

Abstract
Clinically approved antibiotics inhibit only a small number of conserved pathways that are essential for bacterial viability, and the physiological effects of inhibiting these pathways have been studied in great detail. Likewise, characterizing the effects of candidate antibiotics that function via novel mechanisms of action is critical for their development, which is of increasing importance due to the ever-growing problem of resistance. The arylomycins are a novel class of natural-product antibiotics that act via the inhibition of type I signal peptidase (SPase), which is an essential enzyme that functions as part of the general secretory pathway and is not the target of any clinically deployed antibiotic. Correspondingly, little is known about the effects of SPase inhibition or how bacteria may respond to mitigate the associated secretion stress. Using genetically sensitized Escherichia coli and Staphylococcus aureus as model organisms, we examine the activity of arylomycin as a function of its concentration, bacterial cell density, target expression levels, and bacterial growth phase. The results reveal that the activity of the arylomycins results from an insufficient flux of proteins through the secretion pathway and the resulting mislocalization of proteins. Interestingly, this has profoundly different effects on E. coli and S. aureus. Finally, we examine the activity of arylomycin in combination with distinct classes of antibiotics and demonstrate that SPase inhibition results in synergistic sensitivity when combined with an aminoglycoside.
INTRODUCTION
Most clinically deployed antibiotics interfere with the essential cellular processes of DNA synthesis, RNA synthesis, peptidoglycan synthesis, or membrane polarization (43). While these drugs have revolutionized health care, the emergence of multidrug-resistant pathogens necessitates the identification of new classes of antibiotics (50), especially those that act via novel mechanisms of action and thereby minimize the potential for cross-resistance. Bacterial type I signal peptidase (SPase) has long been appreciated as a potential target for antibacterial therapy, because it is conserved, essential, and located in the relatively accessible outer leaflet of the cytoplasmic membrane (29, 31, 46). SPase functions as part of the general secretory pathway to remove the N-terminal signal peptide from membrane-bound preproteins following their translocation across the cytoplasmic membrane, which affords the mature proteins access to their ultimate destinations in the periplasm, outer membrane, or extracellular milieu (7–9, 38). Therefore, the inhibition of SPase is likely to result in the accumulation of preproteins in the cytoplasmic membrane and the misregulation of essential pathways that utilize these proteins. In addition, depending on the protein-protein interactions involved, SPase inhibition could result in the obstruction of secretion channels, which has been linked to the antibiotic activity of some translational inhibitors (52).
The arylomycins (Fig. 1) are a relatively recently discovered class of natural-product antibiotics that were shown to bind and inhibit SPase in vitro (20, 30, 41). Despite the apparent accessibility and essentiality of SPase, initial reports suggested that the arylomycins are active against only a few Gram-positive bacteria, including Streptococcus pneumoniae, Rhodococcus opacus, and Brevibacillus brevis (20, 41), and not against other important Gram-positive pathogens or against any Gram-negative bacteria. However, after reporting the first total synthesis of an arylomycin (35), as well as several derivatives (23, 35, 36, 44, 45), including arylomycin A-C16 (Fig. 1) (previously referred to as arylomycin C16), we found that they have potent antibacterial activity against a wide variety of Gram-positive and Gram-negative bacteria. Moreover, we identified a specific binding-site Pro residue that contributes to the natural resistance of the Gram-positive pathogen Staphylococcus aureus and the Gram-negative pathogens Escherichia coli and Pseudomonas aeruginosa (45). Importantly, for both E. coli and S. aureus, we demonstrated that the Pro mutation contributes to resistance by reducing the affinity with which SPase is bound by the arylomycins, suggesting that arylomycin derivatives that bind SPase with improved affinity could have clinical utility (45). Indeed, several derivatives with improved activity against S. aureus have been identified (34, 36).
Fig 1.

Structure of arylomycin A-C16. Other members of the arylomycin family of natural-product antibiotics are defined by different fatty acid lipid tails or modifications of the central biphenyl core (51).
Due to the novelty of the arylomycin class of antibiotics and of SPase as a target, little is known about their activity, including whether it is bacteriostatic or bactericidal and how it varies as a function of concentration, bacterial density, state of growth, or level of SPase expression. Moreover, it is unknown if the lipid tail contributes to activity, for example, by causing nonspecific membrane depolarization, as has been observed with other lipidated antibiotics (49). Herein, we begin to explore these issues using model strains of E. coli and S. aureus whose SPases have been rendered sensitive to the arylomycins via a single point mutation that removes the resistance-conferring Pro. The data demonstrate that the antibiotic activity of the arylomycins results from insufficient SPase activity and not from blocked secretion channels or from a nonspecific membrane depolarization caused by their lipophilicity. Furthermore, arylomycin activity can be either bacteriostatic or bactericidal, depending on the organism and growth conditions, and the activities against the Gram-negative and Gram-positive organisms are distinctly different, suggesting that secretion plays distinct roles in the viability of these divergent organisms and perhaps suggesting that they have evolved different mechanisms to cope with secretion stress. Finally, while we found that the arylomycins show relatively little synergy or antagonism with most other classes of antibiotics, they do show pronounced synergy with gentamicin, suggesting that SPase inhibitors may be particularly efficacious when coadministered with an aminoglycoside.
MATERIALS AND METHODS
Medium and antibiotics.
Bacteria were routinely grown at 37°C on Mueller-Hinton II agar (MHAII) or in cation-adjusted Mueller-Hinton II broth (MHBII) with shaking at 275 rpm. Stock solutions of antibiotics were prepared in water or dimethyl sulfoxide (DMSO) at the following concentrations: arylomycin A-C16, 10 mg/ml (DMSO); polymyxin B, 1 mg/ml (H2O); vancomycin, 10 mg/ml (H2O); cephalexin, 1 mg/ml (H2O); erythromycin, 15 mg/ml (DMSO); tetracycline, 15 mg/ml (DMSO); rifampin, 15 mg/ml (DMSO); gentamicin, 15 mg/ml (H2O); trimethoprim, 15 mg/ml (DMSO); ciprofloxacin, 1 mg/ml (H2O). Arylomycin A-C16 was synthesized as described previously (35); polymyxin B and vancomycin were obtained from Sigma-Aldrich (St. Louis, MO); all other antibiotics were obtained from MP Biomedicals (Solon, OH). Anhydrotetracycline (aTc; Acros, Morris Plains, NJ) stock solutions were prepared in neat DMSO at a concentration of 100 μg/ml.
Bacterial strains and plasmids.
All strains and plasmids used in this study are listed in Table 1. The effects of inhibiting SPase of E. coli MG1655 and S. aureus NCTC 8325-4 were examined with derivative strains PAS0260 and PAS8001, respectively (45). In these strains, the Pro residue in the SPase responsible for arylomycin resistance was replaced by a residue that confers arylomycin sensitivity; the resulting SPases were LepB(P84L) in E. coli strain PAS0260 and SpsB(P29S) in S. aureus strain PAS8001. To examine the effect of increased expression of arylomycin-susceptible SPases, E. coli strains PAS0275 and PAS0234 were created to allow for the aTc-inducible, ectopic expression of LepB(P84L) and LepB(P84S) from the plasmid pTetBHR2. Briefly, the wild-type E. coli lepB coding sequence and upstream Shine-Dalgarno sequence were amplified using primers lepB+RBS_NF_NdeI and lepB_CR_KpnI, and the product was phosphorylated using T4 polynucleotide kinase (New England BioLabs). The DNA was ligated into the vector pTetBHR2 that had been digested with XhoI and XmaI (New England BioLabs) and treated with Klenow fragment DNA polymerase (New England BioLabs). The resulting vector was subjected to QuikChange mutagenesis (Stratagene) with primers described previously (45) to introduce the LepB(P84L) or LepB(P84S) mutations, resulting in plasmids pTetBHR2-LepB(P84L) and pTetBHR2-LepB(P84S), respectively. The plasmid pTetBHR2-LepB(P84L) was subsequently electroporated into PAS0260 to generate PAS0275 to allow overexpression of LepB(P84L). Plasmid pTetBHR2-LepB(P84S) was transformed into PAS0232, which contains the chromosomal LepB(P84S) mutation. The P84S mutation in SPase also sensitizes E. coli to the arylomycins (45).
Table 1.
Strains used in this study
| Strain | Relevant characteristicsa | Reference or source |
|---|---|---|
| MG1655 | E. coli F− λ− ilvG rbf-50 rph-1 | 2 |
| PAS0162 | MG1655 ΔLepB::Kanr + pTetBHR2-LepB | This work |
| PAS0216 | MG1655 ΔLepB::Kanr + pTetBHR2-LepB(P84S) | This work |
| PAS0232 | MG1655 LepB(P84S)::Kanr | 45 |
| PAS0234 | MG1655 LepB(P84S)::Kanr + pTetBHR2-LepB(P84S) | This work |
| PAS0260 | MG1655 LepB(P84L)::Kanr | 45 |
| PAS0275 | MG1655 LepB(P84L)::Kanr + pTetBHR2-LepB(P84L) | This work |
| PAS8001 | S. aureus NCTC 8325 SpsB(P29S) | 45 |
Kanr, kanamycin resistance.
E. coli strain PAS0162 expresses wild-type SPase solely from a plasmid and requires aTc for SPase expression and wild-type growth rates. To construct this strain, following transformation of E. coli MG1655 with pTetBHR2-LepB, the chromosomal copy of lepB was removed via allelic exchange (28). Briefly, a disruption cassette was assembled using 3-way assembly PCR, in which approximately 1,000-bp regions upstream and downstream of the gene to be deleted were fused to the kanamycin resistance cassette from plasmid pUC4K in place of the lepB gene. The resulting cassette was electroporated into cells expressing SPase from the vector pTetBHR2-LepB and the lambda red recombinase (5), and the resulting chromosomal lepB deletion was transferred into E. coli MG1655 with pTetBHR2-LepB by P1 transduction. Strain PAS0216 was generated similarly using pTetBHR2-LepB(P84S) to complement the chromosomal deletion.
Susceptibility determination.
Antibiotic susceptibilities were determined for PAS0260 or PAS8001 by measuring MICs using the CSLI broth microdilution method (3). Briefly, 2-fold serial dilutions of antibiotics were prepared in 96-well plates containing 100 μl of cation-adjusted MHBII. Bacterial inocula were prepared by suspending colonies grown for 24 h on MHAII to a final density of 1 × 107 CFU per ml in MHBII. Wells containing the antibiotic dilutions were inoculated to a final density of 5 × 105 CFU/ml, and MICs were defined as the lowest arylomycin A-C16 concentration at which no visible growth occurred following 24 h of incubation at 37°C. The contribution of increased levels of SPase expression to arylomycin susceptibilities was determined by inoculating 96-well plates containing a checkerboard of 2-fold dilutions of arylomycin A-C16 and aTc with E. coli strain PAS0275 harboring LepB(P84L) under the control of the aTc-inducible promoter on plasmid pTetBHR2. The contribution of SPase underexpression to the susceptibility of E. coli expressing wild-type and P84S SPases was determined similarly except that bacteria were grown on MHAII containing 64 μg/ml aTc. Colonies were then diluted into MHBII without aTc to an initial optical density (OD) of 0.01 and grown to an OD of 0.5 to dilute out the high levels of SPase produced under inducing conditions on solid media. These cultures were used to inoculate 96-well plates containing a checkerboard of 2-fold dilutions of arylomycin A-C16 and aTc.
Time-kill assays.
Time-kill experiments were performed by suspending bacterial colonies in MHBII and inoculating the suspension into a 50-ml conical tube containing 10 ml of MHBII to a final OD at 600 nm (OD600) of 0.025. Cultures were grown to mid-logarithmic growth phase (OD600 = 0.4 to 0.5) and diluted with prewarmed MHBII to a final density of 1 × 106 or 1 × 108 CFU/ml for normal and high-density kill curves, respectively (1 × 108 CFU/ml corresponds to an OD600 of 0.2 for E. coli and 0.1 for S. aureus). Diluted cultures (3 ml) were aliquoted into 14-ml culture tubes containing arylomycin A-C16 and incubated at 37°C with shaking at 275 rpm. Bacterial viability was quantified by plating serial dilutions onto MHAII and determining viable CFU after 24 h. Additionally, to increase the detection limit at the final 18-h time point, 1 ml of each culture was pelleted, washed once in phosphate-buffered saline (PBS), and plated onto MHAII. Time-kill assays were also performed with S. aureus in the presence of 0.5 μg/ml (2 times the MIC) of the bacteriostatic translational inhibitor tetracycline. Time-kill assays with stationary-phase cells were performed similarly. Briefly, MHBII (10 ml) was inoculated with colonies suspended to a final OD600 of 0.025, and the cultures were grown for 18 h at 37°C with shaking at 275 rpm. The resulting saturated cultures were pelleted and washed twice in PBS, then diluted into PBS to a density of approximately 1 × 106 CFU/ml and incubated for 30 min at 37°C with shaking to provide time to acclimate to the nutrient-depleted conditions. Cultures were then aliquoted into 14-ml culture tubes containing arylomycin A-C16, and CFU were quantified as described above.
Interactions between arylomycin A-C16 and other antibiotics.
Interactions between antibiotics were determined by calculating fractional inhibitory concentration (FIC) indexes from microdilution checkerboard MIC experiments (14, 33). The FIC of an antibiotic is the concentration that kills when used in combination with another antibiotic divided by the concentration that has the same effect when used alone, and the FIC index is defined as the sum of the FICs of the two antibiotics. FIC indexes of ≤0.5 reflect significant synergism, and those that are ≥2 reflect significant antagonism (10, 14). Because interactions between two antibiotics can vary depending on the relative concentration of each agent, the minimum and maximum FIC indexes are reported for each combination of agents. Checkerboard analyses for each antibiotic combination were performed five times to ensure the significance of the results (33), and the average and standard deviation of each FIC index are reported.
RESULTS
Time-kill studies.
To examine whether the arylomycins have bacteriostatic or bactericidal activity, time-kill experiments were performed with actively growing cultures of the arylomycin-susceptible strains E. coli PAS0260 and S. aureus PAS8001 (Fig. 2). Each of these strains differs from its corresponding wild-type strain by a single amino acid substitution that converts the resistance-conferring Pro to either Leu (E. coli strain PAS0260) or Ser (S. aureus strain PAS8001), resulting in increased arylomycin A-C16 sensitivity (45). In the absence of arylomycin A-C16, the growth of these strains is identical to that of the respective wild-type strains. Unless otherwise noted, experiments were conducted with E. coli PAS0260 and S. aureus PAS8001.
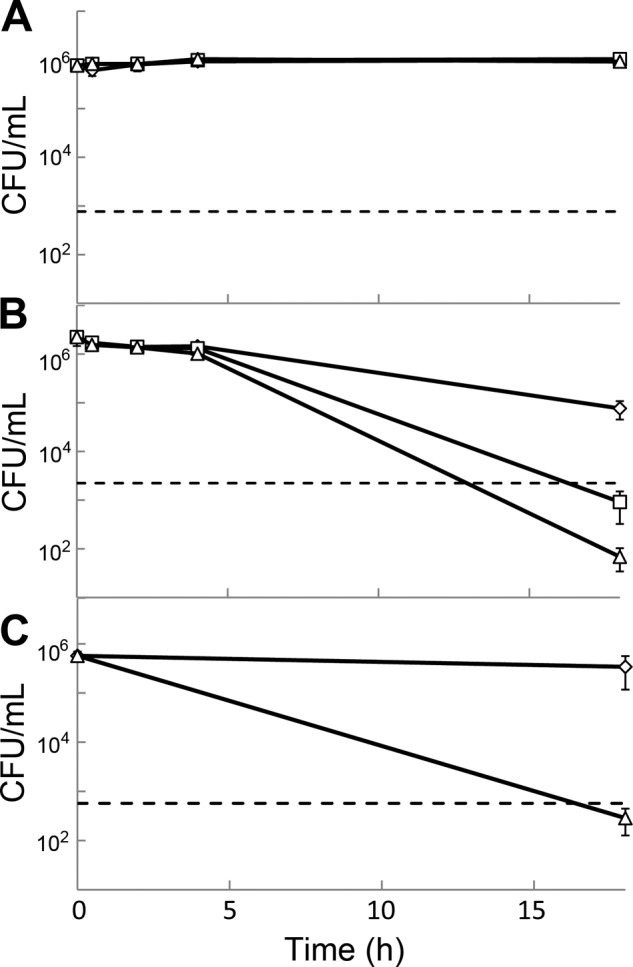
Fig 2.

Viability of exponentially growing E. coli PAS0260 at normal density (A) and high density (B) and exponentially growing S. aureus PAS8001 at normal density (C) and high density (D) in the presence of arylomycin A-C16 at concentrations 2-fold (open squares) or 8-fold (open triangles) above the MIC compared to that of the vehicle control (open diamonds). A 3-log kill is used as the definition of bactericidal activity and is represented by dashed lines.
At a cell density of 1 × 106 CFU/ml, we observed rapid inhibition of growth for both PAS0260 and PAS8001 at arylomycin concentrations 2- and 8-fold above their respective MICs, and this inhibition lasted the entire 18-h course of the experiment. During the initial 4 h of exposure, no decrease in the viability of the S. aureus cultures was observed, and the majority of cells remained viable even after 18 h of exposure to the lower concentration of arylomycin. Only a 20-fold decrease in viability was observed at the higher concentration after 18 h. In contrast, the viability of E. coli cultures was reduced 10- to 100-fold during the initial 4 h, with a slightly greater effect at higher arylomycin concentrations. Moreover, the cultures of E. coli were almost completely sterilized (105-fold reduction in CFU/ml) following the prolonged exposure. Increasing the density of the inocula to 1 × 108 CFU/ml had only modest effects on the kinetics of killing, and at 18 h the viability of S. aureus was unchanged, whereas the viability of E. coli cultures decreased more than 103-fold. Similar to the results observed with lower-density cultures, increasing the concentration of arylomycin had at most a modest effect on the activity against higher-density cultures.
Activity against quiescent cells.
To determine whether the arylomycins have bactericidal activity against nongrowing cells, PAS0260 and PAS8001 cultures were grown to stationary phase in rich media (MHBII), diluted in PBS to a density of 1 × 106 CFU/ml, and then treated with various concentrations of arylomycin A-C16 for a period of 18 h (Fig. 3A and B). Under these conditions, cultures of E. coli maintained full viability in the presence or absence of arylomycin, suggesting that active growth is required for bactericidal activity. In contrast, while the viability of S. aureus cultures decreased slightly (approximately 30-fold) in the absence of arylomycin A-C16, a reduction of over 3 orders of magnitude was observed in the presence of arylomycin A-C16 at both 2 and 8 times the MIC. To examine whether the observed sensitivity of S. aureus results from poor tolerance of prolonged suspension in PBS, we characterized the effect of arylomycin A-C16 on rich-medium cultures of S. aureus treated with the bacteriostatic translational inhibitor tetracycline (Fig. 3C). While tetracycline alone had almost no effect on the viability of S. aureus, tetracycline in combination with arylomycin A-C16 resulted in a greater-than-103-fold reduction in viability.
Fig 3.
Viability of stationary-phase cultures of arylomycin-susceptible E. coli and S. aureus strains. Stationary-phase cultures of E. coli (A) and S. aureus (B) were washed and diluted into PBS and treated with vehicle controls (open diamonds) or with arylomycin A-C16 at concentrations 2-fold (open squares) or 8-fold (open triangles) above the respective MICs. Cultures of S. aureus whose growth had been arrested by the addition of tetracycline were treated with vehicle controls (open diamonds) or with arylomycin A-C16 at 8 times the MIC (open triangles) (C). A 3-log kill is used as the definition of bactericidal activity and is represented by dashed lines.
Contribution of SPase expression levels.
To determine whether SPase expression levels affect arylomycin susceptibility, arylomycin MICs were determined for E. coli underexpressing wild-type (arylomycin-resistant) SPase (PAS0162) or overexpressing either of the arylomycin-sensitive P84L (PAS0234) or P84S (PAS0275) mutants (Table 2). The controlled underexpression of wild-type SPase failed to yield detectable arylomycin sensitivities (MIC > 64 μg/ml), even at levels that reduced the growth rates by over 80% (see Fig. S1 in the supplemental material). However, underexpression of the sensitive P84S mutant resulted in a decrease of the arylomycin A-C16 MIC of up to 8-fold, whereas overexpression of either P84L or P84S mutants resulted in an 8-fold increase of the MIC, demonstrating that target overexpression can yield at least modest levels of resistance. Antibiotics whose activity decreases with target overexpression typically demonstrate dominant resistance when both resistant and susceptible alleles of the target are expressed. Consistently, an E. coli strain expressing a wild-type SPase chromosomally and the susceptible (P84L) SPase ectopically was completely resistant to arylomycin A-C16 (Table 2).
Table 2.
Effects of SPase expression levels on the arylomycin A-C16 sensitivity of E. coli
| aTc concn (ng/ml) | Arylomycin A-C16 MIC (mg/ml) for straina: |
||||||
|---|---|---|---|---|---|---|---|
| MG1655 (NEV/Wt) | PAS0162 (Wt/NCS) | PAS0260 (NEV/P84L) | PAS0275 (P84L/P84L) | PAS0232 (NEV/P84S) | PAS0234 (P84S/P84S) | PAS0216 (P84S/NCS) | |
| 0 | >64 | >64 | 1 | 2 | 8 | 16 | 1 |
| 4 | >64 | >64 | 1 | 2 | 8 | >64 | 2 |
| 8 | >64 | >64 | 1 | 4 | 8 | >64 | 4 |
| 16 | >64 | >64 | 1 | 8 | 8 | >64 | 64 |
| 32 | >64 | >64 | 1 | 16 | 8 | >64 | >64 |
| 64 | >64 | >64 | 1 | 16 | 8 | >64 | >64 |
| 128 | >64 | >64 | 1 | 16 | 8 | >64 | >64 |
For each strain, ectopic/chromosomal SPases are in parentheses. Wt, wild-type SPase; P84L and P84S, arylomycin-sensitive mutants (see the text); NEV, no SPase expression vector; NCS, no chromosomal SPase.
Antibiotic interactions.
To examine how the presence of other antibiotics affects the MIC of arylomycin, checkerboard MIC experiments were performed with PAS0260 and PAS8001 and minimal and maximal FIC indexes were determined (Table 3; see Fig. S2 in the supplemental material). Against E. coli, we observed no significant interactions between arylomycin A-C16 and erythromycin, polymyxin B, trimethoprim, or ciprofloxacin. In contrast, arylomycin A-C16 showed mild synergism with cephalexin, pronounced synergism with rifampin and gentamicin, and antagonism with the translational inhibitor tetracycline. Against S. aureus, tetracycline, erythromycin, and vancomycin each interacted additively with arylomycin A-C16, while rifampin and trimethoprim showed pronounced antagonism. There was mild synergism between arylomycin A-C16 and ciprofloxacin or cephalexin with S. aureus, and as with E. coli, pronounced synergism between arylomycin A-C16 and gentamicin.
Table 3.
MICs of antibiotics alone and FIC indexesa of antibiotics in combination with arylomycin A-C16
| Antibiotic |
E. coli PAS0260 |
S. aureus PAS8001 |
||||
|---|---|---|---|---|---|---|
| MIC (μg/ml) | FICmin | FICmax | MIC (μg/ml) | FICmin | FICmax | |
| Cephalexin | 12 | 0.70 ± 0.19 | 1.14 ± 0.07 | 4 | 0.65 ± 0.06 | 1.38 ± 0.42 |
| Gentamicin | 1 | 0.55 ± 0.11 | 0.91 ± 0.25 | 0.5 | 0.46 ± 0.12 | 0.83 ± 0.22 |
| Tetracycline | 1.5 | 1.10 ± 0.06 | 2.68 ± 0.89 | 0.25 | 0.73 ± 0.06 | 1.36 ± 0.40 |
| Erythromycin | 128 | 0.90 ± 0.21 | 1.28 ± 0.14 | 1 | 0.79 ± 0.26 | 1.15 ± 0.06 |
| Trimethoprim | 0.25 | 0.90 ± 0.21 | 1.58 ± 0.50 | 2 | 1.05 ± 0.03 | 2.55 ± 1.02 |
| Ciprofloxacin | 0.023 | 0.80 ± 0.11 | 1.23 ± 0.06 | 0.25 | 0.63 ± 0.08 | 1.39 ± 0.38 |
| Rifampin | 24 | 0.50 ± 0.00 | 1.03 ± 0.22 | 0.008 | 1.06 ± 0.00 | 2.85 ± 1.25 |
| Polymyxin B | 1 | 0.78 ± 0.30 | 1.15 ± 0.32 | — | — | — |
| Vancomycin | —b | — | — | 2 | 0.80 ± 0.11 | 1.23 ± 0.06 |
| Arylomycin A-C16 | 1 | — | — | 4 | — | — |
FICmin and FICmax, minimum and maximum FIC indexes observed, respectively. Values are averages ± standard deviations.
—, not determined.
DISCUSSION
Protein secretion is an essential process in all bacteria, and the general secretory pathway, consisting of the SecA motor protein and the SecYEG channel, is universally conserved in bacteria (7–9, 38). Due to its essential role in this process (24, 37), as well as its relatively accessible location, SPase has long engendered interest as a potential antibiotic target (29, 31, 46). The arylomycin class of natural products is the only known class of inhibitors of SPase that exhibit significant antibiotic activity. While this activity is limited in many organisms due to a mutation in SPase, the arylomycins are active against representative organisms from four bacterial phyla, including Gram-negative and Gram-positive pathogens, suggesting that with optimization, the arylomycins could have broad-spectrum activity (45), and indeed several derivatives with increased potency against S. aureus have recently been identified (34, 36). To better understand the effects of arylomycin-mediated SPase inhibition, we examined how its activity depends on its concentration, bacterial growth and density, SPase expression levels, and the presence of unrelated classes of antibiotics.
To begin to understand how cells arrested with arylomycin A-C16 cope with the stresses associated with SPase inhibition, we examined the effect of arylomycin A-C16 on cell viability as a function of exposure time. Against rapidly growing S. aureus, arylomycin A-C16 is largely bacteriostatic, but under nutrient-depleted conditions or following growth arrest by tetracycline, exposure to arylomycin A-C16 induces a greater-than-103-fold decrease in viability. It is possible that, even in the presence of tetracycline at twice the MIC or during nutrient starvation, cells still produce proteins at some reduced level and that the secretion of these proteins is essential to tolerating these stresses. Alternatively, tetracycline or nutrient starvation may prevent the production of some protein(s) required to tolerate the inhibition of SPase. Consistent with the latter possibility, we recently demonstrated that arylomycin A-C16 induces the expression of three proteins in S. aureus, PrsA, HtrA, and SAOUHSC_01761 (40). Although little is known about SAOUHSC_01761, PrsA and HtrA are conserved in Gram-positive bacteria and are thought to facilitate the folding and maturation of secreted proteins and the degradation of proteins that misfold during secretion (4, 12, 15, 17, 25, 32, 39, 42, 48). Thus, the activity of the arylomycins may be bacteriostatic against S. aureus due to the production of these, and possibly other, proteins that help mitigate the stress associated with inhibited protein secretion.
Interestingly, the effect of arylomycin A-C16 on viability is notably different with E. coli, where bactericidal activity against rapidly growing cells is observed, but not under nutrient-deprived conditions. It has recently been shown that inhibition of ribosomes involved in cotranslation/translocation induces blockage of SecYEG secretion channels and activates a specific FtsH-mediated cell death pathway (52). Similarly, if SPase directly associates with the SecYEG secretion channel, this pathway could be responsible for the observed bactericidal activity. In this case, increased levels of SPase expression should increase toxicity, as observed with other antibiotics that generate toxic products upon binding their targets (18). In contrast, we find that under- or overexpressing an arylomycin-susceptible SPase in E. coli increases or decreases sensitivity, respectively. Moreover, the resistance phenotype is dominant in cells simultaneously expressing both susceptible and resistant variants of SPase. These data argue against an FtsH-mediated process and suggest that the bactericidal activity of the arylomycins results from protein mislocalization in the cytoplasmic membrane. Indeed, in Gram-negative bacteria, which must maintain a protein-rich periplasm and outer membrane, SPase processes a far greater number of proteins than in Gram-positive organisms (47). It remains to be determined whether cell death ultimately results from an excessive accumulation of unprocessed preproteins in the membrane, possibly eventually causing depolarization, or from misregulation of essential periplasmic and outer-membrane processes. Regardless of the observed differences between E. coli and S. aureus, when bactericidal activity is observed in either organism, the kinetics of cell death are slow and relatively independent of arylomycin A-C16 concentration. This is consistent with the antibiotic activity resulting solely from the inhibition of SPase and argues against the nonspecific membrane depolarization mechanisms that have been associated with other classes of lipopeptide antibiotics (49).
The interactions between two antibiotics can often provide mechanistic insight into their activities that is not apparent when measuring their independent effects on growth (11, 53). Antibiotics that inhibit pathways that mediate the deleterious effects of SPase inhibition are expected to antagonize arylomycin A-C16 activity. In contrast, agents whose mechanisms of action increase the cellular dependence on SPase are expected to result in synergistic interactions with arylomycin A-C16. Indeed, by chemical (16; M. Gallant et al., WIPO patent application WO/2011/112441 [http://patentscope.wipo.int/search/en/WO2011112441]) and genetic (21) methods, SPase inhibition in methicillin-resistant S. aureus (MRSA) has recently been shown to be synergistic with the activity of the carbapenem class of antibiotics, suggesting that increased protein secretion plays a critical role in the MRSA phenotype. With notable exceptions, we find that arylomycin A-C16 shows no interaction or mild antagonism with antibiotics that inhibit DNA, RNA, or protein synthesis. Interestingly, antagonism, where it was observed, was always manifest as an increase in the arylomycin A-C16 MIC in the presence of sublethal concentrations of the second antibiotic (see Fig. S2 in the supplemental material). Sublethal inhibition of DNA, RNA, or protein synthesis induces specific stress response pathways (6), and facets of these responses might help alleviate arylomycin-induced stress. Reduced growth rates induced by these agents could also minimize the production of proteins that require translocation across the cytoplasmic membrane and thereby reduce the burden on the general secretion pathway. Similar antagonism between DNA and protein synthesis inhibitors has been attributed to an inability of cells to properly downregulate protein production in response to DNA damage (1). In contrast to the antagonism observed with S. aureus, the simultaneous inhibition of RNA synthesis and secretion results in synergistic sensitivity in E. coli. Although this synergism could result from an increased penetrance of rifampin following arylomycin-mediated membrane damage, similar interactions were not observed with the hydrophobic antibiotic erythromycin, suggesting that specific target-mediated effects may be involved. The modest synergy observed with arylomycin A-C16 and cell wall synthesis inhibitors is consistent with the significant number of secreted proteins that are known to be required for cell wall biosynthesis (22, 27). However, vancomycin, a Gram-positive organism-specific inhibitor of cell wall biosynthesis, does not interact synergistically with the arylomycins, demonstrating that more-nuanced mechanisms are likely involved.
The strongest interaction with both E. coli and S. aureus was a synergy between arylomycin and the bactericidal aminoglycoside gentamicin. The aminoglycosides bind the 30S ribosomal subunit, and while the antibiotic activity of the aminoglycosides results in part from the inhibition of protein synthesis, they also induce mistranslation, which results in the production of toxic peptides (26). Indeed, aminoglycoside-induced mistranslation of E. coli membrane proteins has been shown to induce malfunctions in the electron transport complex, which in turn induce oxidative damage and a periplasmic membrane stress response (19). Thus, the observed synergy between the arylomycin and the aminoglycoside could result from the compounded stress of mistranslated integral membrane proteins and the accumulation of unprocessed preproteins. The mechanism of arylomycin and aminoglycoside synergy warrants further study, since the use of combinations of antibiotics is attracting increasing attention as a method to improve efficacy and decrease susceptibility to resistance. The latter may be particularly important for antibiotics inhibiting single targets that may be prone to single-point-mutation-mediated resistance (43).
The mechanism of arylomycin activity is complex and not yet fully understood. For example, the activity of the arylomycin against both E. coli and S. aureus is relatively independent of culture density, which is curious given that arylomycin presumably localizes to the bacterial membrane such that higher densities of bacteria should dilute its concentration. Clearly, the parameters affecting arylomycin activity, including its localization, need to be further defined. Despite our incomplete understanding, it appears likely that the antibacterial activity of the arylomycins results from the accumulation of preproteins in the cytoplasmic membrane and/or the depletion of essential proteins in the periplasm, outer membrane, or extracellular milieu and that Gram-negative and Gram-positive bacteria respond very differently to these perturbations. This likely reflects the fundamentally different roles of secretion in these pathogens and, along with previously published phylogenetic data which suggest that E. coli and S. aureus may each have encountered naturally produced arylomycins in the past (45), might also suggest that these bacteria evolved different ways of coping with the associated secretion stress. Moreover, the data suggest that, if developed as a therapeutic, the arylomycin scaffold may have promising activity, especially when coadministered with an aminoglycoside.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Office of Naval Research (awards N000140310126 and N000140810478) and the National Institutes of Health (1R21AI081126).
Footnotes
Published ahead of print 16 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Bollenbach T, Quan S, Chait R, Kishony R. 2009. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell 139:707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cirz RT, et al. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 3:e176 doi:10.1371/journal.pbio.0030176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—8th ed. CLSI document M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 4. Cole JN, et al. 2007. Role of group A Streptococcus HtrA in the maturation of SpeB protease. Proteomics 7:4488–4498 [DOI] [PubMed] [Google Scholar]
- 5. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies J, Spiegelman GB, Yim G. 2006. The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 9:445–453 [DOI] [PubMed] [Google Scholar]
- 7. Desvaux M, Parham NJ, Scott-Tucker A, Henderson IR. 2004. The general secretory pathway: a general misnomer? Trends Microbiol. 12:306–309 [DOI] [PubMed] [Google Scholar]
- 8. Driessen AJ, Nouwen N. 2008. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 77:643–667 [DOI] [PubMed] [Google Scholar]
- 9. du Plessis DJ, Nouwen N, Driessen AJ. 2011. The Sec translocase. Biochim. Biophys. Acta 1808:851–865 [DOI] [PubMed] [Google Scholar]
- 10. Elion GB, Singer S, Hitchings GH. 1954. Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol. Chem. 208:477–488 [PubMed] [Google Scholar]
- 11. Farha MA, Brown ED. 2010. Chemical probes of Escherichia coli uncovered through chemical-chemical interaction profiling with compounds of known biological activity. Chem. Biol. 17:852–862 [DOI] [PubMed] [Google Scholar]
- 12. Foucaud-Scheunemann C, Poquet I. 2003. HtrA is a key factor in the response to specific stress conditions in Lactococcus lactis. FEMS Microbiol. Lett. 224:53–59 [DOI] [PubMed] [Google Scholar]
- 13. Reference deleted.
- 14. Hall MJ, Middleton RF, Westmacott D. 1983. The fractional inhibitory concentration (FIC) index as a measure of synergy. J. Antimicrob. Chemother. 11:427–433 [DOI] [PubMed] [Google Scholar]
- 15. Heikkinen O, et al. 2009. Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA—implications for the catalytic mechanism of parvulins. BMC Struct. Biol. 9:17 doi:10.1186/1472-6807-9-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huber J, et al. 2011. F1-1351—a novel lipoglycopeptide inhibitor of signal peptidase I synergizes with imipenem against MRSA: a new approach for developing an MRSA combination therapy. Annu. Intersci. Conf. Antimicrob. Agents Chemother., Chicago, IL http://www.icaac.org/ [Google Scholar]
- 17. Hyyrylainen HL, et al. 2010. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 77:108–127 [DOI] [PubMed] [Google Scholar]
- 18. Ince D, Hooper DC. 2003. Quinolone resistance due to reduced target enzyme expression. J. Bacteriol. 185:6883–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kulanthaivel P, et al. 2004. Novel lipoglycopeptides as inhibitors of bacterial signal peptidase I. J. Biol. Chem. 279:36250–36258 [DOI] [PubMed] [Google Scholar]
- 21. Lee SH, et al. 2011. Antagonism of chemical genetic interaction networks resensitize MRSA to beta-lactam antibiotics. Chem. Biol. 18:1379–1389 [DOI] [PubMed] [Google Scholar]
- 22. Lewenza S, Gardy JL, Brinkman FS, Hancock RE. 2005. Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 15:321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, et al. 2011. Synthesis and characterization of the arylomycin lipoglycopeptide antibiotics and the crystallographic analysis of their complex with signal peptidase. J. Am. Chem. Soc. 133:17869–17877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luke I, Handford JI, Palmer T, Sargent F. 2009. Proteolytic processing of Escherichia coli twin-arginine signal peptides by LepB. Arch. Microbiol. 191:919–925 [DOI] [PubMed] [Google Scholar]
- 25. Lyon WR, Caparon MG. 2004. Role for serine protease HtrA (DegP) of Streptococcus pyogenes in the biogenesis of virulence factors SpeB and the hemolysin streptolysin S. Infect. Immun. 72:1618–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem. Rev. 105:477–498 [DOI] [PubMed] [Google Scholar]
- 27. Marraffini LA, Dedent AC, Schneewind O. 2006. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70:192–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy KC, Campellone KG, Poteete AR. 2000. PCR-mediated gene replacement in Escherichia coli. Gene 246:321–330 [DOI] [PubMed] [Google Scholar]
- 29. Paetzel M, Dalbey RE, Strynadka NC. 2000. The structure and mechanism of bacterial type I signal peptidases. A novel antibiotic target. Pharmacol. Ther. 87:27–49 [DOI] [PubMed] [Google Scholar]
- 30. Paetzel M, Goodall JJ, Kania M, Dalbey RE, Page MG. 2004. Crystallographic and biophysical analysis of a bacterial signal peptidase in complex with a lipopeptide-based inhibitor. J. Biol. Chem. 279:30781–30790 [DOI] [PubMed] [Google Scholar]
- 31. Paetzel M, Karla A, Strynadka NC, Dalbey RE. 2002. Signal peptidases. Chem. Rev. 102:4549–4580 [DOI] [PubMed] [Google Scholar]
- 32. Poquet I, et al. 2000. HtrA is the unique surface housekeeping protease in Lactococcus lactis and is required for natural protein processing. Mol. Microbiol. 35:1042–1051 [DOI] [PubMed] [Google Scholar]
- 33. Rand KH, Houck HJ, Brown P, Bennett D. 1993. Reproducibility of the microdilution checkerboard method for antibiotic synergy. Antimicrob. Agents Chemother. 37:613–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roberts TC, Schallenberger MA, Liu J, Smith PA, Romesberg FE. 2011. Initial efforts toward the optimization of arylomycins for antibiotic activity. J. Med. Chem. 54:4954–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberts TC, Smith PA, Cirz RT, Romesberg FE. 2007. Structural and initial biological analysis of synthetic arylomycin A2. J. Am. Chem. Soc. 129:15830–15838 [DOI] [PubMed] [Google Scholar]
- 36. Roberts TC, Smith PA, Romesberg FE. 2011. Synthesis and biological characterization of arylomycin B antibiotics. J. Nat. Prod. 74:956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson C, et al. 2011. Transport and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria. Biochim. Biophys. Acta 1808:876–884 [DOI] [PubMed] [Google Scholar]
- 38. Rusch SL, Kendall DA. 2007. Interactions that drive Sec-dependent bacterial protein transport. Biochemistry 46:9665–9673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarvas M, Harwood CR, Bron S, van Dijl JM. 2004. Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim. Biophys. Acta 1694:311–327 [DOI] [PubMed] [Google Scholar]
- 40. Schallenberger MA, Niessen S, Shao C, Fowler BJ, Romesberg FE. 2012. Type I signal peptidase and protein secretion in Staphylococcus aureus. J. Bacteriol. 194:2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schimana J, et al. 2002. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomyces sp. Tu 6075. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. (Tokyo) 55:565–570 [DOI] [PubMed] [Google Scholar]
- 42. Sibbald MJ, et al. 2006. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol. Mol. Biol. Rev. 70:755–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silver LL. 2007. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6:41–55 [DOI] [PubMed] [Google Scholar]
- 44. Smith PA, Powers ME, Roberts TC, Romesberg FE. 2011. In vitro activities of arylomycin natural-product antibiotics against Staphylococcus epidermidis and other coagulase-negative staphylococci. Antimicrob. Agents Chemother. 55:1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith PA, Roberts TC, Romesberg FE. 2010. Broad spectrum antibiotic activity of the arylomycin natural products is masked by natural target mutations. Chem. Biol. 17:1223–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smitha Rao CV, Anné J. 2011. Bacterial type I signal peptidases as antibiotic targets. Future Microbiol. 6:1279–1296 [DOI] [PubMed] [Google Scholar]
- 47. Song C, Kumar A, Saleh M. 2009. Bioinformatic comparison of bacterial secretomes. Genomics Proteomics Bioinformatics 7:37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sriraman K, Jayaraman G. 2008. HtrA is essential for efficient secretion of recombinant proteins by Lactococcus lactis. Appl. Environ. Microbiol. 74:7442–7446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Straus SK, Hancock RE. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 1758:1215–1223 [DOI] [PubMed] [Google Scholar]
- 50. Talbot GH, et al. 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42:657–668 [DOI] [PubMed] [Google Scholar]
- 51. Tan YX, Romesberg FE. Latent antibiotics and the potential of the arylomycins for broad-spectrum antibacterial activity. MedChemComm. doi:10.1039/C2MD20043K [Google Scholar]
- 52. van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yeh P, Tschumi AI, Kishony R. 2006. Functional classification of drugs by properties of their pairwise interactions. Nat. Genet. 38:489–494 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.