experiments.
Selection of viable triple mutants in gt 1b required sequential passage in gradually increasing concentrations of inhibitor as detailed in Materials and Methods.
Triple selection in the gt 1a replicon cells readily yielded a cell line that displayed significant resistance to all three inhibitors. Along with known substitutions at amino acids 155 and 168, several additional substitutions were identified in NS3, and clonal analysis revealed 100% linkage between NS3 D168E and M179T, R155K and M179A, and Q80R and Y134H, as well as between T389I and A421V in NS5B (Table 6). The Q89R, Y134H, and M179A/T NS3 mutations showed no resistance to ASV and did not significantly affect resistance levels in combination with other substitutions except M179A, which slightly (3- to 5-fold) enhanced R155K resistance in the context of the triple mutant (Table 6). Alone, the Q89R change significantly enhanced replication (2 to 6× the efficiency of the WT) and may play a similar role in the fitness of triple mutants to allow replication of these highly altered replicons. Along with substitutions at amino acid 495, a new combination of NS5B mutations, T389I-A421V, that conferred low-level (3- to 8-fold) resistance to BMS-791325 was identified in the triple selection. It also appears that high-level resistance to NS3 and NS5B inhibitors was not selected in the same clone. High-level NS5B resistance due to the P495L substitution is associated with low-level NS3 resistance from Q80R, and, conversely, high-level NS3 resistance (Q89R-R155K-M179A) is linked to T389I-A421V changes in NS5B which confer low-level resistance. Levels of NS5A resistance remain consistent in all three triple mutants. Table 7 provides a summary of the resistant substitutions observed during treatment with combinations of DAAs in these studies. However, as reported here, additional replication-enhancing substitutions, particularly during triple-DAA treatment, were also observed.
Table 6.
Characterization of gt 1a variants identified in the triple selection using transient replication assays
| Selection condition | Varianta |
Fold resistanceb |
Replication efficiency relative to WT level | ||||
|---|---|---|---|---|---|---|---|
| NS3 | NS5A | NS5B | Asunaprevir (ASV) | Daclatasvir (DCV) | BMS-791325 | ||
| 30× triple | Q80R-Y134H | Q30H-K68R | P495L | 10–12 | 1,000 | 25–70 | 0.03–0.07 |
| Q89R-D168E-M179T | Q30H-K68R | A421V | 15–35 | 1,500–2,200 | 2 | 0.05–0.12 | |
| Q89R-R155K-M179A | Q30H-K68R | T389I-A421V | 67–170 | 800–1,375 | 5–8 | 0.72–1.9 | |
| Site-directed mutant | — | Q30H-K68R | T389I-A421V | 2 | 1,400 | 4–5 | 0.2–0.5 |
| Site-directed mutant | Q80R | — | — | 6–11 | 2 | 2 | 0.4–0.6 |
| Site-directed mutant | Q89R | — | — | 2 | 1 | 1 | 2.0–6 |
| Site-directed mutant | Y134H | — | — | 2 | 2 | 2–3 | 0.2–0.3 |
| Site-directed mutant | D168E | — | — | 47–74 | 1 | 1 | 0.6–1.2 |
| Site-directed mutant | M179A | — | — | 1 | 1 | 1 | 0.6–1.2 |
| Site-directed mutant | M179T | — | — | 1 | 1 | 1 | 0.3–0.7 |
| Site-directed mutant | Q89R-R155K | — | — | 26–67 | 2 | 1 | 0.4–1.1 |
| Site-directed mutant | — | — | T389I-A421V | 1 | 1 | 3–5 | 0.5–1.2 |
| Site-directed mutant | — | — | T389I | 1 | 1 | 1 | 1.1–2.6 |
Replicons containing specific substitution(s) identified from selections were tested for resistance and replication fitness. Major selected variants are in bold font. Site-directed mutants were not selected but were generated as controls in the WT backbone to assess impact on phenotype. For each selection condition, each row represents a unique variant that includes any linked mutations. —, WT sequence.
Fold resistance was determined by dividing the mutant HCV replicon EC50 by the WT replicon EC50. WT EC50s were 0.7 ± 0.3, 0.006 ± 0.002, and 2.2 ± 0.9 nM for ASV, DCV, and BMS-791325, respectively. Results represent the range of values determined from two or three independent experiments.
Table 7.
Summary of predominant resistance substitutions selected during inhibitor treatment
| Target(s) of inhibitors used for selection | Resistance substitution(s) |
|||||
|---|---|---|---|---|---|---|
| Genotype 1b |
Genotype 1a |
|||||
| NS3 | NS5A | NS5B | NS3 | NS5A | NS5B | |
| NS3 | D168V | R155K | ||||
| NS5A | L31M, Y93H | Q30H | ||||
| NS5B | P495A/S/L | P495L | ||||
| NS3, NS5A | N77S, R155Q | R30Q/H, L31M, Y93H | R155K, D168N/G | Q30H, M28T | ||
| NS5A, NS5B | R30Q/H, L31M/F, Y93H | P495A/S/L | Q30H | L392I, P495S/L | ||
| NS3, NS5A, NS5B | Q41R, Q80R, R155Q | R30Q/H, L31M, Y93H | P495A | Q80R, R155K, D168E | Q30H | P495L, T389I-A421V |
DISCUSSION
Having shown promising antiviral activity in early clinical trials (15), the picomolar NS5A replication complex inhibitor DCV was examined in combination with other DAAs targeting distinct steps in the replication process. Since it has yet to be established whether efficacy and resistance profiles for combinations of DAAs reflect the sum of the profiles of the individual drugs, in vitro replicon studies may provide insight into clinical outcomes for combinations of non-cross-resistant DAAs in terms of impact on viral replication and emergent resistance pathways.
In 72-h replicon assays, combinations of DCV with NS3 protease and/or NS5B polymerase inhibitors demonstrated additive to synergistic inhibitory effects on replicon activity. Replicon curing performed over a 7-day treatment period provided evidence of a similar, greater-than-additive effect from combining these inhibitors. Clearance from gt 1b replicons was generally twice as efficient as from gt 1a, a genotype difference that appears to extend to clinical outcomes for DCV (15, 34). Moreover, a low EC50 multiple of DCV coupled with either the NS3 or NS5B-targeted DAA had a much more profound impact on limiting resistance development than did a low EC50 multiple of either NS3 or NS5B inhibitor combined with DCV. The impressive dose-response curve for curing with DCV compared to the other inhibitors suggests that NS5A inhibition more readily attenuates the ability of HCV replicons to survive. Since data suggest homotypic oligomeric interactions of the NS5A protein (7, 35, 59), it is possible that binding of a single NS5A inhibitor molecule may induce conformational changes that translate to adjacent NS5A molecules, cooperatively impacting the functionality of the entire replication complex. Higher concentrations of NS5A would not have such an impact, as the system may become saturated. Such a model could explain the steep dose-response curve of clearance with DCV and suggests an advantage of inclusion of DCV in DAA combination regimens.
It is known that minor changes in selection conditions can affect the pattern of resistance observed. Here we performed selections with single and multiple inhibitors in parallel to compare resistance emergence results between monotherapy and combination DAA treatment under the same selective conditions. In general, the resistant variants observed in this study were all at reported or predicted positions but were distinct in terms of frequency and emergence among different DAA combinations. In multiple experiments, the NS3 substitution N77S emerged in gt 1b replicons during two-DAA treatment. Substitution at this residue has been detected in gt 1a replicons selected with a close analog of ASV (39) and in gt 1b replicons selected with a combination of a protease inhibitor (boceprevir) and nucleoside analog NS5B inhibitor, without an apparent phenotype, but to our knowledge N77S has not previously been reported as the predominant resistance variant in either gt 1b or gt 1a with any protease inhibitor. This substitution is located at the end of the enzyme's EF loop and may interact with the bulky P2 moiety of inhibitors such as ASV. Although it is a low-level-resistance substitution, N77S may influence subsequent changes in the population, as it was observed first under conditions of low dual-selective pressure and was 98% linked to R155Q in populations selected under conditions of higher selective pressure with the two DAAs. In contrast, populations of cells treated with any level of ASV pressure alone were homogeneous for D168V. In these studies, the opposite was observed in gt 1a, where substitution at amino acid 168 occurred only during combination treatment whereas monotherapy elicited a R155K substitution. In NS5A, the mix of amino acid changes at positions 30, 31, and 93 differed in gt 1b populations emerging under conditions of single- and dual-drug selection. The new R30H substitution in gt 1b was observed only in the NS5A/NS3 dual selection, and while it showed minimal resistance itself, it enhanced resistance of L31M ∼100-fold. In contrast, position 30 plays an important direct role in resistance in gt 1a NS5A, where the change from WT glutamine to histidine (Q30H) conferred significant (>1,000-fold) resistance to DCV, highlighting the genotype context-dependent role of this residue. In general, resistance in NS5B for both genotypes arose predominantly at amino acid 495, where the different relative frequencies of S/L/A variants correlated with different levels of resistance. However, an additional NS5B substitution (L392I) occurred in the gt 1a NS5A/NS5B dual selection, generating a variant with low-level resistance and suggesting that alterations to NS5A, either through inhibitor interactions or via resistance substitutions, can affect emergence of resistance in NS5B, consistent with reports of NS5A-NS5B interactions (48, 55).
From the mutations that emerged only in dual selections, we can infer that cross-target influences allowed the different substitutions to emerge. Given that the nonstructural proteins targeted by these DAAs are believed to function together as a multicomponent complex, one can envision that simultaneously targeting multiple proteins may necessitate additional changes to allow formation of an active replicase complex. Others have also reported novel mutations (5) or increased prevalence of minor variants (29) in dual-DAA-selected populations. Although the overall differences between singly and dually treated populations in the sequence and resistance levels we observed were not dramatic, they alert us to the potential for unexpected outcomes during combination treatment. The results indicate that the replicon may not tolerate more-dramatic differences, given that the target proteins still need to associate into an active replication complex. Whether the mutations that emerged in vitro as a result of dual- and triple-DAA challenges would also be observed in patients treated with the same combinations is to be determined as clinical data are reported. The HCV replicon system should be extremely useful for analyzing samples from subjects treated with combinations of DAAs.
Interesting differences between monotherapy and dual-therapy treatment in replication fitness were also observed. For example, in both gt 1a and gt 1b, selection with ASV alone elicited the NS3 resistance substitution, which is “less costly” in terms of fitness. However, in the presence of both DCV and ASV, the more costly substitution appears to have been compensated for by the presence of additional substitutions in the second protein. For example, in gt 1b, N77S-R155Q replicates with ≤10% the efficiency of the WT replicon but, when linked with NS5A substitutions, the dually resistant variants replicated at least 3× more efficiently. Such compensatory changes could explain why cells treated with either DCV or ASV developed >100-fold resistance whereas cultures treated in parallel with both inhibitors developed only 5- to 10-fold resistance and required additional passages to develop a high level of resistance. More time may be needed to derive the correct mix of compatible substitutions whose combined fitness costs are not overwhelming. Likewise, the poor (<10%) replication of the Q30H-K68R variant selected in gt 1a with DCV treatment suggests that an additional change outside NS5A must have been present in the selected cell line to enhance replication. The increased (30% to 70% compared to the WT level) replication of the dually selected D168V/Q30H-K68R variant compared to variants with the same changes in the individual NS3 and NS5A proteins (both at <10% of the WT level) was noteworthy. This finding suggests the presence of NS3-NS5A cross-target influences that enhance efficiency of the replication complex.
The high clearance efficiency of the triple combination probably reflects the reduced frequency of the multiple genetic events necessary to generate a viable triply resistant HCV replicon. However, variants resistant to all three inhibitors were eventually selected, and these had some of the same mutations that had been selected during dual-combination studies, as well as new ones. In NS3, Q80R was found to be linked to either R155Q or Q41R in gt 1b cultures maintained in the presence of the three DAAs but not in parallel cultures selected with monotherapy or 2-DAA combinations. These mutations, both previously identified as NS3 resistance sites, conferred low-level resistance on their own, and Q80R has been shown to have an auxiliary role in enhancing the resistance of R155 or D168 mutations (28, 49; F. McPhee et al., poster 1223, presented at the 46th EASL Congress, Berlin, Germany, 30 March to 3 April 2011). In gt 1a, D168E and M179T substitutions were 100% linked, as were R155K and M179A in a more highly resistant variant. In addition, both clones contained a Q89R substitution. This substitution on its own greatly enhanced replicon replication and may be essential to permit replication of these triple mutants during the increased pressure from selection with three inhibitors. A new combination of low-level NS5B resistance variants was also detected specifically in the gt 1a triple selection (T389I-A421V). The fact that the T389I-A421V substitutions enhanced (5- to 10-fold) replication of the Q30H-K68R NS5A variant suggests that, together, changes in these two proteins can form a replicase that is more active than one with only NS5A substitutions. It is of interest that, in gt 1a clones demonstrating resistance to all three inhibitors, high-level resistance to NS3 and high-level resistance to NS5B were not detected in the same clone. This may indicate that the fitness cost associated with highly resistant substitutions in three key proteins of the HCV replication machinery is too great to yield a genome capable of high replicative fitness.
Collectively, these results indicate that challenging HCV with multiple DAAs had a compelling suppressive effect on replication and elicited novel mutations and combinations of mutations. Since IFN-α and unrelated HCV inhibitor chemotypes retained activity against the multi-DAA-resistant populations generated in these experiments (data not shown), further complementation or follow-up to a combination DAA regimen should be feasible if required to achieve SVR. If the synergies demonstrated here for combinations of DAA inhibitors are predictive of reduced resistance and enhanced viral clearance in patients, a triple-combination regimen targeting three different proteins of HCV replication, and particularly including an NS5A inhibitor, promises to markedly improve the rates of SVR over alfa/RBV regimens, regimens that include a single DAA with or without RBV, and many dual-DAA combination regimens. Such combinations have the potential to quickly reduce viral load and limit opportunities for the emergence of multidrug resistance, helping to achieve SVR. Given the altered patterns of resistance observed during these combination studies, these results provide guidance for resistance monitoring during ongoing combination trials.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the technical contributions of Lourdes Valera and Bernadette Kienzle.
Footnotes
Published ahead of print 30 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164 [DOI] [PubMed] [Google Scholar]
- 2. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 3. Brown NA. 2009. Progress towards improving antiviral therapy for hepatitis C with hepatitis C virus polymerase inhibitors. Part I: nucleoside analogues. Expert Opin. Invest. Drugs 18:709–725 [DOI] [PubMed] [Google Scholar]
- 4. Burton JR, Jr, Everson GT. 2009. HCV NS5B polymerase inhibitors. Clin. Liver Dis. 13:453–465 [DOI] [PubMed] [Google Scholar]
- 5. Chase R, et al. 2009. A novel HCV NS3 protease mutation selected by combination treatment of the protease inhibitor boceprevir and NS5B polymerase inhibitors. Antiviral Res. 84:178–184 [DOI] [PubMed] [Google Scholar]
- 6. Chou T. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58:621–681 [DOI] [PubMed] [Google Scholar]
- 7. Dimitrova M, Imbert I, Kieny MP, Schuster C. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 77:5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Egger D, et al. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Failla C, Tomei L, De Francesco R. 1994. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 68:3753–3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flint M, et al. 2009. Selection and characterization of hepatitis C virus replicons dually resistant to the polymerase and protease inhibitors HCV-796 and boceprevir (SCH 503034). Antimicrob. Agents Chemother. 53:401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foote BS, Spooner LM, Belliveau PP. 9 August 2011, posting date Boceprevir: a protease inhibitor for the treatment of chronic hepatitis C. Ann. Pharmacother. 45:1085–1093 [DOI] [PubMed] [Google Scholar]
- 12. Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the HCV NS5A inhibitor BMS-790052 in the in vitro replicon system. Antimicrob. Agents Chemother. 54:3641–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fridell RA, et al. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935 [DOI] [PubMed] [Google Scholar]
- 14. Gane EJ, et al. 2010. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 376:1467–1475 [DOI] [PubMed] [Google Scholar]
- 15. Gao M, et al. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gentile I, Carleo MA, Borgia F, Castaldo G, Borgia G. 2010. The efficacy and safety of telaprevir—a new protease inhibitor against hepatitis C virus. Expert Opin. Invest. Drugs 19:151–159 [DOI] [PubMed] [Google Scholar]
- 17. Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. 1993. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. U. S. A. 90:10583–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobson IM, et al. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 19. Kieffer TL, Kwong AD, Picchio GR. 2010. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J. Antimicrob. Chemother. 65:202–212 [DOI] [PubMed] [Google Scholar]
- 20. Kneteman NM, et al. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752 [DOI] [PubMed] [Google Scholar]
- 21. Kronenberger B, Zeuzem S. 2008. Future treatment options for HCV: double, triple, what is the optimal combination? Best Pract. Res. Clin. Gastroenterol. 22:1123–1136 [DOI] [PubMed] [Google Scholar]
- 22. Kronenberger B, Zeuzem S. 2009. Treatment of chronic hepatitis C: anticipated impact of resistance in patients treated with protease inhibitors. Curr. Gastroenterol. Rep. 11:15–21 [DOI] [PubMed] [Google Scholar]
- 23. Kukolj G, et al. 2005. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260–39267 [DOI] [PubMed] [Google Scholar]
- 24. Kwong AD, Kim JL, Rao G, Lipovsek D, Raybuck SA. 1999. Hepatitis C virus NS3/4A protease. Antiviral Res. 41:67–84 [PubMed] [Google Scholar]
- 25. Lamarre D, et al. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189 [DOI] [PubMed] [Google Scholar]
- 26. Legrand-Abravanel F, Nicot F, Izopet J. 2010. New NS5B polymerase inhibitors for hepatitis C. Expert Opin. Invest. Drugs 19:963–975 [DOI] [PubMed] [Google Scholar]
- 27. Lemm JA, et al. 2010. Identification of hepatitis C virus NS5A inhibitors. J. Virol. 84:482–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenz O, Verbinnen, et al. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Pogam S, et al. 2006. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 80:6146–6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin K. 2010. Development of novel antiviral therapies for hepatitis C virus. Virol. Sin. 25:246–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin K, Perni RB, Kwong AD, Lin C. 2006. VX-950, a novel hepatitis C virus (HCV) NS3-4A protease inhibitor, exhibits potent antiviral activities in HCV replicon cells. Antimicrob. Agents Chemother. 50:1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liverton NJ, et al. 2010. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 54:305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lohmann V, Koch JO, Bartenschlager R. 1996. Processing pathways of the hepatitis C virus proteins. J. Hepatol. 24(2 Suppl):11–19 [PubMed] [Google Scholar]
- 34. Lok AS, et al. 2012. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 366:216–224 [DOI] [PubMed] [Google Scholar]
- 35. Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83:4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu L, et al. 2004. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 48:2260–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Macdonald A, Harris M. 2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 85(Pt 9):2485–2502 [DOI] [PubMed] [Google Scholar]
- 38. McPhee F, et al. 2012. Resistance analysis of the hepatitis C virus NS3 protease inhibitor asunaprevir. Antimicrob. Agents Chemother. 56:3670–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McPhee F, et al. 2009. The discovery and early development of the HCV NS3 protease inhibitor BMS-605339. Global Antiviral J. 5(Suppl 1):51 [Google Scholar]
- 40. Miyanari Y, et al. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 41. Neddermann P, Clementi A, De Francesco R. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J. Virol. 73:9984–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nettles RE, et al. 2011. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology 54:1956–1965 [DOI] [PubMed] [Google Scholar]
- 43. O'Boyle DR, Jr, et al. 2005. Development of a cell-based high-throughput specificity screen using a hepatitis C virus-bovine viral diarrhea virus dual replicon assay. Antimicrob. Agents Chemother. 49:1346–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohara E, et al. 2011. Elimination of hepatitis C virus by short term NS3-4A and NS5B inhibitor combination therapy in human hepatocyte chimeric mice. J. Hepatol. 54:872–878 [DOI] [PubMed] [Google Scholar]
- 45. Pasquinelli C, et al. 2012. Single- and multiple-ascending-dose studies of the NS3 protease inhibitor asunaprevir in subjects with or without chronic hepatitis C. Antimicrob. Agents Chemother. 56:1838–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pierra C, et al. 2006. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. J. Med. Chem. 49:6614–6620 [DOI] [PubMed] [Google Scholar]
- 47. Poordad F, et al. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qin W, et al. 2001. Mutational analysis of the structure and functions of hepatitis C virus RNA-dependent RNA polymerase. Hepatology 33:728–737 [DOI] [PubMed] [Google Scholar]
- 49. Romano KP, Ali A, Royer WE, Schiffer CA. 2010. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U. S. A. 107:20986–20991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rydberg EH, et al. 2009. Structural basis for resistance of the genotype 2b hepatitis C virus NS5B polymerase to site A non-nucleoside inhibitors. J. Mol. Biol. 390:1048–1059 [DOI] [PubMed] [Google Scholar]
- 51. Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 52. Sarrazin C, Hézode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56(Suppl):S88–S100 [DOI] [PubMed] [Google Scholar]
- 53. Seiwert SD, et al. 2008. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 52:4432–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567 [DOI] [PubMed] [Google Scholar]
- 55. Shirota Y, et al. 2002. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 277:11149–11155 [DOI] [PubMed] [Google Scholar]
- 56. Soriano V, Peters MG, Zeuzem S. 2009. New therapies for hepatitis C virus infection. Clin. Infect. Dis. 48:313–320 [DOI] [PubMed] [Google Scholar]
- 57. Susser S, et al. 2009. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50:1709–1718 [DOI] [PubMed] [Google Scholar]
- 58. Tellinghuisen T, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032 doi:10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thompson AJ, McHutchison JG. 2009. Antiviral resistance and specifically targeted therapy for HCV (STAT-C). J. Viral Hepatol. 16:377–387 [DOI] [PubMed] [Google Scholar]
- 61. Tong X, et al. 2008. Characterization of resistance mutations against HCV ketoamide protease inhibitors. Antiviral Res. 77:177–185 [DOI] [PubMed] [Google Scholar]
- 62. Zheng X, et al. 2011. Syntheses and initial evaluation of a series of indolo-fused heterocyclic inhibitors of the polymerase enzyme (NS5B) of the hepatitis C virus. Bioorg. Med. Chem. Lett. 21:2925–2929 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.