

Abstract
Alisporivir is the most advanced host-targeting antiviral cyclophilin (Cyp) inhibitor in phase III studies and has demonstrated a great deal of promise in decreasing hepatitis C virus (HCV) viremia in infected patients. In an attempt to further elucidate the mechanism of action of alisporivir, HCV replicons resistant to the drug were selected. Interestingly, mutations constantly arose in domain II of NS5A. To demonstrate that these mutations are responsible for drug resistance, they were reintroduced into the parental HCV genome, and the resulting mutant viruses were tested for replication in the presence of alisporivir or in the absence of the alisporivir target, CypA. We also examined the effect of the mutations on NS5A binding to itself (oligomerization), CypA, RNA, and NS5B. Importantly, the mutations did not affect any of these interactions. Moreover, the mutations did not preserve NS5A-CypA interactions from alisporivir rupture. NS5A mutations alone render HCV only slightly resistant to alisporivir. In sharp contrast, when multiple NS5A mutations are combined, significant resistance was observed. The introduction of multiple mutations in NS5A significantly restored viral replication in CypA knockdown cells. Interestingly, the combination of NS5A mutations renders HCV resistant to all classes of Cyp inhibitors. This study suggests that a combination of multiple mutations in domain II of NS5A rather than a single mutation is required to render HCV significantly and universally resistant to Cyp inhibitors. This in accordance with in vivo data that suggest that alisporivir is associated with a low potential for development of viral resistance.
INTRODUCTION
Hepatitis C virus (HCV) is the major causative agent of acute and chronic liver diseases (9). Nearly 200 million people worldwide (3% of the population), including 4 to 5 million in the United States, are chronically infected with HCV, and 4 million new infections occur every year (2, 48). In the developed world, HCV accounts for 2/3 of all cases of liver cancer and transplants (45), and in the United States, ∼12,000 people are estimated to die from HCV each year (3).
The introduction of alpha interferon (IFN-α) and the nucleoside analog ribavirin (RBV) greatly increased the percentage of chronically HCV-infected patients able to reach a sustained antiviral response (SVR) (49, 51). However, the current standard PEGylated IFN-α-plus-RBV (pIFNα/RBV) therapy has a success rate of ∼80% in patients with genotypes 2 (GT2) and 3 (GT3) and only ∼50% in patients with GT1 (8, 47), and it causes severe side effects (35). Not only is GT1 the most prevalent HCV genotype in Europe, North and South America, China, and Japan, it is also the most difficult to treat (56). Although the recent approval of the protease inhibitors boceprevir and telaprevir should improve the efficacy of the previous standard of care, there is an urgent need for the development of additional anti-HCV agents with novel mechanisms of action (MOA) in order to provide alternative treatments for the increasing number of patients who are unresponsive to the pIFNα/RBV treatment (2, 9).
A novel class of HCV inhibitors that have great potential for the treatment of HCV has recently emerged: the host-targeting antiviral (HTA) cyclophilin (Cyp) inhibitors (12, 16, 31, 40, 41, 52, 53). To date, three Cyp inhibitors, alisporivir, NIM811, and SCY-635, have demonstrated safety and efficacy in patients with HCV in phase I and II studies (13, 24, 29). Alisporivir is the most advanced Cyp inhibitor in phase III studies and has demonstrated a great deal of promise in decreasing HCV viremia in infected patients, but its MOA remains to further be elucidated.
MATERIALS AND METHODS
Compounds.
Telaprevir, cyclosporine (CsA), sanglifehrin A (SfA), sanglifehrin B (SfB), and BC556 were synthesized by and obtained from Biotica, NIM811 and alisporivir were synthesized by and obtained from Novartis, and SCY-635 was synthesized by and obtained from Scynexis.
Development of alisporivir-resistant HCV replicons.
Huh-7 cells expressing the subgenomic Con1 (GT1b) replicon (Huh7-Luc/Neo ET cells) were incubated with 0.5 μM alisporivir (∼10-fold excess above the 50% effective concentration [EC50]) and 100 μg/ml of G418 in 10-cm CellBind plates (Corning). Cells were split every 3 days at 1:6, and the G418 concentration was increased sequentially to 200 and 300 μg/ml. Most of the cells were dying 3 weeks after drug incubation. At this point, medium was changed every 3 to 4 days until emergence of alisporivir-resistant clones. Clones were individually transferred to 24-well CellBind plates using cloning disks. Each clone was split into 2 identical plates and separately maintained under 0.5 or 1.0 μM alisporivir and 100 μg/ml of G418. Once the colonies were established, the G418 concentration was increased to 300 μg/ml. Clones resistant to 1 μM alisporivir were further screened for resistance to 2 μM alisporivir. Total RNA was prepared using the RNeasy Plus minikit per the manufacturer's recommendations. Two hundred ng of total RNA was used for cDNA production using primers specific to NS4B, NS5B, or oligo(dT)18 using an AccuScript high-fidelity first-strand cDNA synthesis kit (Stratagene). The amplicons were gel purified, quantitated, and sequenced. Sequences were assembled and analyzed using the ClustalW algorithm within the MacVector program.
HCV RNA replication monitored by RT-qPCR.
The in vitro transcription of Con1 (GT1b) RNA was accomplished using the T7 MEGAscript kit (Ambion) by following the manufacturer's instructions. In vitro-transcribed RNAs were introduced into parental or CypA knockdown (KD) Huh7.5.1 cells by electroporation. The establishment of the CypA-KD cells was described previously (4). Trypsinized cells were washed twice with and resuspended in phosphate-buffered saline (PBS) (calcium-free and magnesium-free) at 1 × 107 cells per ml. Ten micrograms of RNA for each mutant was mixed with 0.4 ml of cells in a 4-mm cuvette, and a Bio-Rad Gene Pulser system was used to deliver a single pulse at 0.27 kV, 100 Ω, and 960 μF; the cells were then plated in 12-well dishes. RNA transfection efficiency and HCV subgenomic replication were assessed by reverse transcription-quantitative PCR (RT-qPCR) and presented as genome equivalents (GE) per microgram of total RNA as described previously (4).
HCV RNA replication monitored by FACS.
A subgenomic Con1-NS5A-YFP replicon plasmid was established by insertion of the yellow fluorescent protein (YFP) into the C terminus of the NS5A gene (MVSKGEELF-YFP-TLGMDELYK) as described previously (27, 36) via homologous recombination using the In-Fusion HD cloning kit (Clontech). A Huh7.5.1 Con1-NS5A-YFP stable cell line was achieved by electroporation of Con1-NS5A-YFP RNA into Huh7.5.1 cells. In brief, RNA was synthesized from SpeI-linearized Con1-NS5A-YFP DNA using the T7 Megascript kit (Ambion), and Huh7.5.1 cells (4 × 106) were electroporated with viral RNA (10 μg) and selected with G418 (50 μg/ml) for 3 weeks. Huh7.5.1 Con1-NS5A-YFP replicon cells were enriched using a BD FACSAria sorter. For drug inhibition, compounds were added to Huh7.5.1 Con1-NS5A-YFP replicon cells (500,000) 24 h postseeding. Replicon replication was analyzed via fluorescence-activated cell sorting (FACS) 1, 2, 3, and 4 days after drug treatment. At the time of YFP expression analysis, cells were trypsinized, washed twice with PBS, and resuspended in 500 μl of sorting buffer (PBS supplemented with 1 mM EDTA, 25 mM HEPES, pH 7.0, and 1% fetal bovine serum [FBS]). FACS analysis was performed using a BD LSR II flow cytometer system and FACSDiva software. Gates for the fluorescein isothiocyanate A (FITC-A) and Pacific Blue channels were set using empty Huh7.5.1 cells as negative controls, and YFP expression was measured within the FITC-A-positive gate. Results (triplicates) were all normalized to dimethylsulfoxide (DMSO) controls. Con1-NS5A-D320E/Y321N-YFP mutant replicons were generated via site-directed mutagenesis utilizing the Phusion site-directed mutagenesis kit (NEB). D320E/Y321N mutant replicons were generated from the wild-type Con1-NS5A-YFP replicon. Con1-NS5A-D320E-YFP, Con1-NS5A-Y321N-YFP, and Con1-NS5A-D320E/Y321N-YFP were generated using the following forward and reverse phosphorylated (p) primer sets, respectively: (p)GGGCACGCCCGGAATACAACCCTCCACTGT and (p)ATATGGGCATCGCTCGAGGGAATTTCCTGG, (p)GATAACAACCCTCCACTGTTAGAGTCCTGGAAGGA and (p)CGGGCGTGCCCATATGGGCATCGCTCGAGGGAATT, and (p)GAGAACAACCCTCCACTGTTAGAGTCCTGGAAGGA and (p)CGGGCGTGCCCATATGGGCATCGCTCGAGGGAATT. PCR products were circularized with Quick T4 DNA ligase (NEB) for 5 min and transformed into NEB 10-beta competent cells. The NS5A gene of all established cell lines was sequenced to eliminate the possibility of mutation emergence during stable cell line selection.
HCV RNA replication monitored by luciferase activity.
The Huh7.5.1 Con1-NS5A-YFP stable cell lines established above that contain both NS5A-YFP and luciferase (Luc) reporter genes were treated as described above, but instead of being analyzed for YFP expression by FACS, cells were analyzed for luciferase activity in cell lysates as described previously (6).
Confocal analyses.
Live Con1-NS5A-YFP Huh-7.5.1 cells were fixed in 4% (wt/vol) paraformaldehyde. Cells were examined with a Zeiss LSM 710 laser-scanning confocal microscope using a 63× objective with the 488-nm laser to detect NS5A-YFP. Images were analyzed using Zeiss Zen software.
Production of recombinant proteins.
Recombinant glutathione S-transferase (GST)-CypA, GST-NS5BΔ21, GST-NS5A, and full-length NS5A proteins (pET-Ub-NS5A Con1-His) were produced and purified as described previously (1, 5, 58). NS5A mutants were created by PCR mutagenesis as described previously (5).
NS5A protein interactions.
Glutathione beads were incubated for 2 h in dialysis buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.5% NP-40, 1 mM dithiothreitol [DTT]) with 5 mg/ml bovine serum albumin (BSA) and washed twice at 4°C in binding buffer (20 mM Tris, pH 7.9, 0.5 M NaCl, 10% glycerol, 10 mM DTT, and 1% NP-40). Meanwhile, GST-CypA, GST-NS5BΔ21, or GST-NS5A (10 μg) was mixed with NS5A-His (200 ng) in a total volume of 200 μl of binding buffer for 3 h at 4°C on a wheel. Glutathione beads (25 μl) were added to the GST protein-NS5A mixture for 30 min at 4°C and washed 3 times with 400 μl of binding buffer. Beads were pelleted for 30 s at 2,000 × g in a microcentrifuge, and bound material was eluted with 25 μl of 2× SDS sample buffer, heated for 5 min, and frozen at −20°C. Bound material was analyzed by Western blotting using anti-GST and anti-His antibodies. Note that the antiviral FACS assay is more sensitive than the pulldown assay, likely because we had to use significant amounts of GST-CypA (10 μg) to observe detectable amounts of pulled-down NS5A proteins.
NS5A binding to RNA.
Recombinant NS5A-His proteins (10 nM) were incubated with 32P-labeled 3′ untranslated region (UTR) RNA (1 nM) for 30 min in binding buffer as described previously (14). Reaction mixtures were then added to filters for binding. Filters were then extensively washed, dried, and placed in a scintillation counter for radioactivity counting.
Statistic analyses.
Data were analyzed using the Graph Pad Prism software program (La Jolla, CA), and t tests were used to evaluate the statistical significance of values obtained using all mutant cell lines against wild-type virus (*, P < 0.05; **, P < 0.01; and ***, P < 0.001).
RESULTS
Selection of alisporivir-resistant replicons.
In an attempt to further delineate the MOA of the highly potent Cyp inhibitor alisporivir, we selected in vitro HCV replicons for alisporivir resistance. Con1-Huh7 cells were grown in the presence of increasing concentrations of alisporivir and G418 for an extended period of time (>13 weeks) until the appearance of alisporivir-resistant colonies. Single colonies were further expanded for 4 to 6 additional weeks. Sequencing of the complete viral genome of the alisporivir-resistant replicon colonies revealed that two adjacent mutations in the NS5A gene, D320E and Y321N, constantly emerged. Specifically, nine alisporivir-resistant replicon colonies contained the D320E mutation, five contained the Y321N mutation, and two contained both D320E and Y321N mutations. The D320 residue is conserved among HCV genotypes (GT1a, GT1b, GT2a, GT3, GT4, and GT6) except for GT2b (D320E) and GT5 (D320G), whereas the Y321 residue is highly conserved among all genotypes (Fig. 1A).
Fig 1.

(A) NS5A sequences from different genotypes (GT) surrounding D320/Y321 residues, which are mutated to E320/N321 residues under alisporivir selection, were aligned using the ClustalW algorithm within MacVector (version 12.5.1). The sequences used for the alignment were based on those available in the NCBI database. The genotypes (and accession numbers) are GT1a (AF009606), GT1b (AJ238799), GT2a [J6] (AF177036), GT2a [JFH] (BAF34893), GT2b (AY232730), GT3 (NC_009824), GT4 (NC_009825), GT5 (NC_009826), and GT6 (NC_009827). (B) Secondary-structure predictions of the NS5A 300-350 segment are indicated as helical (h; blue), extended (e; red), turn (t; green), or undetermined (coil [c]; yellow). Predictions were made by using the web-based algorithms DSC, HNNC, MLRC, PHD, Predator, and SOPM, which are available at the NPSA website (http://npsa-pbil.ibcp.fr) (7). Positions 320 and 321, exhibiting alisporivir-resistant mutations, are highlighted in green. Sec.Cons., secondary structure consensus.
Multiple rather than single NS5A mutations mediate alisporivir resistance.
We showed above that adjacent mutations in domain II of NS5A arose under alisporivir selection. We then examined whether these mutations truly confer resistance to alisporivir. To address this issue, we established a flow cytometry live cell-based assay that permits the measurement of the potency of anti-HCV agents. Specifically, we created a stable cell line that harbors a subgenomic Con1 replicon encoding an NS5A-YFP fusion protein (Fig. 2A), which allows viral replication to be quantified by FACS (Fig. 2B) and to be visualized by confocal microscopy (Fig. 2C). We used this stable fluorescent system to measure HCV replication inhibition by alisporivir (Fig. 2D). We introduced D320E and Y321N NS5A mutations into Con1 NS5A-YFP plasmid and generated stable cell lines expressing the following replicons: Con1 D320E NS5A-YFP, Con1 Y321N NS5A-YFP, and Con1 D320E/Y321N NS5A-YFP. We next examined the resistance of the replicons to alisporivir. We first found that alisporivir profoundly blocks wild-type Con1 replication (Fig. 2D), confirming that this Cyp inhibitor efficiently inhibits HCV replication in vitro (6, 11, 18, 19, 25, 26, 33, 37, 39, 44, 55). Importantly, the D320E or Y321N mutation alone confers only a slight resistance to the Cyp inhibitor, with the Y321N mutation having a more profound resistance impact (Fig. 2D). Remarkably, the combination of the mutations renders HCV highly resistant to alisporivir (Fig. 2D). We obtained EC50s of 52, 107, 179, and 2,379 nM for the wild-type, D320E, Y321N, and D320E/Y321N proteins, respectively (Fig. 2D). We obtained similar results by measuring the luciferase activity in the cell lysates (Fig. 2D). Note that the NS5A-YFP reporter assay always gave minimal standard errors between triplicates compared to the luciferase reporter assay (Fig. 2D). Moreover, we found that the protease inhibitor telaprevir blocks the replication of all viruses (Fig. 2E), demonstrating that the D320E/Y321N NS5A mutations render HCV specifically resistant to alisporivir. These data suggest not only that the mutations found in NS5A of alisporivir-resistant replicons are truly responsible for the observed drug resistance but also that a combination of mutations, rather than a single NS5A mutation, is a precondition for alisporivir resistance.
Fig 2.

(A) Schematic diagram of the NS5A-YFP reporter subgenomic Con1 replicon. NPTII, neomycin phosphotransferase gene; EMCV IRES, encephalomyocarditis virus internal ribosomal entry site. (B) The YFP-positive cell population was enriched using the BD FACSAria sorter. (C) Con1-NS5A-YFP Huh-7.5.1 cells were visualized via confocal analysis for 5 days. (D) Con1-WT NS5A-YFP, Con1-WT D320E NS5A-YFP, Y321N Con1-WT, and D320E/Y321N NS5A-YFP Huh-7.5.1 cells (500,000) were exposed to increasing concentrations of alisporivir and analyzed for YFP content 3 days after drug treatment. The percentage of YFP-positive cells treated with the DMSO control was arbitrarily fixed at 100. Results are representative of 3 independent experiments. Error bars represent standard errors from triplicates. (E) Same as panel D, except that luciferase activity in cell lysates was quantified; the percentage of luciferase activity in cells treated with the DMSO control was arbitrarily fixed at 100. Error bars represent standard errors from triplicates. Statistical significance was measured between each mutant construct in relation to Con1-WT NS5A-YFP for the following drug concentrations: 32, 63, and 125 nM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The alisporivir resistance mutations do not render CypA-NS5A interactions impervious to alisporivir.
We and others showed that CypA, via its isomerase pocket, directly binds NS5A (4, 10, 22, 28, 32). We and others also showed that Cyp inhibitors, including CsA, alisporivir, sanglifehrins, and sangamides, totally disrupt NS5A-CypA interactions (5, 6, 10, 14, 19, 22, 54, 57). Thus, one possibility explaining how D320E and Y321N mutations render HCV resistant to alisporivir is that the mutations render NS5A-CypA interactions resistant to the drug-mediated dissociation. To address this issue, we generated and produced the following proteins: wild type, D320E, Y321N, and D320E/Y321N Con1 NS5A. We then tested the capacities of these NS5A proteins to interact with CypA in the presence or absence of increasing concentrations of alisporivir. We found that all NS5A proteins bind similarly to CypA in the absence of the drug (Fig. 3A). However, all NS5A-CypA interactions were abolished by alisporivir (>1.25 μM) (Fig. 3A). This finding indicates that the alisporivir resistance mutations do not render NS5A-CypA interactions impervious to alisporivir.
Fig 3.

(A) GST-CypA (10 μg) was mixed with wild-type, D320E, Y321N, and D320E/Y321N NS5A Con1-His proteins (200 ng) for 3 h at 4°C in the presence or absence of increasing concentrations of alisporivir. Glutathione beads were added to the GST-CypA–NS5A mixture for 30 min at 4°C and washed. Bound material was eluted and analyzed by Western blotting using anti-His antibodies. Results are representative of two independent experiments. (B) Same as panel A, except that GST, GST-CypA, or GST-NS5B (10 μg) was mixed with wild-type, D320E, Y321N, and D320E/Y321N NS5A Con1-His proteins (200 ng) for 3 h at 4°C in the presence or absence of 1.25 μM alisporivir. (C) Same as panel A, except that GST or GST-NSS5A (10 μg) was mixed with wild-type, D320E, Y321N, and D320E/Y321N NS5A Con1-His proteins (200 ng) for 3 h at 4°C in the presence or absence of 1.25 μM alisporivir.
The alisporivir resistance NS5A mutations do not influence NS5A binding to NS5B.
If the alisporivir resistance mutations do not affect NS5A-CypA contacts, they could influence interactions of NS5A with other binding partners. We thus first asked whether the alisporivir resistance mutations affect the binding of NS5A to NS5B, since previous work showed that both viral proteins interact (46). To address this issue, we examined the direct binding of D320E, Y321N, and D320E/Y321N NS5A proteins to NS5B using GST-NS5B as bait. We used GST and GST-CypA as negative and positive controls, respectively. Importantly, we found that all NS5A proteins behave similarly in terms of NS5B binding (Fig. 3B). In contrast to NS5A-CypA interactions, NS5A-NS5B interactions were insensitive to alisporivir (Fig. 3B). These data confirm that both proteins interact directly (46) and also suggest that the alisporivir resistance NS5A mutations do not affect NS5A-NS5B interactions.
The alisporivir resistance NS5A mutations do not influence NS5A dimerization.
Previous studies suggested that NS5A forms dimers (30, 50). We thus asked whether the alisporivir resistance NS5A mutations influence NS5A dimerization. To address this issue, we examined the capacity of GST-NS5A to dimerize with wild-type, D320E, Y321N, and D320E/Y321N NS5A proteins. We used GST as a negative control. Importantly, we found that all NS5A proteins form dimers (Fig. 3C). Alisporivir has no effect on NS5A-NS5A interactions (Fig. 3C). These data suggest that the alisporivir resistance NS5A mutations do not influence NS5A dimerization.
The alisporivir resistance NS5A mutations do not influence NS5A binding to RNA.
Recombinant wild-type, D320E, Y321N, and D320E/Y321N NS5A proteins (10 nM) were incubated with 32P-labeled 3′ UTR RNA (1 nM) for 30 min in binding buffer as described previously (14). Reaction mixtures were then added to filters for binding. Filters were then extensively washed, dried, and placed in a scintillation counter for radioactivity counting. All NS5A proteins, including those that contain the alisporivir resistance NS5A mutations, similarly bind RNA (Fig. 4). Alisporivir has no effect on NS5A binding to RNA (Fig. 4). These data suggest that alisporivir resistance NS5A mutations do not significantly affect NS5A binding to RNA, at least in this in vitro assay.
Fig 4.

Recombinant wild-type and mutant NS5A-His proteins (10 nM) were incubated with 32P-labeled 3′ UTR RNA (1 nM) for 30 min in binding buffer in the presence or absence of 1.25 μM alisporivir. Reaction mixtures were added to filters for binding. Filters were extensively washed, dried, and placed in a scintillation counter for radioactivity measurement. Results expressed as percentages of RNA bound are representative of two independent experiments. Error bars represent standard errors from triplicates.
Multiple rather than single NS5A mutations render HCV CypA independent.
One explanation for the ability of replicons to replicate even in the presence of alisporivir is that D320E/Y321N mutations render HCV less dependent on CypA. To test this hypothesis, we examined the capacity of wild-type, D320E, Y321N, and D320E/Y321N replicons to replicate in CypA-KD cells that we previously established (4). Specifically, viral RNAs were electroporated into parental or CypA-KD Huh7.5.1 cells, and replication was monitored over time by RT-qPCR. All replicons replicate equally well in parental cells (Fig. 5), suggesting that the NS5A mutations do not significantly influence viral RNA growth. In sharp contrast, the wild-type replicon poorly replicates in CypA-KD cells (Fig. 5), confirming that HCV highly relies on CypA to replicate (4, 15, 17, 28, 38, 57). Both D320E and Y321N replicons replicate weakly but significantly in CypA-KD cells compared to the wild-type replicon (Fig. 5). Remarkably, the combination of NS5A mutations (D320E/Y321N) allows the replicon to replicate in CypA-KD cells at levels almost identical to those observed in parental cells (Fig. 5). As expected, alisporivir has a minimal effect on viral replication in CypA-KD cells (Fig. 5). Together, these data suggest that multiple NS5A mutations rather than a single mutation render HCV significantly CypA independent.
Fig 5.
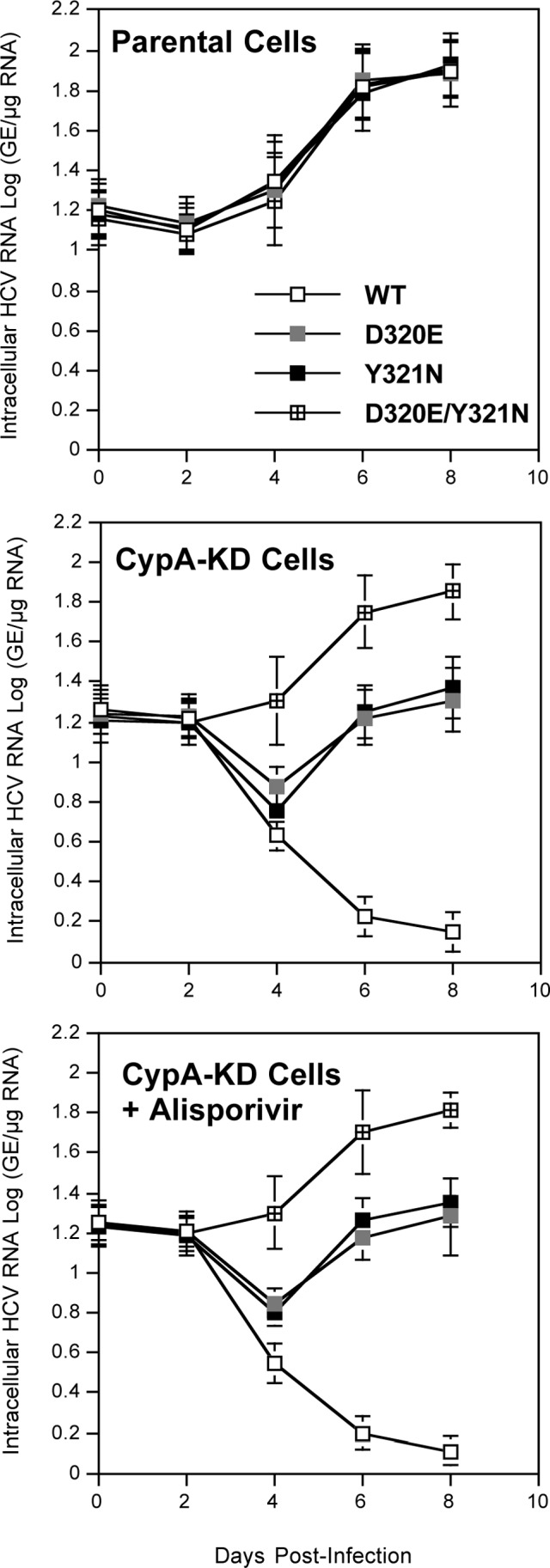
Ten micrograms of in vitro-transcribed subgenomic wild-type, D320E, Y321N, or D320E/Y321N Con1 RNA was electroporated into parental or CypA-KD Huh7.5.1 cells. At the indicated time points, intracellular HCV RNA was analyzed via RT-qPCR and is presented as genome equivalents (GE) per microgram total RNA. CypA-KD Huh7.5.1 cells were treated with or without 1.25 μM alisporivir. These results are representative of three independent experiments. Error bars represent standard errors from triplicates. Statistical significance was measured between each mutant construct in relation to Con1-WT NS5A-YFP after 8 days postinfection. **, P < 0.01; ***, P < 0.001.
A combination of NS5A mutations governs universal resistance to Cyp inhibitors.
If the D320E/Y321N mutations truly render HCV less CypA dependent (Fig. 5), they should offer resistance to all classes of Cyp inhibitors, which are structurally diverse, including the immunosuppressive CsA, nonimmunosuppressive CsA derivatives (i.e., alisporivir, NIM811, and SCY-635), immunosuppressive sanglifehrins (i.e., SfA and SfB), and nonimmunosuppressive sanglifehrin derivatives (i.e., the sangamide BC556). Indeed, all classes of Cyp inhibitors target and neutralize the isomerase pocket of CypA. We tested this hypothesis by examining the effect of the Cyp inhibitors mentioned above on wild-type, D320E, Y321N, and D320E/Y321N replicon replication. We used the protease inhibitor telaprevir as a positive control. We found that telaprevir inhibits the replication of wild-type and mutant replicons equally (Fig. 6). In contrast, Cyp inhibitors block wild-type and mutant replicons to different degrees. Importantly, all Cyp inhibitors block the replication of wild-type replicon more efficiently than that of mutant replicons (Fig. 6A). We obtained various degrees of sensitivity for the wild-type replicon to Cyp inhibitors. The D320E/Y321N replicon was the most resistant replicon, whereas D320E and Y321 replicons exhibited low resistance profiles (Fig. 6A). Interestingly, the D320E/Y321N replicon was completely impervious to CsA and highly resistant to SfA, NIM811, and SCY-635 (Fig. 6A). The EC50 for each Cyp inhibitor was calculated (Fig. 6B), and EC50 fold changes to telaprevir and Cyp inhibitors for the D320E/Y321N mutant relative to the wild-type replicon were the following: telaprevir (∼1-fold), CsA (>10-fold), NIM811 (12-fold), SCY-635 (19-fold), alisporivir (∼2-fold), SfA (>5-fold), SfB (∼4-fold), and BC556 (∼2.5-fold). Together, these data suggest that a combination of mutations in domain II of NS5A renders HCV resistant to all classes of Cyp inhibitors.
Fig 6.

Empty, Con1 wild-type NS5A-YFP, Con1 D320E NS5A-YFP, Con1 Y321N, or Con1 D320E/Y321N NS5A-YFP Huh7.5.1 cells (500,000) were exposed to increasing concentrations of telaprevir or a panel of Cyp inhibitors, including CsA, alisporivir, NIM811, SCY-635, SfA, SfB, and BC556, and analyzed for YFP content 3 days after drug treatment. The percentage of YFP-positive cells treated with the DMSO control was arbitrarily fixed at 100. Results are representative of 3 independent experiments. Error bars represent standard errors from triplicates. EC50s were calculated for each Cyp inhibitor. Statistical significance was measured between each mutant construct in relation to Con1-WT NS5A-YFP for the 500 nM drug concentration. **, P < 0.01; ***, P < 0.001.
DISCUSSION
To further elucidate the MOA of alisporivir, drug-resistant replicons were selected. In contrast to short direct-acting antiviral (DAA)-resistant replicon selections (2 to 3 weeks), an extended period of time was necessary to obtain alisporivir-resistant replicons (∼15 to 20 weeks), further suggesting that alisporivir offers a much higher genetic barrier to resistance than protease and nonnucleoside NS5B inhibitors (6). One possibility to explain this high barrier to resistance is that alisporivir, in contrast to DAA, targets a host protein, CypA. Thus, HCV cannot develop mutations which prevent the binding of the drug to its own proteins but instead has to develop mutations which bypass the need for the host protein CypA. This is perfectly in accordance with our data that showed that the NS5A mutations that arose under alisporivir selection render HCV CypA independent. Nevertheless, the fact that the time needed for Cyp inhibitor resistance selection is extremely long strongly suggests that the development of CypA independence is a challenging task for the virus.
We identified a specific motif, D320E/Y321N, in domain II of NS5A that renders HCV considerably more resistant to alisporivir. It is important to note that we identified mutations in genes other than NS5A. However, these mutations were only present in single colonies. Except for the D320E and Y321N mutations, we did not find other mutations repeated in more than one colony. For that reason, we did not further analyze these mutations in the present study. During the time of this study, other groups also identified the D320E mutation as a spot of resistance to Cyp inhibitors (6, 10, 17, 42). The Y321N mutation was also recently linked to CsA resistance in vitro (10). One group very recently identified D317E/Y318N NS5A mutations that confer JFH-1 resistance to CsA (20, 57). Note that all D320E, Y321N, D317E, and Y318N mutations result from a single-nucleotide change of T to A (6, 20). The positions of D317/Y318 residues in JFH-1 (GT2a) coincide perfectly with the positions of D320/Y321 in Con1 (GT1b). As mentioned above, D320 and Y321 residues locate in a region of NS5A that is well conserved among genotypes (Fig. 1A), suggesting that this domain plays a functional role in HCV replication. Secondary-structure analyses of NS5A by various methods did not allow us to identify the presence of a canonical secondary structure in the D320-Y321 region (Fig. 1B), which is predicted as a coil (F. Penin, personal communication). The D320 and Y321 residues reside in domain II of NS5A. Domain II, which contains D320 and Y321 residues, was shown to be globally unstructured (21). Size-exclusion chromatography, circular dichroism, homonuclear nuclear magnetic resonance spectroscopy, and high-resolution triple-resonance spectroscopy analyses allowed the identification of a small region of residual structure in domain II of NS5A (21). This region, which contains D320 and Y321 residues, corresponds to the most conserved sequence for various genotypes (21), suggesting a functional importance of this region for the virus. Supporting this notion, several lines of evidence suggest that this NS5A region serves as an anchoring point for CypA (21). Thus, our finding that adjacent mutations emerge under alisporivir selection in this specific region of NS5A correlates well with the assumption that this region serves as a main locus for CypA binding in HCV.
It is important to emphasize that we found that a single D320E or Y321N change offers only weak resistance to alisporivir, whereas the dual D320E/Y321N change offers significant resistance to alisporivir. One thus can envision that multiple (>2) mutations in this region will be necessary to offer robust resistance to alisporivir, at least in vitro. This may explain why only a few viral breakthroughs occurred in alisporivir-treated patients (K. Lin, personal communication). Importantly, population sequencing of the HCV genome did not identify any genotypic change consistently associated with viral breakthrough, further confirming the high barrier to resistance that alisporivir offers. The remarkable in vivo resistance profile of alisporivir makes it an attractive candidate for future anti-HCV regimens. It also may suggest that HCV will be unable to develop any alisporivir resistance in vivo if it cannot develop a combination of multiple mutations in domain II of NS5A.
A critical issue remains to be elucidated, namely, how these relatively minor changes, D320E and Y321N, render HCV CypA independent. It has been proposed that CypA, by interacting with domain II of NS5A, accelerates the rotation of peptidyl-prolyl bonds in the proline-rich region that surrounds the D320/Y321 residues (6, 22). In this model, E320 and N321 changes would substitute for the CypA-mediated isomerase action in the D320/Y321 region. It is important to emphasize that the isomerase activity of CypA on NS5A was demonstrated in vitro using NS5A domains or peptides rather than full-length NS5A (6, 22, 56). Thus, definitive proof that CypA isomerizes NS5A in a cellular context remains to be provided.
Several hypotheses could explain how the alisporivir resistance NS5A mutations render HCV CypA independent. However, our present study excluded several of them. Specifically, we showed that the D320E/Y321N NS5A protein behaves like wild-type NS5A in terms of (i) CypA binding, (ii) CypA-NS5A complex sensitivity to alisporivir, (iii) RNA binding, (iv) dimerization, and (v) NS5B binding. Evidently, one cannot exclude the possibility that the assays used in this study are not physiologically relevant to a cellular context. Nevertheless, our data suggest that D320E/Y321N mutations influence NS5A functions or features distinct from those examined in this study. NS5A has been shown to bind to a broad number of host proteins, including protein kinase R (PKR), Src kinases, human vascular adhesion protein (hVAP), phosphatidylinositol 4,5-bisphosphate, etc. (23, 34, 43). One thus cannot also exclude the possibility that NS5A in the context of the replicon binds to viral proteins other than NS5B. In this scenario, D320E/Y321N mutations modulate the contact between NS5A and any of the putative viral and cellular ligands described above. Another possibility is that CypA is critical for the proper binding of NS5A to the viral RNA within the HCV replication complex. Supporting this hypothesis, a recent study showed that the RNA binding of D320E NS5A domain II, but not wild-type NS5A domain II, was unaffected by CypA (14), suggesting that the D320E mutation bypasses the need for CypA, in this case binding tightly to the RNA. Nevertheless, further work is required to determine why NS5A requires CypA binding and/or isomerization for its functions. Moreover, further work is necessary to delineate the true functions of NS5A in HCV replication. The identification of specific alisporivir resistance mutations within NS5A will be of great help to elucidate the functions of both CypA and NS5A in the HCV replication cycle and will also help to further unravel the MOA of Cyp inhibitors such as alisporivir.
Taken together, these data suggest that a combination of multiple mutations rather than a single mutation in domain II of NS5A is required to render HCV significantly and universally resistant to Cyp inhibitors. These in vitro data are perfectly in accordance with in vivo data that suggest that Cyp inhibitors, especially alisporivir, are associated with a low potential for development of viral resistance in drug-treated HCV patients. Further work is required to determine how this short stretch of mutations in domain II of NS5A bypasses the need for CypA in HCV replication.
ACKNOWLEDGMENTS
We thank Janet Kuhns for secretarial assistance, C. Cameron for the pET26Ub-NS5A-His Con1 plasmid, G. Luo for the GST-NS5BΔ21 plasmid, K.-L. Hoe for the GST-NS5A plasmid, F. Chisari for Huh7.5.1 cells, R. Bartenschlager for the Con1 plasmid and Huh7-Luc/Neo ET cells, and F. Penin for the preparation of Fig. 1B and careful reading of the manuscript.
We acknowledge financial support from the U.S. Public Health Service (grant no. AI087470) (P.A.G.).
Footnotes
Published ahead of print 16 July 2012
This is publication no. 21743 from the Department of Immunology & Microbial Science, The Scripps Research Institute, La Jolla, CA.
REFERENCES
- 1. Ahn J, et al. 2004. Systematic identification of hepatocellular proteins interacting with NS5A of the hepatitis C virus. J. Biochem. Mol. Biol. 37:741–748 [DOI] [PubMed] [Google Scholar]
- 2. Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armstrong GL, et al. 2006. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann. Intern. Med. 144:705–714 [DOI] [PubMed] [Google Scholar]
- 4. Chatterji U, et al. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284:16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chatterji U, et al. 2010. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 53:50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coelmont L, et al. 2010. DEB025 (alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5:e13687 doi:10.1371/journal.pone.0013687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Combet C, Blanchet C, Geourjon C, Deleage G. 2000. NPS@: network protein sequence analysis. Trends Biochem. Sci. 25:147–150 [DOI] [PubMed] [Google Scholar]
- 8. Cross TJ, Antoniades CG, Harrison PM. 2008. Current and future management of chronic hepatitis C infection. Postgrad. Med. J. 84:172–176 [DOI] [PubMed] [Google Scholar]
- 9. Dienstag JL, McHutchison JG. 2006. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 130:214–264 [DOI] [PubMed] [Google Scholar]
- 10. Fernandes F, Ansari IU, Striker R. 2010. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5:e9815 doi:10.1371/journal.pone.0009815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernandes F, et al. 2007. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 46:1026–1033 [DOI] [PubMed] [Google Scholar]
- 12. Fischer G, Gallay P, Hopkins S. 2010. Cyclophilin inhibitors for the treatment of HCV infection. Curr. Opin. Investig. Drugs 11:911–918 [PubMed] [Google Scholar]
- 13. Flisiak R, et al. 2008. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47:817–826 [DOI] [PubMed] [Google Scholar]
- 14. Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 85:7460–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaither LA, et al. 2010. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 397:43–55 [DOI] [PubMed] [Google Scholar]
- 16. Gallay PA. 2011. Cyclophilin inhibitors: a novel class of promising host-targeting anti-HCV agents. Immunol. Res. 52:200–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goto K, Watashi K, Inoue D, Hijikata M, Shimotohno K. 2009. Identification of cellular and viral factors related to anti-hepatitis C virus activity of cyclophilin inhibitor. Cancer Sci. 100:1943–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goto K, et al. 2006. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 343:879–884 [DOI] [PubMed] [Google Scholar]
- 19. Gregory MA, et al. 2011. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob. Agents Chemother. 55:1975–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grise H, Frausto S, Logan T, Tang H. 2012. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates alisporivir susceptibility. J. Virol. 86:4811–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hanoulle X, Badillo A, Verdegem D, Penin F, Lippens G. 2010. The domain 2 of the HCV NS5A protein is intrinsically unstructured. Protein Peptide Lett. 17:1012–1018 [DOI] [PubMed] [Google Scholar]
- 22. Hanoulle X, et al. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284:13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He Y, Staschke KA, Tan SL. 2006. HCV NS5A: a multifunctional regulator of cellular pathways and virus replication. In Tan SL. (ed), Hepatitis C viruses: genomes and molecular biology. Horizon Bioscience, Norfolk, United Kingdom: [PubMed] [Google Scholar]
- 24. Hopkins S, et al. 2012. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. 57:47–54 [DOI] [PubMed] [Google Scholar]
- 25. Hopkins S, et al. 2010. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 54:660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishii N, et al. 2006. Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 80:4510–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones DM, Gretton SN, McLauchlan J, Targett-Adams P. 2007. Mobility analysis of an NS5A-GFP fusion protein in cells actively replicating hepatitis C virus subgenomic RNA. J. Gen. Virol. 88:470–475 [DOI] [PubMed] [Google Scholar]
- 28. Kaul A, et al. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546 doi:10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lawitz E, et al. 2011. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res. 89:238–245 [DOI] [PubMed] [Google Scholar]
- 30. Lemm JA, et al. 2011. Discovery of potent hepatitis C virus NS5A inhibitors with dimeric structures. Antimicrob. Agents Chemother. 55:3795–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin K. 2010. Development of novel antiviral therapies for hepatitis C virus. Virol. Sinica 25:246–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Z, Yang F, Robotham JM, Tang H. 2009. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 83:6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma S, et al. 2006. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 50:2976–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macdonald A, Harris M. 2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 85:2485–2502 [DOI] [PubMed] [Google Scholar]
- 35. Manns MP, Wedemeyer H, Cornberg M. 2006. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55:1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moradpour D, et al. 2004. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 78:7400–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakagawa M, et al. 2004. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 313:42–47 [DOI] [PubMed] [Google Scholar]
- 38. Nakagawa M, et al. 2005. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology 129:1031–1041 [DOI] [PubMed] [Google Scholar]
- 39. Paeshuyse J, et al. 2006. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 43:761–770 [DOI] [PubMed] [Google Scholar]
- 40. Pawlotsky JM. 2012. New antiviral agents for hepatitis C. F1000 Biol. Rep. 4:5 doi:10.3410/B4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pockros PJ. 2010. New direct-acting antivirals in the development for hepatitis C virus infection. Therap. Adv. Gastroenterol. 3:191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Puyang X, et al. 2010. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob. Agents Chemother. 54:1981–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reiss S, et al. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robida JM, Nelson HB, Liu Z, Tang H. 2007. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J. Virol. 81:5829–5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567 [DOI] [PubMed] [Google Scholar]
- 46. Shirota Y, et al. 2002. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 277:11149–11155 [DOI] [PubMed] [Google Scholar]
- 47. Simmonds P, et al. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 48. Soriano V, et al. 2008. Emerging drugs for hepatitis C. Expert Opin. Emerg. Drugs 13:1–19 [DOI] [PubMed] [Google Scholar]
- 49. Sy T, Jamal MM. 2006. Epidemiology of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 3:41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tong MJ, et al. 1997. Treatment of chronic hepatitis C with consensus interferon: a multicenter, randomized, controlled trial. Consensus Interferon Study Group. Hepatology 26:747–754 [DOI] [PubMed] [Google Scholar]
- 52. Vermehren J, Sarrazin C. 2011. New hepatitis C therapies in clinical development. Eur. J. Med. Res. 16:303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. von Hahn T, Ciesek S, Manns MP. 2011. Arrest all accessories–inhibition of hepatitis C virus by compounds that target host factors. Discov. Med. 12:237–244 [PubMed] [Google Scholar]
- 54. Waller H, Chatterji U, Gallay P, Parkinson T, Targett-Adams P. 2010. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J. Virol. Methods 165:202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. 2003. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38:1282–1288 [DOI] [PubMed] [Google Scholar]
- 56. WHO 1999. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J. Viral Hepatitis 6:35–47 [PubMed] [Google Scholar]
- 57. Yang F, et al. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 6:e1001118 doi:10.1371/journal.ppat.1001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang C, et al. 2005. Stimulation of hepatitis C virus (HCV) nonstructural protein 3 (NS3) helicase activity by the NS3 protease domain and by HCV RNA-dependent RNA polymerase. J. Virol. 79:8687–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]