

Abstract
CB-183,315 is a novel lipopeptide antibiotic structurally related to daptomycin currently in phase 3 clinical development for Clostridium difficile-associated diarrhea (CDAD). We report here the in vitro mechanism of action, spontaneous resistance incidence, resistance by serial passage, time-kill kinetics, postantibiotic effect, and efficacy of CB-183,315 in a hamster model of lethal infection. In vitro data showed that CB-183,315 dissipated the membrane potential of Staphylococcus aureus without inducing changes in membrane permeability to small molecules. The rate of spontaneous resistance to CB-183,315 at 8× the MIC was below the limit of detection in C. difficile. Under selective pressure by serial passage with CB-183,315 against C. difficile, the susceptibility of the bacteria changed no more than 2-fold during 15 days of serial passages. At 16× the MIC, CB-183,315 produced a ≥3-log reduction of C. difficile in the time-kill assay. The postantibiotic effect of CB-183,315 at 8× the MIC was 0.9 h. At 80× the MIC the postantibiotic effect was more than 6 h. In the hamster model of CDAD, CB-183,315 and vancomycin both demonstrated potent efficacy in resolving initial disease onset, even at very low doses. After the conclusion of dosing, CB-183,315 and vancomycin showed a similar dose- and time-dependent pattern with respect to rates of CDAD recurrence.
INTRODUCTION
Clostridium difficile is a spore-forming, Gram-positive, anaerobic bacterium that can colonize the intestinal tracts of humans (2, 14). C. difficile is thought to colonize the colon after ingestion of acid-resistant spores that pass through the stomach to germinate in the small bowel and eventually colonize the colon. Following treatment with various broad-spectrum antibiotics that alter the normal intestinal microbial environment, C. difficile can overgrow and populate the colon. Finally, toxin production leads to intestinal inflammation and damage, diarrhea, and sometimes life-threatening disease, such as pseudomembranous colitis, toxic megacolon, and sepsis (2). C. difficile produces disease primarily by the production of two toxins, toxin A and toxin B. These toxins invade epithelial cells, disrupting their cytoskeleton, which results in damage to the epithelial barrier and promotes mucosal inflammation (14).
C. difficile infection is the most common cause of health care-related infectious diarrhea in developed countries, accounting for up to 20% of the cases of antibiotic-associated diarrhea and nearly all cases of antibiotic-associated colitis (14). C. difficile-associated diarrhea (CDAD) is a major cause of morbidity and mortality in elderly patients in the hospital setting, responsible for 20% to 25% of all cases of antibiotic-associated diarrhea (2). CDAD can vary from mild to severe, and the severity of disease has been shown to correlate with different clinical and microbiologic cure rates (1, 29). In patients with mild CDAD, vancomycin and metronidazole have been found to be equivalent, resulting in clinical cure rates between 90% and 98% (29). In contrast, in patients with severe CDAD, vancomycin has been shown to be superior to metronidazole (7, 29). The superiority of vancomycin in treating patients with severe CDAD has been called into question, however, because the definition of cure has been based to a degree on persistence of the C. difficile toxin in the stool. Since C. difficile toxin is known to persist in stool samples after resolution of diarrhea, repeat stool toxin testing is of no value for assessing response to treatment (15). High recurrence rates (20% to 30%) associated with metronidazole, coupled with increased frequency and severity of the disease and the emergence of a highly virulent strain (referred to as BI, NAP1, or 027), have increased the urgency of developing new therapeutic options (6, 16).
Many agents are at various stages of development for the treatment of CDAD, but currently there are few data from clinical trials to evaluate their therapeutic potential. Important data are being reported on some anti-C. difficile antibiotics, such as rifaximin, ramoplanin, nitazoxanide, teicoplanin, oritavancin, and tigecycline (12, 19, 23). Fidaxomicin is the drug most recently approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of CDAD (16, 18).
CB-183,315 (Fig. 1) is an orally available lipopeptide antibiotic that is structurally related to daptomycin. It has the same peptide sequence as daptomycin but has an aromatic ring containing an unsaturated lipid tail [(E)-3-(4-pentylphenyl)but-2-enoic acid tail]. Based on the rapid in vitro bactericidal activity of daptomycin against Gram-positive pathogens, including C. difficile (28), we investigated the activity of structurally related lipopeptides against C. difficile strains. CB-183,315 was chosen for further evaluation based upon potent MICs (MIC90 = 0.5 μg/ml) and its bactericidal activity (3-log10-unit reduction at 4× and 8× the MIC after 24 h) versus a collection of C. difficile strains, including the newer, highly virulent (NAP1) strain (5, 26).
Fig 1.

Structure of CB-183,315. CB-183,315 is a lipopeptide antibiotic that is structurally related to daptomycin.
CB-183,315 successfully completed a phase 2 clinical trial and was demonstrated to be safe and well tolerated in patients with C. difficile infection. In addition, better sustained cure rates were achieved with CB-183,315 than with vancomycin. Importantly, reduction and delay in recurrence of CDAD episodes were observed with CB-183,315 compared with vancomycin (4, 21). CB-183,315 is now in phase 3 development.
The aims of this study were to evaluate the mechanism of action (MOA), spontaneous resistance incidence (RI), resistance by serial passage, time-kill kinetics, and postantibiotic effect (PAE) of CB-183,315 in vitro to gain a better understanding of its potential as a therapeutic agent for the treatment of C. difficile infection. Its efficacy compared with that of vancomycin in a lethal hamster model of CDAD was also assessed.
(Portions of this work were presented at the 50th Interscience Conference on Antimicrobial Agents and Chemotherapy.)
MATERIALS AND METHODS
Strains, media, and antibiotics.
The following strains were used in the in vitro studies. Staphylococcus aureus (ATCC 29213) was used in the fluorescent-activated cell sorting (FACS) assay. For the resistance and PAE studies, nontoxigenic C. difficile (ATCC 700057) was used. In the time-kill study, three C. difficile strains were tested. C. difficile ATCC 700057 (CB-183,315 MIC, 0.25 μg/ml) is a nontoxigenic strain used for susceptibility testing. C. difficile ATCC 43596 (CB-183,315 MIC, 0.06 μg/ml) is a toxigenic clinical isolate from the feces of a patient in Belgium with pseudomembranous colitis. Lastly, C. difficile RMA 19139 (CB-183,315 MIC, 0.06 μg/ml) is a NAP1 (REA group BI) strain, purchased from RM Alden Laboratory, which was isolated in 2006 from C. difficile clinical trials where patients had had at least three watery stools in the preceding 24 h and the stools were toxin positive. All C. difficile work was performed in a Bactron I anaerobic chamber (Sheldon Manufacturing) infused with anaerobic mixed gas (5% H2, 10% CO2, balance N2; catalog no. Z03NI8532002103; Airgas, Salem, NH) using reduced media.
In the in vivo studies all experiments used C. difficile ATCC 43596. The C. difficile spores were prepared by a method developed at Ilypsa, Inc. (Santa Clara, CA). In brief, C. difficile bacteria were grown on brucella blood agar plates at 37°C under anaerobic conditions for 10 days. C. difficile lawns were scraped from plates with inoculating loops and transferred to 5 ml saline. Cell/spore suspensions were heat-shocked in a 56°C water bath for 10 min and then centrifuged. Supernatants were drawn off; pellets containing spores were resuspended in saline, enumerated by serial dilution into deionized water (dH2O), and plated onto C. difficile selective agar (VWR, Pittsburgh, PA). Spore suspensions were stored at −20°C prior to use.
All CB-183,315 (lot numbers 8, 9, 10, and LC-01-004) and daptomycin (lot number 4) lyophilized powders used were from Cubist Pharmaceuticals, Inc. (Lexington, MA). Rifampin and clindamycin were purchased from Sigma (St. Louis, MO). Vancomycin hydrochloride powder (NDC 0409-6509-01) was purchased from Hospira, Inc. (Lake Forest, IL).
Mueller-Hinton broth (MHB) was prepared and sterilized according to the manufacturer's specifications. For Mueller-Hinton Agar (MHA) 18 g/liter of BD BactoAgar was added to MHB and steam sterilized. In time-kill and PAE studies, viable C. difficile organisms were quantified on brucella agar (28 g/liter BBL brucella broth [catalog no. 211088] with 15 g/liter BD BactoAgar [catalog no. 214040] and steam sterilized) from BD Diagnostic Systems (Sparks, MD). Calcium-supplemented media (MHBc, MHAc, and brucella+c) were prepared by addition of a 10 mg/ml stock of CaCl2 (Sigma) to a final concentration of 50 mg/liter Ca2+. Prepared brucella agar plates supplemented with 5% sheep blood, hemin, and vitamin K1 (BBHK; catalog no. 297716) were purchased from BD Diagnostic Systems. Tryptic soy agar fortified with 5% sheep blood (TSAB) was purchased from bioMérieux (Durham, NC).
Mechanism-of-action (MOA) flow cytometry studies.
To investigate the MOA of CB-183,315, changes in membrane potential and permeability of S. aureus ATCC 29213 were monitored using FACS, as described previously (20, 25). Overnight cultures of S. aureus were diluted 1:10,000 in MHBc and incubated at 35 to 37°C and 200 rpm for 3 h before treatment. Samples were taken at various time points from cultures left untreated (control) or treated with nisin (20 μg/ml), daptomycin (MIC = 0.5 μg/ml), or CB-183,315 (MIC = 0.25 μg/ml). The membrane potential was detected by 30 μM 3,3′-diethyloxacarbocyanine iodide (DiOC2), and membrane permeability was detected by 100 nM TO-PRO-3 iodide. Samples were also serially diluted and plated on TSAB to determine viability.
Spontaneous resistance incidence (RI) evaluations.
We measured the RI for CB-183,315 at 8× the MIC for C. difficile ATCC 700057 using the following procedures. C. difficile suspended to ∼0.5 McFarland standard (McF) in MHBc and incubated at 37°C for ∼4 h was used as the inoculum (1 ml) on MHAc selection plates (150 mm) containing CB-183,315 (MIC = 0.5 μg/ml) or rifampin (MIC = 0.25 μg/ml) at 2×, 4×, and 8× the MIC and incubated at 37°C for up to 5 days. CFU per milliliter of each inoculum were counted, and the RI was calculated as the colony frequency (i.e., the number of colonies on drug-containing plates divided by the total number of colonies plated). Colonies recovered on drug-containing plates were all evaluated by MIC to confirm elevated MICs compared with naïve colonies of C. difficile.
Serial passage resistance studies.
Serial passage of C. difficile was performed in the presence of increasing concentrations of CB-183,315 or rifampin as previously described (24). Cultures of C. difficile suspended in MHBc to equal ∼1 McF and incubated at 37°C for ∼3 h were inoculated into media containing various concentrations of drug around the MIC and grown overnight at 37°C for 20 to 24 h. Each day, wells with the highest concentration of drug (μg/ml) permitting C. difficile growth equal to ∼1 McF were used to inoculate the next day's drug series; this procedure was repeated for 15 days. The starting MIC for CB-183,315 was 0.5 μg/ml, and the starting MIC for rifampin was 0.25 μg/ml.
Time-kill studies.
For time-kill analyses, individual C. difficile colonies were isolated by streaking onto rich, nonselective BBHK agar with anaerobic incubation at 35 to 37°C for 48 h. On the day of testing, cultures were scraped off plates, suspended in reduced MHBc to equal approximately 0.5 McF (∼106 CFU/ml), either left untreated (no-drug control) or treated with 16× the MIC of CB-183,315, and incubated at 35 to 37°C. Cell viability was determined at 0, 1, 2, 4, 6, and 24 h.
Postantibiotic effect (PAE) studies.
PAE values were determined as described previously (9). Briefly, C. difficile was suspended in MHBc to equal ∼1 McF, incubated at 37°C for 1 h prior to addition of 0, 2, or 20 μg/ml CB-183,315 (0, 8×, or 80× the MIC, respectively), and then incubated at 37°C for 30 min. Viability was measured, and samples were diluted 1,000-fold to remove the drug and sampled for viability over time. Samples were incubated at 37°C between time points. The PAE was calculated as T − C, where T is the time required for the count of CFU in the treated culture to increase 1 log10 (10-fold) above the count observed immediately after dilution and C is the time required for the control culture to increase 1 log10 (10-fold) above the count observed immediately after dilution.
Hamster efficacy model.
A hamster model of CDAD was developed at Cubist Pharmaceuticals, Inc., on the basis of several hamster models presented in the literature (11, 27). Male Syrian golden hamsters (Mesocricetus auratus) were purchased from Charles River Laboratories (Wilmington, MA) and housed at 1 animal per cage in a disposable cage rack system (Innocage IVC Rat; Innovive, San Diego, CA), with each cage being independently ventilated and kept at a negative pressure relative to the local environment. Water and Agway rodent chow were provided ad libitum throughout the study. All work with laboratory animals was performed in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and commenced following review and approval by the Cubist Institutional Animal Care and Use Committee.
Spore suspensions, stored at −20°C, were thawed at room temperature and diluted to final concentrations with sterile saline immediately prior to inoculation. C. difficile infections were established in three replicate studies in hamsters by pretreatment with a subcutaneous dose of 10 mg/kg clindamycin, to disrupt normal intestinal flora, followed by an oral inoculation (via gavage) of approximately 20 C. difficile spores in 0.5 ml sterile saline 1 day later. A CB-183,315 or vancomycin dose of 2, 10, or 25 mg/kg (n = 8 per treatment group) was administered by oral gavage to hamsters in 0.5 ml deionized water (dH2O); infected control animals were given 0.5 ml dH2O (n = 5), and uninfected sentinels (n = 5 or 6) were sham dosed with an empty gavage needle and syringe. All infected control animals died by day 2 in this animal model of lethal C. difficile infection. All uninfected sentinels survived to the end of the study. All treatments were given twice a day (BID) for 5 days, starting at 12 h after inoculation with C. difficile.
Clinical observations of hamsters were made twice daily at dosing time points, and then daily after the end of the dosing period for 30 days. Clinical observations included survival, observations of diarrhea in the cage, wet tail (moisture around the base of the tail), and other standard clinical signs such as lethargy or ruffled fur. Any hamsters found to be moribund were humanely euthanized, and that day was recorded. All hamsters that died during the study were examined externally and by cecal necropsy to confirm cecitis as the cause of death. Animals surviving to 35 days after inoculation were humanely euthanized with carbon dioxide, according to American Veterinary Medical Association Guidelines, and their ceca were visually inspected to confirm normal gross anatomy.
Compound efficacies were evaluated on the basis of two end points: (i) resolution of initial disease and (ii) prevention of recurrence. Any death occurring on study days 0 to 6 (i.e., from the time of inoculation to 1 day after the completion of dosing on day 6) was interpreted as a failure to resolve initial CDAD; any death taking place on study days 7 to 35 (i.e., from 2 or more days after the end of dosing to the end of the study) was viewed as failure to prevent a recurrence of CDAD. A prerequisite for both of these interpretations was the presence of gross clinical anomalies (external and/or internal) associated with CDAD infection.
Survival rates per treatment group against initial and recurring CDAD were determined and subjected to Kaplan-Meier survival analysis for each study and for the pooled survival data from three separate studies (8, 13). Any animal censored prior to the end of a study (due to an escape from its controlled cage environment) was accounted for in Kaplan-Meier group percent survival estimates up through the time of the censoring event. Log-rank test (Mantel-Cox) statistical comparisons of treatment group survival data for each separate study and using treatment-matched, pooled survival data from three separate studies were performed (GraphPad Prism version 5.02). A P value of <0.05 was considered statistically significant.
RESULTS
MOA flow cytometry studies.
Given that daptomycin and CB-183,315 are structurally similar and that daptomycin is known to act via depolarization of the S. aureus membrane (25), CB-183,315 was analogously tested in the same system to determine if its MOA was also membrane depolarization. Figure 2 shows the membrane potential of daptomycin-treated and CB-183,315-treated S. aureus. At time zero (control), cultures were homogeneous and fully polarized. Treatment with pore-forming antibiotic nisin resulted in immediate (<10 min) depolarization. Daptomycin or CB-183,315 depolarized cells more slowly, suggesting a common mechanism. After 15 min of treatment, the population was highly heterogeneous. After 30 min of treatment, the majority of the population was depolarized. After 60 min, the peaks overlapped those of the nisin samples almost completely.
Fig 2.

CB-183,315, like daptomycin, dissipates membrane potential in S. aureus, as measured by flow cytometry. S. aureus cell samples were incubated for 0 (control), 15, 30, or 60 min with 2.5 μg/ml of daptomycin or 1.25 μg/ml CB-183,315 (both at 5× the MIC). Additional cell samples were incubated for 10 min with 20 μg/ml of nisin. Changes in membrane potential were measured by the ratio of fluorescence emission intensities at 610 and 530 nm with 30 μM 3,3′-diethyloxacarbocyanine iodide (DiOC2; Molecular Probes, Eugene, OR). The membrane potential of S. aureus cells become more depolarized over time with daptomycin and CB-183,315, in contrast to the rapid membrane depolarization in the presence of nisin.
As daptomycin has previously been shown to depolarize without permeabilizing membranes (25), the effect of CB-183,315 on membrane permeability was also examined. Figure 3 shows the membrane permeability of daptomycin-treated and CB-183,315-treated S. aureus. Control populations were highly homogeneous, but with a reduced fluorescence relative to nisin-treated, permeabilized cells. Treatment with CB-183,315 or daptomycin resulted in a reduction in fluorescence below that of control cells, consistent with a shared mechanism. The reduction in fluorescence suggests that CB-183,315 and daptomycin do not permeabilize the cytoplasmic membrane of S. aureus and may inhibit some level of energy-dependent uptake of the indicator.
Fig 3.

CB-183,315, like daptomycin, has little or no effect on S. aureus cell permeability, as measured by flow cytometry. S. aureus cell samples were incubated at 0 (control), 15, 30, or 60 min with 2.5 μg/ml daptomycin or 1.25 μg/ml CB-183,315 (both at 5× the MIC). Additional cell samples were incubated for 10 min with 20 μg/ml of nisin. Changes in membrane permeability were measured by the uptake of 100 nM the membrane-impermeant fluorescent indicator TO-PRO-3 iodide.
Figure 4 shows the comparison of the percent polarized and percent viable daptomycin-treated and CB-183,315-treated S. aureus. Changes in viability were monitored in parallel with changes in membrane potential. These results indicate that depolarization and bactericidal activity are closely linked for both CB-183,315 and daptomycin.
Fig 4.
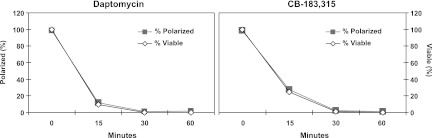
Changes in membrane potential and cell viability decrease in parallel for both daptomycin and CB-183,315. Changes in S. aureus membrane potential (as measured by the ratio of fluorescence emission intensities at 610 and 530 nm) and cell viability (from samples serially diluted and plated on TSAB) were plotted over time following incubation with 2.5 μg/ml daptomycin or 1.25 μg/ml CB-183,315 (both at 5× the MIC). Values are expressed as percentages of the untreated control cells.
Resistance development evaluations.
Observed RI values of CB-183,315 and rifampin against C. difficile are shown in Table 1. RI determinations were performed in duplicate and confirmed as a ≥8-fold change in MIC compared with naïve control isolates. As the levels of CB-183,315 found in the gastrointestinal tracts of healthy volunteers during phase 1 were quite high (>100× the MIC90; internal data on file), the use of a ≥8-fold change for RI determination enabled examination of only large MIC shifts. RI values for CB-183,315 at 8× the MIC fell below the limit of detection (<4.1 × 10−8 to < 7.4 × 10−8). RI values of rifampin at 8× the MIC ranged from ∼1.6 × 10−7 to ∼9.6 × 10−7.
Table 1.
Resistance incidence (RI) values of CB-183,315 and rifampin against C. difficile ATCC 700057
| Compound | MIC (μg/ml) | MIC multiple used | RI at no. of cells plated |
|
|---|---|---|---|---|
| 2.5E+07 (CFU/ml) | 1.4E+07 (CFU/ml) | |||
| CB-183,315 | 0.5 | 4 | <4.1E−08 | 1.2E−07 |
| CB-183,315 | 0.5 | 8 | <4.1E−08 | <7.4E−08 |
| Rifampin | 0.25 | 8 | 1.6E−07a | 9.6E−07a |
Only single colonies plus unquantifiable growth on the rim were obtained, so the RI was calculated using countable colonies only.
Figure 5 displays C. difficile serial passage results in CB-183,315 and rifampin. The highest concentration of drug permitting growth was determined daily for each sample. None of the CB-183,315 samples grew in >1 μg/ml of drug; the susceptibility against CB-183,315 changed 2-fold at day 14 to 15 compared with day 0. Organisms grew in >200 μg/ml rifampin, resulting in a >4,000-fold change at day 14 to 15 compared with day 0.
Fig 5.

Daily changes in the concentration of drug permitting growth during serial passage studies of CB-183,315 (in triplicate) and rifampin (in duplicate) against C. difficile (ATCC 700057). Cells grown in MHBc were inoculated into media containing various concentrations of drug around the MIC and grown overnight at 37°C for 20 to 24 h. Each day, wells with the highest concentration of drug (μg/ml) permitting C. difficile growth equal to ∼1 McF were used to inoculate the next day's drug series; this procedure was repeated for 15 days. The starting MIC for CB-183,315 was 0.5 μg/ml, and that for rifampin was 0.25 μg/ml.
Time-kill assay.
In the time-kill study (Fig. 6), the starting inocula for all three strains were between 106 and 107 CFU/ml. CB-183,315 at 16× the MIC was bactericidal against all three strains, producing a ≥3-log reduction. For the NAP1 (REA group BI) and nontoxigenic strains, the bactericidal endpoint was achieved within 6 h. For the toxigenic strain, the 3-log reduction was achieved within 24 h.
Fig 6.

Time-kill studies of CB-183,315 at 16× the MIC against three strains of C. difficile, including RMA 19139, a NAP1 strain from RM Alden Laboratory, ATCC 43596, a toxigenic clinical isolate, and ATCC 700057, a nontoxigenic strain used for susceptibility testing. MICs against CB-183,315 for these strains were 0.06 μg/ml for the NAP1 strain, 0.06 μg/ml for the toxigenic strain, and 0.25 μg/ml for the nontoxigenic strain. CB-183,315 was bactericidal against all three strains of C. difficile.
PAE studies.
Figure 7 demonstrates the PAE of CB-183,315. C. difficile was exposed to CB-183,315 at 2 μg/ml (8× the MIC) and 20 μg/ml (80× the MIC) for 30 min prior to removal of drug by 1:1,000 dilution. The calculated PAE, or the ability to suppress bacterial growth after removal of CB-183,315, was 0.9 h at 8× the MIC and >6 h at 80× the MIC.
Fig 7.

Postantibiotic effect of CB-183,315 at 8× and 80× the MIC against C. difficile ATCC 700057 (MIC = 0.25 μg/ml). CB-183,315 was incubated with C. difficile at the given concentration for 30 min. Viability was measured, and samples were diluted 1,000-fold to remove the drug and sampled for viability over time.
Hamster efficacy model.
Measured MICs of CB-183,315 and vancomycin against C. difficile ATCC 43596 were 0.063 and 0.25 to 0.5 μg/ml, respectively. CB-183,315 and vancomycin displayed dose- and time-dependent efficacy in protecting hamsters from lethal inocula of this C. difficile clinical isolate in three separate studies. A representative set of dose- and time-dependent survival curves from a single protection study (study 2) is presented in Fig. 8.
Fig 8.

Dose- and time-dependent efficacy of CB-183,315 (A) and vancomycin (B) against C. difficile ATCC 43596 in the hamster model of C. difficile-associated diarrhea (CDAD). Note that this animal model of CDAD is rapidly lethal for 100% of control hamsters dosed with deionized water; all five hamsters dosed with this vehicle were found dead on day 2. CB-183,315 and vancomycin produced similar patterns of dose- and time-dependent survival in this model. These data are from one of three studies comparing doses of CB-183,315 and vancomycin in this hamster model. Data from all three hamster studies are compared and compiled in Tables 2 and 3.
The Kaplan-Meier survival rates (%) against initial or recurrent CDAD, for each treatment group by study or from treatment-matched, pooled survival data collected from three repeat studies, are summarized in Table 2 and Table 3. In all three studies, all inoculated hamsters dosed with dH2O died from CDAD on day 2 (n = 15 total), whereas uninoculated sham-dosed animals survived to day 35 and never developed CDAD (n = 17).
Table 2.
Therapeutic efficacy of CB-183,315 and vancomycin against initial hamster CDAD evaluated on day 6 (1 day after the last dose)a
| Study | % (no.) of hamsters surviving initial CDAD after receiving: |
||||||
|---|---|---|---|---|---|---|---|
| Sterile water (0.5 ml) | CB-183,315 |
Vancomycin |
|||||
| 2 mg/kg | 10 mg/kg | 25 mg/kg | 2 mg/kg | 10 mg/kg | 25 mg/kg | ||
| 1 | 0 | 100 | 100 | 100 | 100 | 100 | 100 |
| 2 | 0 | 100 | 87.5 | 100 | 100 | 100 | 100 |
| 3b | 0 | 100 | 100 | Not done | 100 | 100 | Not done |
| Overall | 0 (15) | 100 (32) | 97 (32) | 100 (16) | 100 (24) | 100 (24) | 100 (16) |
All hamsters were dosed twice daily by oral gavage for 5 days, starting 12 h after inoculation with C. difficile. Sentinel hamsters (n = 17) were not inoculated with C. difficile spores and received sham dosing with empty needles and syringes; survival of sentinels was 100% in all studies.
In study 3, the initial number was 16 each for the 2-mg/kg and 10-mg/kg CB-183,315 treatment groups.
Table 3.
Efficacy of CB-183,315 and vancomycin against CDAD recurrence in hamsters evaluated on day 35 (4 weeks postdosing)a
| Study | % (no.) of hamster surviving CDAD recurrence after receiving: |
|||||
|---|---|---|---|---|---|---|
| CB-183,315 |
Vancomycin |
|||||
| 2 mg/kg | 10 mg/kg | 25 mg/kg | 2 mg/kg | 10 mg/kg | 25 mg/kg | |
| 1 | 75.0 | 100 | 85.7 | 31.3b | 100 | 83.3 |
| 2 | 41.7c | 75.0 | 87.5 | 50.0 | 75.0 | 75.0 |
| 3d | 50.0 | 68.8 | Not done | 87.5 | 87.5 | Not done |
| Overall | 56.0 (16) | 78.1 (25) | 86.7 (13) | 57.0 (13) | 87.5 (21) | 78.6 (11) |
All hamsters were dosed twice daily by oral gavage for 5 days, starting 12 h after inoculation with C. difficile. Sentinel hamsters (n = 17) were not inoculated with C. difficile spores and received sham dosing with empty needles and syringes; survival of sentinels was 100% in all studies. Survival rates were subjected to Kaplan-Meier survival analysis.
In study 1, 2 mg/kg vancomycin was significantly less protective than 10 and 25 mg/kg vancomycin (P < 0.05).
In study 2, 2 mg/kg CB-183,315 trended toward less protection than 25 mg/kg CB-183,315 (P = 0.052).
In study 3, the initial number was 16 each for the 2-mg/kg and 10-mg/kg CB-183,315 treatment groups.
CB-183,315 and vancomycin were both consistently successful in protecting hamsters from initial CDAD at all doses tested (2, 10, or 25 mg/kg BID) (Table 2). One hamster out of 32 total receiving 10 mg/kg CB-183,315 BID was found dead prior to the end of therapy, at 4 days postinoculation. All other hamsters dosed BID with 2, 10, or 25 mg/kg CB-183,315 or vancomycin survived through at least 1 day after dosing, or day 6.
During the CDAD recurrence phase, from day 6 though day 35, hamsters dosed BID with 2, 10, or 25 mg/kg CB-183,315 or vancomycin showed a dose- and time-dependent survival (Table 3). At doses of 2, 10, and 25 mg/kg BID, CB-183,315 yielded overall (pooled) survival rates of 56.0%, 78.1%, and 86.7%, respectively, whereas vancomycin produced overall survival rates of 57.0%, 87.5%, and 78.6%, respectively. Overall, 2 mg/kg CB-183,315 BID was significantly less protective than 25 mg/kg CB-183,315 BID (P < 0.05) and trended toward less protection than 10 mg/kg CB-183,315 BID (P = 0.062). Additionally, 2 mg/kg vancomycin BID was significantly less protective than 10 and 25 mg/kg vancomycin BID (P < 0.05).
At all doses, CB-183,315 and vancomycin resulted in statistically significant protection both from initial CDAD and from CDAD recurrence up to 4 weeks postdosing compared with vehicle controls (P < 0.05). At matched doses, there were no statistically significant differences in overall survival between hamsters dosed with CB-183,315 and those dosed with vancomycin.
DISCUSSION
CDAD is a widespread disease with considerable morbidity and mortality, and its incidence continues to rise. Current treatments, although often effective for the initial episode, remain inadequate, since relapse rates remain unacceptably high. In the United States, CDAD is not only growing in frequency but also increasing in disease severity and mortality (10, 17).
CB-183,315 is structurally related to the lipopeptide antibiotic daptomycin and appears to share its MOA. In our in vitro examination of CB-183,315-treated S. aureus, CB-183,315 dissipated the membrane potential of target cells without inducing changes in membrane permeability to small molecules. Although this specific MOA has not been demonstrated in C. difficile, it is reasonable to assume that the MOA in C. difficile may be similar.
With regard to RI, the rate of spontaneous resistance to CB-183,315 at 8× the MIC was below the limit of detection in C. difficile. In a phase 1 randomized, double-blind, placebo-controlled, multiple-dose study in healthy volunteers, stool samples were collected from all subjects in the 1,000-mg-BID CB-183,315 dose group on day 5. CB-183,315 reached mean concentrations of 6,394 ± 3,104 μg/g (Cubist Pharmaceuticals, Inc., data on file). Although the phase 1 dose was 4- to 8-fold higher than the clinical dose of CB-183,315 during the phase 2 clinical trial, the average concentration of CB-183,315 was 12,000-fold greater than the MIC90 of CB-183,315 for C. difficile, and therefore, the emergence of resistance to CB-183,315 against C. difficile is likely to be a rare event. In our serial passage resistance study, under selective pressure with CB-183,315, C. difficile susceptibility changed no more than 2-fold over 15 days of serial passage, again suggesting that emergence of resistance should be infrequent.
Against toxigenic and nontoxigenic isolates of C. difficile and a NAP1 (REA group BI) clinical isolate, 16× the MIC of CB-183,315 reached the bactericidal endpoint between 6 and 24 h. Bactericidal activity against this range of C. difficile strains, including the hypervirulent NAP1 strain, has rarely been achieved in vitro. Vancomycin, one of the standard treatments for CDAD, has been shown in vitro to be bacteriostatic but not bactericidal against many strains of C. difficile (5, 16, 28).
The PAE of CB-183,315 at 8× the MIC is relatively short, 0.9 h, and is not likely to be relevant in clinical settings. The PAE of CB-183,315 at 80× the MIC is >6 h. A significant amount of the bacterial population was killed (∼3 log units) during the 30 min of exposure before being diluted 1,000-fold, suggesting that, when present in high concentrations, CB-183,315 is rapidly bactericidal, extending the time required for C. difficile cultures to rebound after drug removal.
In our hamster model of CDAD, CB-183,315 and vancomycin both demonstrated potent efficacy in protecting hamsters from lethal C. difficile infections during the dosing period, even at very low doses. In addition, after the conclusion of dosing, CB-183,315 and vancomycin showed a similar dose- and time-dependent pattern with respect to rates of CDAD recurrence in the hamster model. Since this hamster infection model of CDAD has been shown previously to resemble many, but not all, aspects of CDAD in humans (3, 22), and as vancomycin is the recommended treatment option for severe CDAD (7), the potent efficacy of CB-183,315 demonstrates that this novel lipopeptide warrants further investigation as a promising new therapeutic agent for CDAD.
In related studies, CB-183,315 has shown good potency against C. difficile isolates, including isolates with elevated MICs of metronidazole, moxifloxacin, and vancomycin (5, 26). Additionally, the lack of activity of CB-183,315 against Enterobacteriaceae and species of the Bacteroides fragilis group (MIC90, >8,192 μg/ml) strongly suggests that CB-183,315 will minimize disruption and favor rapid restoration of the normal gut flora (5). These properties are needed to achieve high rates of clinical cure and lower risks of recurrence. Taken together, these results suggest that CB-183,315, which has recently completed phase 2 clinical trials, may be suitable for further clinical development as a treatment for C. difficile infection.
ACKNOWLEDGMENTS
We thank Nancy Perlmutter and Howard Shapiro for their assistance with flow cytometry experiments and Xi-Xian Zhang for technical assistance relating to the in vivo hamster studies. Under the direction of the authors, Cynthia L. Kryder, agent of PharmaWrite, LLC (Princeton, NJ), provided professional medical writing assistance in preparing and editing the manuscript. We thank Catherine Whalen Kuczewski, of PharmaWrite, LLC (Princeton, NJ), for her assistance with medical editing and logistical support.
Carmela T. M. Mascio, Lawrence I. Mortin, Karen T. Howland, Shuxin Zhang, Anu Arya, Cun Lan Chuong, Chunfeng Kang, Tongchuan Li, and Jared A. Silverman are employees of and stockholders in Cubist Pharmaceuticals, Inc. During the study, Andrew D. G. Van Praagh was an employee of and stockholder in Cubist Pharmaceuticals, Inc.; he is no longer an employee of or stockholder in Cubist.
Cubist Pharmaceuticals, Inc., provided funding for professional medical writing assistance.
Footnotes
Published ahead of print 16 July 2012
REFERENCES
- 1. Al-Nassir WN, et al. 2008. Comparison of clinical and microbiological response to treatment of Clostridium difficile-associated disease with metronidazole and vancomycin. Clin. Infect. Dis. 47:56–62 [DOI] [PubMed] [Google Scholar]
- 2. Bartlett JG. 2007. Clostridium difficile: old and new observations. J. Clin. Gastroenterol. 41(Suppl 1):S24–S29 [Google Scholar]
- 3. Bartlett JG, Onderdonk AB, Cisneros RL, Kasper DL. 1977. Clindamycin-associated colitis due to a toxin-producing species of Clostridium in hamsters. J. Infect. Dis. 136:701–705 [DOI] [PubMed] [Google Scholar]
- 4. Chesnel L, et al. 2012. Treatment of CDAD with oral CB-183 315: time to recurrence, relapse and re-infection rates compared with vancomycin. Clin. Microbiol. Infect. 18(Suppl 3):380 [Google Scholar]
- 5. Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ. 2012. In vitro activities of CB-183,315, vancomycin, and metronidazole against 556 strains of Clostridium difficile, 445 other intestinal anaerobes, and 56 Enterobacteriaceae species. Antimicrob. Agents Chemother. 56:1613–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cloud J, Kelly CP. 2007. Update on Clostridium difficile associated disease. Curr. Opin. Gastroenterol. 23:4–9 [DOI] [PubMed] [Google Scholar]
- 7. Cohen SH, et al. 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31:431–455 [DOI] [PubMed] [Google Scholar]
- 8. Collett D. 1994. Modeling survival data in medical research. Chapman and Hall, London, United Kingdom [Google Scholar]
- 9. Craig WA, Gudmundsson S. 1996. Postantibiotic effect, p 296–329 In Lorian V. (ed), Antibiotics in laboratory medicine, 4th ed Williams & Wilkins, Baltimore, MD [Google Scholar]
- 10. Dallal RM, et al. 2002. Fulminant Clostridium difficile: an underappreciated and increasing cause of death and complications. Ann. Surg. 235:363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freeman J, Baines SD, Jabes D, Wilcox MH. 2005. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J. Antimicrob. Chemother. 56:717–725 [DOI] [PubMed] [Google Scholar]
- 12. Johnson AP. 2010. New antibiotics for selective treatment of gastrointestinal infection caused by Clostridium difficile. Expert. Opin. Ther. Patents 20:1389–1399 [DOI] [PubMed] [Google Scholar]
- 13. Kaplan EL, Meier P. 1958. Nonparametric estimation from incomplete observation. J. Am. Stat. Assoc. 53:457–481 [Google Scholar]
- 14. Kelly CP, Pothoulakis C, LaMont JT. 1994. Clostridium difficile colitis. N. Engl. J. Med. 330:257–262 [DOI] [PubMed] [Google Scholar]
- 15. Lawrence SJ, Dubberke ER, Johnson S, Gerding DN. 2007. Clostridium difficile-associated disease treatment response depends on definition of cure. Clin. Infect. Dis. 45:1648–1651 [DOI] [PubMed] [Google Scholar]
- 16. Louie TJ, et al. 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 364:422–431 [DOI] [PubMed] [Google Scholar]
- 17. McDonald LC, Owings M, Jernigan DB. 2006. Clostridium difficile infection in patients discharged from US short-stay hospitals, 1996–2003. Emerging Infect. Dis. 12:409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mullane KM, Gorbach S. 2011. Fidaxomicin: first-in-class macrocyclic antibiotic. Expert Rev. Anti-infect. Ther. 9:767–777 [DOI] [PubMed] [Google Scholar]
- 19. Nelson RL, et al. 2011. Antibiotic treatment for Clostridium difficile-associated diarrhea in adults. Cochrane Database Syst. Rev. 7:CD004610 doi:10.1002/14651858.CD004610.pub4 [DOI] [PubMed] [Google Scholar]
- 20. Novo D, Perlmutter NG, Hunt RH, Shapiro HM. 1999. Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry 35:55–63 [DOI] [PubMed] [Google Scholar]
- 21. Patino H, et al. 2011. Efficacy and safety of the lipopeptide CB-183,315 for the treatment of Clostridium difficile infection, abstr. K-205a. Abstr. 51st Intersci. Conf. Antimicrob. Agents Chemother., September 17–20, Chicago, IL [Google Scholar]
- 22. Razaq N, et al. 2007. Infection of hamsters with historical and epidemic BI types of Clostridium difficile. J. Infect. Dis. 196:1813–1819 [DOI] [PubMed] [Google Scholar]
- 23. Shah D, Dang MD, Hasbun R, Koo HL, Jiang DuPont Z-DHL, Garey KW. 2010. Clostridium difficile infection: update on emerging antibiotic treatment options and antibiotic resistance. Expert Rev. Anti-infect. Ther. 8:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silverman JA, Oliver N, Andrew T, Li T. 2001. Resistance studies with daptomycin. Antimicrob. Agents Chemother. 45:1799–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Silverman JA, Perlmutter NG, Shapiro HM. 2003. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 47:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snydman DR, Jacobus NV, McDermott LA. 2012. Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob. Agents Chemother. 56:3448–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swanson RN, et al. 1991. In vitro and in vivo evaluation of tiacumicins B and C against Clostridium difficile. Antimicrob. Agents Chemother. 35:1108–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tyrrell KL, et al. 2006. In vitro activities of daptomycin, vancomycin, and penicillin against Clostridium difficile, C. perfringens, Finegoldia magna, and Propionibacterium acnes. Antimicrob. Agents Chemother. 50:2728–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. 2007. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin. Infect. Dis. 45:302–307 [DOI] [PubMed] [Google Scholar]