

Abstract
The health threat caused by multiresistant bacteria has continuously increased and recently peaked with pathogens resistant to all current drugs. This has triggered intense research efforts to develop novel compounds to overcome the resistance mechanisms. Thus, antimicrobial peptides (AMPs) have been intensively studied, especially the family of proline-rich AMPs (PrAMPs) that was successfully tested very recently in murine infection models. PrAMPs enter bacteria and inhibit chaperone DnaK. Here, we studied the toxicity of intracellular PrAMPs in HeLa and SH-SY5Y cells. As PrAMPs cannot enter most mammalian cells, we coupled the PrAMPs with penetratin (residues 43 to 58 in the antennapedia homeodomain) via a C-terminally added cysteine utilizing a thioether bridge. The resulting construct could transport the covalently linked PrAMP into mammalian cells. Penetratin ligation reduced the MIC for Gram-negative Escherichia coli only slightly (1 to 8 μmol/liter) but increased the activity against the Gram-positive Micrococcus luteus up to 32-fold (MIC ≈ 1 μmol/liter), most likely due to more effective penetration through the bacterial membrane. In contrast to native PrAMPs, the penetratin-PrAMP constructs entered the mammalian cells, aligned around the nucleus, and associated with the Golgi apparatus. At higher concentrations, the constructs reduced the cell viability (50% inhibitory concentration [IC50] ≈ 40 μmol/liter) and changed the morphology of the cells. No toxic effects or morphological changes were observed at concentrations of 10 μmol/liter or below. Thus, the IC50 values were around 5 to 40 times higher than the MIC values. In conclusion, PrAMPs are in general not toxic to mammalian cells, as they do not pass through the membrane. When shuttled into mammalian cells, however, PrAMPs are only slightly cross-reactive to mammalian chaperones or other intracellular mammalian proteins, providing a second layer of safety for in vivo applications, even if they can enter some human cells.
INTRODUCTION
Since Alexander Fleming discovered penicillin in 1928, a wide variety of antibiotics, including β-lactams and quinolones, has been developed to fight bacterial infections. Thus, large bacterial epidemics have been prevented and millions of lives have probably been rescued in the following decades. Despite this overwhelming success, the prolonged global application of antibiotics has increased the evolutionary pressure on bacteria, favoring the generation of pathogens increasingly resistant to antibiotic drugs. Resistances first developed in species in which single mutations were sufficient to cause clinically important levels of resistance (e.g., Staphylococcus aureus and Pseudomonas aeruginosa), followed by bacteria in which multiple mutations were required (e.g., Escherichia coli). This was mainly due to the broad use of fluoroquinolones (13). The greatest source of resistance against β-lactam-based drugs is represented by β-lactamases (21), i.e., enzymes that inactivate this class of antibiotics by opening the β-lactam ring. Important causes of Gram-negative resistance include extended-spectrum β-lactamases (ESBL) in E. coli and broad-range β-lactamases like Klebsiella pneumoniae carbapenemase (KPC) (3, 14, 27). Thus, novel treatment options are required, using innovative antimicrobials, preferably with novel modes of action and/or belonging to novel drug classes (2).
Apart from their immunostimulatory activities, it has been recognized that many gene-encoded host defense peptides (HDPs) also possess antimicrobial activities. These antimicrobial peptides (AMPs) act by disrupting the integrity of the bacterial membrane and/or by translocating through the bacterial membranes and inhibiting internal targets. AMPs inhibiting defined bacterial targets appear especially favorable for medical applications, provided that they possess no or only weak cross-reactivity toward the human analogues. Short proline-rich AMPs (PrAMPs), isolated from both mammals and insects, represent such a promising class of compounds (23). Insect-derived PrAMPs, e.g., abaecin, apidaecin, drosocin, and pyrrhocoricin (4, 5, 7, 23), are typically 20 to 35 residues long and have a relatively high proportion of basic amino acid residues. Recently, several laboratories have started to optimize PrAMPs for clinical applications, e.g., A3-APO (25), Bac7(1-35) (12), oncocin (16), and Api88 (9), which proved successful in rodent infection models (1, 15, 32). Mechanistically, PrAMPs and the related designer peptides enter the bacteria and kill them, most likely by inhibiting the 70-kDa bacterial chaperone DnaK (17, 20, 24). The low toxicity toward mammalian cell lines is attributed to the fact that short PrAMPs appear to enter only some cells of the immune system, without inhibiting their intracellular chaperones, but are excluded from other mammalian cells (18, 26, 33).
This report focuses on the intracellular toxicity of different PrAMPs delivered into human cell lines via a cell-penetrating peptide (CPP). Apidaecin 1b, drosocin, pyrrhocorocin, and oncocin were covalently attached to penetratin (residues 43 to 58 in the antennapedia homeodomain), and their antimicrobial activities and uptake evaluated. Fluorescein-labeled PrAMP-penetratin constructs showed that uptake into bacterial and human cells was fast and efficient. Unlabeled constructs retained the full antibacterial activity and were only mildly toxic to human cells, indicating that PrAMPs are to a minor extent cross-reactive toward human chaperones.
MATERIALS AND METHODS
Water was purified in-house with a Purelab ultra analytic system (18.2 MΩ-cm; Elga, Berkefeld GmbH, Ransbach, Germany). All reagents used in this study were purchased from the following companies.
Dichloromethane (DCM, peptide synthesis grade), dimethyl sulfoxide (DMSO; puriss. absolute ≥ 99.5%), iodoacetic acid (99%), trifluoroacetic acid (TFA; UV grade, for high-performance liquid chromatography [HPLC]), and tryptic soy broth (TSB) from Sigma-Aldrich GmbH (Taufkirchen, Germany); 9-fluorenylmethoxycarbonyl (Fmoc)-protected amino acids from MultiSynTech GmbH (Witten, Germany) or Orpegen Pharma GmbH (Heidelberg, Germany); O-benzotriazole-N,N,N′,N′-tetramethyl-uroniumhexafluoro-phosphate (HBTU), Rink amide 4-methylbenzhydrylamine (MBHA) resin and 4-benzyloxybenzyl alcohol (Wang) resin from MultiSynTech GmbH; 5(6)-carboxyfluorescein (for fluorescence), N,N′-diisopropylcarbodiimide (DIC; >98% pure by gas chromatography [GC]), N,N′-diisopropylethylamine (DIPEA; >98% pure by GC), ethandithiole (purum ≥ 98%), 1-hydroxy-benzotriazole (HOBt; purum > 98%), thioanisole (purum ≥ 99.0%), TFA (purum, for peptide synthesis), and Hoechst 33342 from Fluka Chemie GmbH (Buchs, Switzerland); 5(6)-carboxytetramethylrhodamine (TAMRA) from Merck KGaA (Darmstadt, Germany); Jupiter C18 columns for preparative (21.2-mm internal diameter, 250-mm length, 15-μm particle size, 30-nm pore size) and analytical (4.6-mm internal diameter, 150-mm length, 5-μm particle size, 30-nm pore size) scale from Phenomenex, Inc. (Torrance, CA); acetonitrile (HPLC-S gradient grade) and N,N-dimethylformamide (DMF, peptide synthesis grade) from Biosolve (Valkenswaard, Netherlands); α-cyano-4-hydroxycinnamic acid (CHCA) from Bruker (Bremen, Germany); fetal bovine serum (FBS), nonessential amino acids (NEAA), penicillin-streptomycin, Dulbecco's modified Eagle's medium (DMEM)-Ham's F-12 medium, and MEM-Earle's salt medium with stabile glutamine from PAA Laboratories (Pasching, Austria); cell proliferation kit I and FuGENE-6 from Roche Diagnostics GmbH (Mannheim, Germany); MitoTracker Red CMXRos from Invitrogen (Darmstadt, Germany); and vector pDsRed-Monomer-Golgi (hereinafter pDsRed-Golgi) from Clontech-TaKaRa Bio Europe (Saint-Germain-en-Laye, France).
Peptide synthesis.
All peptides were synthesized in a 25-μmol scale on a multiple-peptide synthesizer (SYRO2000; MultiSynTech GmbH, Witten, Germany) using the Fmoc–tert-butyl strategy with in situ activation with DIC in the presence of HOBt (17). Peptides were cleaved with TFA containing 12.5% (vol:vol) scavenger mixture (ethandithiole, m-cresole, thioanisole, and water, 1:2:2:2) and precipitated with cold diethyl ether. The crude peptides were purified by reversed-phase (RP)-HPLC using a linear acetonitrile gradient in the presence of 0.1% TFA. The purity of the peptides was judged by analytical RP-HPLC using a Jupiter C18 column (Phenomenex, Inc., Torrance, CA), and sequences were confirmed by matrix-assisted laser desorption ionization–tandem time of flight mass spectrometry (MALDI–TOF-TOF MS) (4700 proteomic analyzer; Applied Biosystems GmbH, Darmstadt, Germany), using CHCA as the matrix.
Peptides modified at the N terminus with iodoacetic acid or 5(6)-carboxyfluorescein were synthesized by the same protocol, except for the modification of the unprotected N terminus after completion of the synthesis and before TFA cleavage. A solution of iodoacetic acid (8 eq) in HOBt-DMF (0.5 mol/liter) was added to the swollen resin, activated with DIC (8 eq), and incubated in the dark for 16 h. Alternatively, a solution of 5(6)-carboxyfluorescein (5 eq) and HBTU (5 eq) in DMF was added to the peptide-resin mixture before DIPEA (10 eq) was added. The mixture was incubated in the dark for 2 h. The reaction mixture was discarded. The peptide-resin mixture was washed four times with DMF-DCM and then dried before the peptide was cleaved with TFA.
Two peptides were dimerized by mixing the cysteine-containing peptide with the iodoacetylated (4 eq) peptide in degassed phosphate-buffered saline (PBS, pH 7.4) under nitrogen atmosphere at 4°C until the cysteine peptide was fully converted. The reaction was monitored by RP-HPLC and MALDI–TOF-TOF MS.
Antibacterial activity.
The MICs were determined in triplicates by a liquid broth microdilution assay in sterile 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany) using a total volume of 100 μl per well (16). Aqueous peptide solutions were serially diluted 2-fold in either 1% or 3% aqueous TSB (wt:vol, corresponding to 33% or 100% TSB medium), starting from a concentration of 128 μg/ml. An overnight culture of bacteria was diluted with 33% (or 100%) TSB medium to 1.5 × 108 cells/ml, and 50 μl was added to each well, gaining a starting concentration of 7.5 × 106 cells/well. The plates were incubated at 37°C and the absorbance of each well was measured after 20 ± 2 h at 595 nm (Sunrise Microtiter plate reader; Tecan Trading AG). The MIC was defined as the lowest peptide concentration (in μmol/liter) where the absorbance did not exceed that of the medium.
Cell cultures.
Cell lines were cultured in 25-cm2 tissue culture flasks equipped with a filter cap (Greiner Bio-One GmbH) in cell culture medium (37°C, 5% CO2 with 95% relative humidity). SH-SY5Y and HeLa cell lines were grown in DMEM-Ham's F-12 medium and MEM-Earle's salt medium with stabile glutamine medium, respectively, containing FBS (5%), NEAA (1%), and streptomycin-penicillin (1%).
Proliferation assay.
Cytotoxicity was determined with the cell proliferation kit I (Roche), based on the reduction of methylthiazolyldiphenyl-tetrazolium bromide (MTT) by cellular oxidoreductases of viable cells, which yields a crystalline blue formazan product. Cells were seeded (2 × 104 cells/well) in 96-well plates (Greiner Bio-One GmbH). After one night (HeLa and SH-SY5Y cells) or after 5 days of differentiation by trans-retinoic acid (19 μmol/liter, SH-SY5Y cells), the cells were rinsed with PBS and the peptides were added as a solution in fresh medium to a final volume of 0.1 ml/well. PBS and DMSO (12%, vol:vol) were used as the positive and negative control, respectively. After an incubation period of 2 h or 24 h, the MTT labeling reagent (10 μl) was added to obtain a final concentration of 0.5 g/liter and incubated for 4 h. The solubilization solution (0.1 ml) was added to lyse the cells and to dissolve the formazan crystals. After 16 h, the absorbance was recorded at 590 nm and 650 nm (Paradigm microplate reader; Beckman Coulter, Waals, Austria).
Fluorescence microscopy.
HeLa and SH-SY5Y cells (2 × 104 cells/well) were grown in 96-well plates (Greiner Bio-One GmbH), incubated with fluorescein-labeled peptides for 2 h, imaged with a Leica DMI6000 B fluorescence microscope (EL6000 light source, N PLAN L 20×/0.40 CORR objective; Leica Microsystems, Wetzlar, Germany), and analyzed with LAS 2.1.8 and Adobe Photoshop CS software packages.
Lipotransfection and confocal laser scanning microscopy.
For confocal laser scanning microscopy, HeLa and SH-SY5Y cells were cultured on glass-bottom culture dishes (MatTek Corporation, Ashland, MA). The Golgi apparatus was stained by lipotransfection. Therefore, a solution of serum-free DMEM (0.1 ml) and FuGENE-6 (6 μl) was added dropwise to pDsRed-Golgi (2 μg) and incubated for 15 min. The solution was added to the cells and incubated overnight. The peptide uptake was studied for concentrations of 7 μmol/liter (HeLa) and 10 μmol/liter (SH-SY5Y) fluorescein-labeled peptide in fresh medium, respectively. After 2 h or 24 h the cells were rinsed three times with PBS, medium (2 ml) was added and the nucleus was stained optionally by incubating the cells with Hoechst 33342 (2 μmol/liter, 15 min). The images were recorded on a TCS SP5 inverted confocal laser scanning microscope [405 nm for Hoechst 33342, 488 nm for 5(6)-carboxyfluorescein, 561 nm for pDsRed-Golgi; objective HCX PL APO lambda blue 63×/1.40-0.60 OIL; Leica Microsystems, Wetzlar, Germany] in sequential scan mode. Image stacks were processed and analyzed with the LAS AF 1.7.1 software (Leica Microsystems) and Adobe Photoshop CS. Uptake of peptides in bacteria was studied by mixing the fluorescein-labeled peptides (50 μmol/liter) with a cell suspension to a final volume of 63 μl (1.5 × 108 cells/ml) and adding 41 μl of a saturated aqueous TAMRA solution (2 g/liter) to quench the extracellular 5(6)-carboxyfluorescein in the medium.
Statistical analysis.
Results are expressed as means ± standard deviations. Significant differences between groups were calculated using the unpaired Mann-Whitney test.
RESULTS
Peptide synthesis.
The four antimicrobial peptides apidaecin 1b, drosocin, pyrrhocoricin, and oncocin, the cell-penetrating peptide penetratin, and a Tau peptide (residues 191 to 202 of human Tau elongated N terminally with two aminocaproic acid residues; control) were synthesized in good yields and obtained in high purities after preparative RP-HPLC (Table 1). The products were confirmed by MALDI MS. To study the uptake and distribution of all peptides in bacterial and mammalian cells, the peptides were additionally synthesized with 5(6)-carboxyfluorescein at the N terminus (Table 1). For the four PrAMPs to be delivered into mammalian cells, it was necessary to elongate the AMPs with the sequence of a cell-penetrating peptide. To keep the synthesis simple, we selected the 16-residue-long penetratin, which has been used in many studies before (10, 19). We synthesized the PrAMPs with an N-terminal iodoacetyl group and penetratin with a C-terminal cysteine to ligate them as thioether after purification (Table 1; also see Fig. S1 in the supplemental material). Penetratin was quantitatively ligated with apidaecin 1b and pyrrhocoricin within 6 h, as monitored by RP-HPLC. Conjugations of the corresponding 5(6)-carboxyfluorescein-labeled peptides were completed within 16 h (see Fig. S2 in the supplemental material), whereas the ligation of oncocin was finished only after 48 h. After RP-HPLC, the ligation products were obtained in high purities of >95% and reasonable yields of around 55% (Table 2; also see Fig. S3 in the supplemental material).
Table 1.
Sequences and analytical data of all synthesized peptides
| Peptidea | Sequencea | tRb (min) | Monoisotopic massc |
|
|---|---|---|---|---|
| Calc | Exp | |||
| Apidaecin 1b | H-GNNRPVYIPQPRPPHPRL-OH | 17.1 | 2,108.16 | 2,108.15 |
| Drosocin | H-GKPRPYSPRPTSHPRPIRV-OH | 15.4 | 2,198.23 | 2,198.28 |
| Oncocin | H-VDKPPYLPRPRPPRRIYNR-NH2 | 16.0 | 2,389.38 | 2,389.35 |
| Pyrrhocoricin | H-VDKGSYLPRPTPPRPIYNRN-NH2 | 16.0 | 2,339.27 | 2,339.27 |
| Penetratin | H-RQIKIWFQNRRMKWKK-OH | 17.9 | 2,246.29 | 2,246.23 |
| Tau-peptide | H-ffSGDRSGYSSPGS-OH | 15.7 | 1,381.48 | 1,381.60 |
| Pen-Cys | H-RQIKIWFQNRRMKWKKC-OH | 18.2 | 2,349.31 | 2,349.51 |
| IAc-apidaecin 1b | IAc-GNNRPVYIPQPRPPHPRL-OH | 17.8 | 2,276.06 | 2,276.11 |
| IAc-drosocin | IAc-GKPRPYSPRPTSHPRPIRV-OH | 15.8 | 2,366.14 | 2,366.20 |
| IAc-pyrrhocoricin | IAc-VDKPPYLPRPRPPRRIYNR-NH2 | 17.7 | 2,507.17 | 2,507.15 |
| IAc-oncocin | IAc-VDKGSYLPRPTPPRPIYNRN-NH2 | 17.8 | 2,557.28 | 2,557.19 |
| IAc-Tau-peptide | IAc-ffSGDRSGYSSPGS-OH | 13.3 | 1,548.94 | 1,548.32 |
| Cf-apidaecin 1b | Cf-GNNRPVYIPQPRPPHPRL-OH | 19.5 | 2,467.48 | 2,467.25 |
| Cf-drosocin | Cf-GKPRPYSPRPTSHPRPIRV-OH | 18.1 | 2,556.55 | 2,556.40 |
| Cf-pyrrhocoricin | Cf-VDKPPYLPRPRPPRRIYNR-NH2 | 19.7 | 2,697.59 | 2,697.42 |
| Cf-oncocin | Cf-VDKGSYLPRPTPPRPIYNRN-NH2 | 19.8 | 2,747.70 | 2,747.55 |
| Cf-penetratin | Cf-RQIKIWFQNRRMKWKK-OH | 21.9 | 2,604.61 | 2,605.01 |
| Cf-penetratin-Cys | Cf-RQIKIWFQNRRMKWKKC-OH | 22.0 | 2,707.63 | 2,708.02 |
IAc, Cf, and f denote the iodoacetyl group, 5(6)-carboxyfluorescein, and aminocaproic acid, respectively.
tR denotes the retention time on RP-HPLC.
Calculated (Calc) and experimentally determined (Exp) monoisotopic masses (MALDI–TOF-MS) of the protonated quasimolecular ion [M+H]+.
Table 2.
Analytical data and yields of the ligated peptides and the dimerized penetratin-Cys
| Peptidea | tRb (min) | Monoisotopic massc (g/mol) |
Yield (%) | |
|---|---|---|---|---|
| Calc | Exp | |||
| Pen-apidaecin 1b | 18.8 | 4,496.45 | 4,496.91 | 56 |
| Pen-drosocin | 18.2 | 4,586.53 | 4,586.40 | 56 |
| Pen-pyrrhocoricin | 18.4 | 4,727.56 | 4,728.00 | 61 |
| Pen-oncocin | 18.6 | 4,777.67 | 4,778.13 | 54 |
| Pen-Tau | 18.0 | 3,769.28 | 3,769.62 | 54 |
| (Penetratin-Cys)2d | 19.2 | 4,695.62 | 4,695.99 | 20–25e |
Pen denotes penetratin-Cys.
tR denotes the retention time on RP-HPLC.
Calculated and experimentally determined monoisotopic masses (MALDI-TOF-MS) of the protonated quasimolecular ion [M+H]+.
Disulfide-bridged dimer.
Yields of this by-product depended on the ligation reaction.
Antibacterial activity.
As expected, all four PrAMPs investigated were active against both tested E. coli strains, with MIC values ranging from 0.5 μmol/liter (or 8 μmol/liter) for apidaecin 1b to 8 μmol/liter (or >32 μmol/liter) for pyrrhocoricin in 33% (or 100%) TSB medium (Table 3). The peptides were also active against the Gram-positive bacterium Micrococcus luteus, although the MIC of pyrrhocoricin was relatively high, indicating a weak antibacterial activity. Apidaecin 1b and oncocin were also less active, with MICs of 16 μmol/liter and 8 μmol/liter, respectively. Drosocin, however, was about four times more active against M. luteus than against the two E. coli strains (Table 3). Importantly, all penetratin-PrAMP ligation constructs were active against all three strains, whereas the penetratin dimer was inactive. For E. coli, the coupling to penetratin decreased the activity of a PrAMP by about 2- to 4-fold in 33% TSB medium and increased it slightly for apidaecin 1b and drosocin in 100% TSB medium. In contrast, the ligation increased the activity against M. luteus for the weakly active peptides apidaecin 1b, oncocin, and pyrrhocoricin by around 16- to 32-fold, whereas the activity of the active drosocin sequence remained unchanged. Thus, all penetratin-PrAMP constructs were equally active against M. luteus, with MICs of around 1 μmol/liter, indicating that the PrAMP sequences were predominantly responsible for the antibacterial activity within the coupled products.
Table 3.
MICs of apidaecin 1b, drosocin, oncocin, and pyrrhocoricin, as well as the corresponding ligation products with penetratin-Cys
| Peptidea | MIC (μmol/liter) in medium with indicated concn of TSB medium |
|||||
|---|---|---|---|---|---|---|
|
E. coli BL21AI |
E. coli MC4100 |
M. luteus 10240 (33%) | S. aureus DSM 6247 (33%) | |||
| 33% | 100% | 33% | 100% | |||
| Apidaecin 1b | 0.5 | 8 | 1 | 32–64c | 16 | >32 |
| Pen-apidaecin 1b | 2 | 4 | 1 | 4 | 1–2 | 8 |
| Drosocin | 2 | 16–32 | 2 | >32 | 0.5 | >32 |
| Pen-drosocin | 4 | 8 | 8 | >32 | 0.125–2 | 4 |
| Oncocin | 4 | 8 | 2 | 16–32 | 8 | >32 |
| Pen-oncocin | 8 | 16 | 8 | 16 | 0.5–1 | 4b |
| Pyrrhocoricin | 8 | >32 | 4 | >32 | 32–64 | >32 |
| Pen-pyrrhocoricin | 8 | >16 | 8 | >16 | 1 | 4 |
| (Penetratin-Cys)2 | >32 | 16–32 | >32 | >16 | >32 | 8 |
Pen denotes penetratin-Cys coupled as thioether to the corresponding peptide.
MICs of Pen-oncocin against S. aureus DSM 6247 were from a single experiment in triplicates.
Ranges indicate the lowest and highest MIC values recorded for two or three independent experiments performed in triplicate.
Uptake studies in bacteria.
All 5(6)-carboxyfluorescein-labeled PrAMPs led to a strong fluorescence of the E. coli BL21AI strain, whereas the corresponding penetratin-labeled constructs yielded significantly weaker fluorescence signals in the confocal laser scanning microscopy images, as shown for oncocin (Fig. 1). The penetratin dimer did not stain the cells within 90 min (Fig. 1). The lower fluorescence intensities observed for the penetratin-PrAMP constructs fit with their reduced antibacterial activities against E. coli, as described above. This effect might be attributed to penetratin, which can shuttle the PrAMPs into cells but also out of the cells again, whereas free PrAMPs are enriched in the bacteria.
Fig 1.

Confocal laser scanning microscopy images of E. coli BL21AI (A to F) and M. luteus 10240 (G to P) incubated with fluorescein-labeled peptides (30 μmol/liter). Phase contrast (top rows) and fluorescence images (bottom rows) were taken of E. coli BL21AI cells incubated with 5(6)-carboxyfluorescein (Cf)-oncocin (A, B) and Cf-penetratin-oncocin (C, D) for 50 min and with the Cf-penetratin-Cys dimer for 90 min (E, F) or M. luteus 10240 cells incubated with Cf-pyrrhocoricin (G, H), Cf-penetratin-pyrrhocoricin (I, J), Cf-drosocin (K, L), CF-penetratin-drosocin (M, N), and the Cf-penetratin-Cys dimer (O, P). TAMRA was added to quench the fluorescence background of the medium. Bars equate to 5 μm.
For M. luteus 10240, the fluorescence uptake was basically reversed. Here, the native carboxyfluorescein-labeled PrAMPs stained the cells weakly (drosocin) or not at all (pyrrhocoricin), whereas the corresponding penetratin constructs provided equally strong fluorescent images (Fig. 1). This resembles the MICs nicely, with pyrrhocoricin being almost inactive against M. luteus (MIC ≥ 32 μmol/liter) and all penetratin-PrAMP constructs being highly active (MIC ∼ 1 μmol/liter) (Table 3). Thus, penetratin appeared to improve the internalization of apidaecin 1b, oncocin, and pyrrhocoricin for M. luteus and therefore allowed the PrAMPs to kill the bacteria efficiently. Only the antibacterial activity of native drosocin, which was able to enter the bacteria at significant levels, was not improved by penetratin, confirming that, indeed, the bacterial uptake of the PrAMP represents the rate-limiting step. Therefore, we also tested the antimicrobial activity of all peptide constructs against Staphylococcus aureus, which is an important Gram-positive pathogen (Table 3). Whereas all PrAMPs were inactive (MIC > 32 μmol/liter), the MICs of the penetratin-PrAMPs ranged from 4 to 8 μmol/liter, which indicated antibacterial activities at least 8 time times higher. It should be noted, however, that the penetratin-Cys dimer was also active against S. aureus (MIC = 8 μmol/liter). Therefore, it is not clear how much the PrAMPs contributed to the activities of the constructs.
Cytotoxicity.
The cell viability of HeLa cells and differentiated and undifferentiated SH-SY5Y cells after treatment with PrAMPs and penetratin-PrAMP constructs was studied in an MTT assay. As expected, apidaecin 1b, drosocin, oncocin, and pyrrhocoricin did not affect the cell viability at a concentration of 600 μg/ml compared to the results with PBS buffer as negative control (Fig. 2). These results are in full agreement with the results in the literature (16, 23) and indicate that neither intracellular chaperones nor receptors on the surface of either cell line were inhibited. The penetratin-PrAMP constructs, however, reduced the viability of HeLa cells at higher concentrations of 400 μg/ml (∼100 μmol/liter) to around 30% to 60%, whereas no effect was observed for concentrations of 100 μg/ml and below (Fig. 2, top). The toxicity increased slightly in the following order: drosocin < pyrrhocoricin ≈ apidaecin 1b < oncocin. Neither penetratin nor the penetratin-tau construct influenced the cell viability in the concentration range studied. This indicates that the observed toxic effects can be attributed to the PrAMP sequences but not to either the penetratin sequence or the thioether linker.
Fig 2.
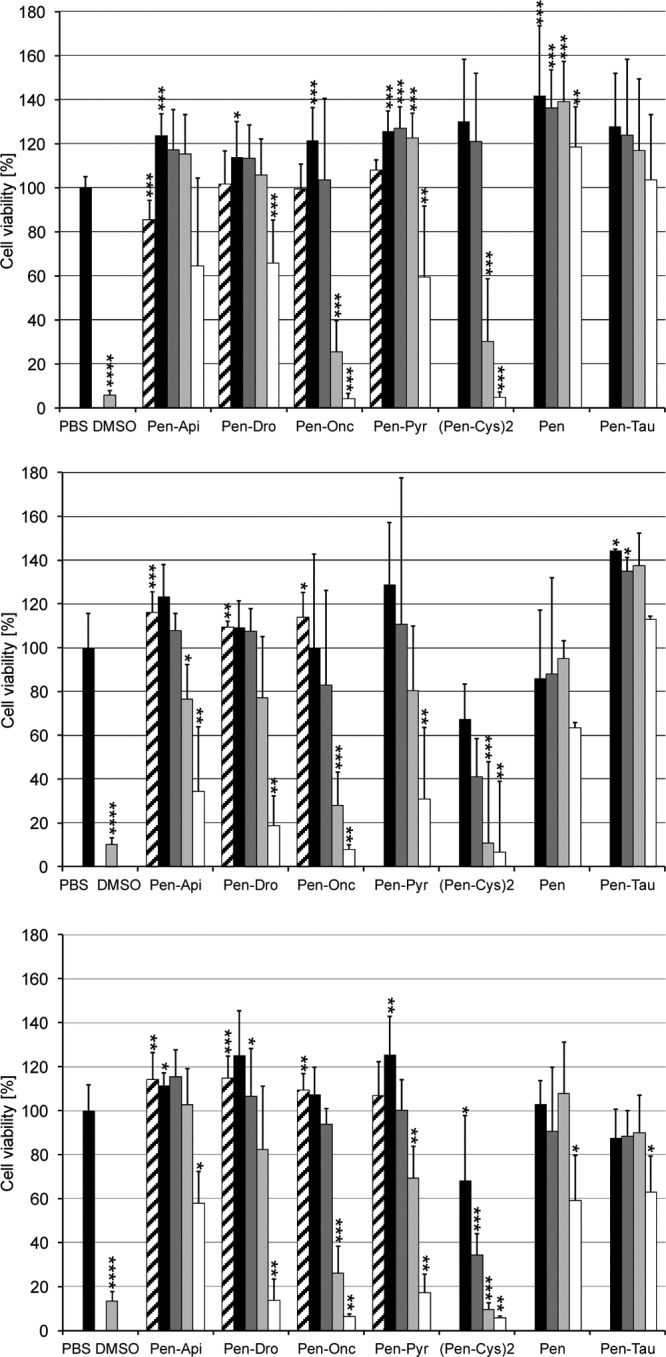
Cell viability of HeLa (top), undifferentiated SH-SY5Y (middle), and differentiated SH-SY5Y cells (bottom) incubated with penetratin constructs with apidaecin 1b (Pen-Api), drosocin (Pen-Dro), oncocin (Pen-Onc), pyrrhocoricin (Pen-Pyr), and Tau peptide (Pen-Tau), dimerized penetratin-Cys [(Pen-Cys)2], and free penetratin (Pen) after a incubation period of 24 h. Peptide concentrations were 50 μg/ml (black), 100 μg/ml (dark gray), 200 μg/ml (light gray), and 400 μg/ml (white) for the penetratin constructs. Results for the corresponding unligated peptides apidaecin 1b, drosocin, oncocin, and pyrrhocoricin (600 μg/ml), used as controls, are shown (hatched). Further controls were PBS and DMSO (12% [vol/vol] in medium). Shown are the results typically obtained from two or three independent experiments conducted in triplicates or duplicates. Pyrrhocoricin was not tested against undifferentiated SH-SY5Y cells. The absorption value determined for each peptide was normalized to that of the corresponding PBS control, and the mean overall determination calculated is shown in the diagram. For statistical analysis, the unpaired Mann-Whitney test was used. ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05.
Differentiated SH-SY5Y cells were more vulnerable, with the viability being decreased by around 10 to 30% for most penetratin-PrAMP constructs at a concentration of 200 μg/ml, whereas no influence was detected for lower concentrations (Fig. 2, bottom). This stronger toxic effect, however, might be attributed to the penetratin sequence, as both free penetratin and the penetratin-tau construct reduced the viability to ∼70% at the highest tested concentration, with the penetratin-Cys dimer being the most toxic peptide. Thus, each PrAMP reduced the viability approximately 5-fold compared to the results with the two control peptides. Undifferentiated SH-SY5Y cells were less affected, with the viabilities ranging from ∼30% for the oncocin construct to ∼80% for the apidaecin 1b and pyrrhocoricin constructs at a concentration of 200 μg/ml (Fig. 2, middle). Interestingly, this was in the same range as the results with the free penetratin. Taken together, the IC50s were around 250 μg/ml (∼60 μmol/liter) for all peptides in SH-SY5Y cells. In HeLa cells, the same IC50 was obtained for penetratin-oncocin, whereas the other three constructs were less toxic (IC50 ∼ 380 μg/ml). It should be noted that the toxicities of all four PrAMPs tested were very similar, with only slight changes among the three mammalian cell systems.
The reversibility of the cell damage was tested by incubating the cells with a high concentration of the penetratin-PrAMP construct for 2 h and then replacing the supernatant in fresh medium without the peptide construct (see Fig. S4 in the supplemental material). After 2 h, only the viability of HeLa cells was reduced to ∼50% when incubated with penetratin-oncocin. For this construct, the HeLa cells did not recover, while HeLa cells treated with apidaecin 1b and pyrrhocoricin constructs recovered afterwards. The viability of the SH-SY5Y cells decreased further during the recovery phase, with the oncocin construct showing the strongest effect, similar to the results with the penetratin-Cys dimer.
Uptake studies in mammalian cells.
Having shown that the penetratin-PrAMP constructs reduced the viability of HeLa and SH-SY5Y cells at higher concentrations, we tested whether the constructs entered the cells and, thus, might have blocked intracellular proteins. By applying confocal microscopy and N-terminal 5(6)-carboxyfluorescein-labeled PrAMP constructs, the uptake could be monitored. As expected, the fluorescein-labeled native PrAMPs were not internalized in detectable quantities (data not shown). The corresponding penetratin constructs, however, entered the cells in significant amounts when added at concentrations of 40 μmol/liter (∼160 μg/ml), i.e., approximately at their IC50 value (Fig. 3). At this high concentration, the free PrAMPs did not alter the morphology of the cells, whereas the penetratin constructs changed the cell morphology significantly (Fig. 3). The morphological changes were not observed when the peptide concentration was reduced well below the IC50 values, i.e., 10 μmol/liter for SH-SY5Y cells and 7 μmol/liter for HeLa cells. Importantly, the cells maintained strong fluorescence at these lower concentrations, indicating that the peptide constructs entered the cells at significant levels.
Fig 3.

Wide-field fluorescence microscopy images of SH-SY5Y (A to D) and HeLa cells (E to H) after incubation with 5(6)-carboxyfluorescein (Cf)-oncocin (A, B, E, and F), and Cf-penetratin-oncocin (C, D, G, and H) at a concentration of 40 μmol/liter. The upper row shows the phase contrast and the lower row the fluorescence images. Bars equate to 20 μm.
To further determine the intracellular localization of the conjugates, the nuclei and mitochondria of SH-SY5Y and HeLa cells were additionally stained with Hoechst 33342 (blue fluorescence) and MitoTracker Red CMXRos (red fluorescence). The green fluorescence of the labeled penetratin-PrAMP constructs was not colocalized with the blue or red fluorescence, indicating that the constructs did not enter the nuclei and mitochondria (data not shown). The peptides, however, were perinuclear aligned in small distinct spots after 2 h and in a more dense cloud after 24 h of incubation (Fig. 4 left). Interestingly, penetratin and the penetratin-Cys dimer showed the same intracellular distribution (Fig. 4, middle and right; also see Fig. S5 in the supplemental material). After 24 h, the localization site was in the area of the Golgi apparatus. Thus, a lipotransfection of the HeLa cells was performed and the Golgi stained with pDsRed-Golgi. The overlay of the fluorescence images yielded a yellow area where the peptides were associated with the Golgi apparatus but, also, further particles in the surrounding area that showed only the green fluorescence of the peptide construct (see Fig. S6 in the supplemental material). This pattern is very similar to the distribution of 5(6)-carboxyfluorescein-labeled penetratin itself, as reported by Fischer et al. (11), which shows that the intracellular distribution is determined by the penetratin sequence.
Fig 4.
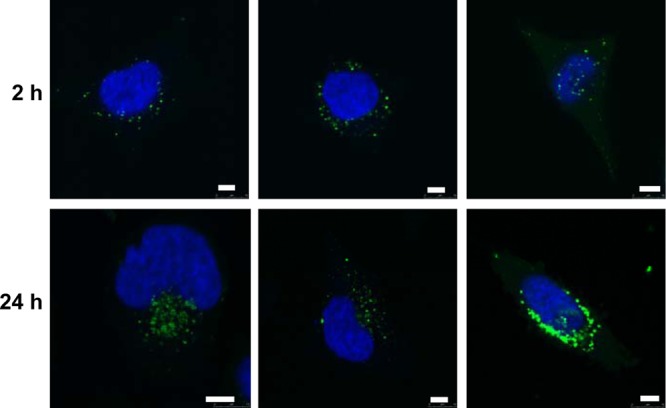
Confocal laser scanning microscopy images of HeLa cells incubated with 5(6)-carboxyfluorescein (Cf)-penetratin-pyrrhocoricin (left), Cf-penetratin (middle), and dimerized Cf-penetratin-Cys (right) for 2 h and 24 h. The nuclei were stained with Hoechst 33342. Bars equate to 5 μm.
DISCUSSION
Native PrAMPs appear to kill bacteria by a general mechanism that consists of three consecutive steps, which are most likely independent of each other. First, the peptides permeate and traverse the outer membrane to enter the periplasmic space (6, 8). This process is reversible, although the outward diffusion appears to be slow compared to the invasion (6). The second step resembles the stereospecific and irreversible translocation into the cytoplasm by a permease/transporter-mediated uptake (6) which involves SbmA as part of an ABC transporter (22). This uptake is crucial for PrAMP activity and might be a major cause of possible resistance mechanisms (6, 22). After entering the bacteria, the peptides encounter their ultimate target, the 70-kDa chaperon DnaK (24), although recent evidence indicates that additional and/or alternative targets might exist, such as chaperonin GroEL and its cofactor GroES (24, 34). Api88, for example, was equally active against E. coli strain MC4100 and a DnaK-null mutant obtained from this E. coli strain (9). Moreover, an alternative killing mechanism was suggested recently, based on the observation that E. coli incubated with apidaecin 1b produced the protease FtsH at higher quantities (35). The authors speculated that the increased expression of FtsH, which can degrade cytoplasmic UDP-3-O-acyl-N-acetylglucosamine deacetylase, might result in an unbalanced synthesis of lipopolysaccharide and phospholipids. Whereas these targets have to be confirmed, it is well established that PrAMPs bind to the substrate binding cleft of DnaK and, thus, contribute at least partially to the killing mechanisms in Enterobacteriaceae (e.g., E. coli and K. pneumoniae) and nonfermenting species (e.g., Pseudomonas aeruginosa and Acinetobacter baumannii). Tempst's research group already showed in an elegant publication that the bacterial uptake of PrAMPs is a prerequisite for their antibacterial effect (6). Thus, the antibacterial activity can be explained by the peptide uptake and the target inhibition.
This mode of action is also likely for Gram-positive bacteria, which are targeted by some PrAMPs, although most PrAMPs kill Gram-negative bacteria much more efficiently in vitro. Interestingly, even small sequence changes can significantly alter the activity, often in opposite directions for different bacteria (6). Most likely, these activities relate directly to transporter and DnaK recognition, i.e., sequence variations among different bacteria, including reduced susceptibilities for certain strains.
As penetratin, a highly cationic CPP, had no antibacterial activity against E. coli and M. luteus in our assays, the influence obtained on the activity of the ligated PrAMPs could be explained either by a better cellular uptake or an improved target binding. As PrAMPs bind to DnaK with only a 7- to 8-residue-long sequence stretch (9, 17, 20), whereas both termini protrude out of the PrAMP-DnaK complex, an influence of the penetratin sequence on the inhibition of DnaK appears unlikely. Furthermore, the intracellular distributions of PrAMPs and penetratin-PrAMP conjugates appeared to be identical. Thus, the significantly lower MICs against M. luteus and the slightly reduced activity against E. coli can most probably be attributed to the translocation through the membrane, as indicated by the fluorescence intensities of the bacteria. As it appears unlikely that penetratin improves the transporter-mediated uptake of PrAMPs, it is probably the passive or active penetratin-based uptake through the bacterial membrane that determines the activities of the ligated PrAMPs. The slightly reduced activity against E. coli can be explained in two different ways. First, the penetratin-PrAMP conjugate might not be trapped within the cells, as proposed for PrAMPs (6), but might leave the cells (28) and thus kill the bacteria less efficiently due to a reduced internal peptide concentration. This also fits the relatively high dissociation constants of the PrAMP-DnaK complex studied, which are in the low micromolar range (17). Second, conjugation to penetratin might reduce the cellular uptake by transporters, such as SbmA, due to a reduced binding to the transporter for sterical reasons or binding of penetratin to other cellular components within the periplasmic space or intracellular membrane.
The very low toxicities of PrAMPs obtained in vitro and in vivo were always attributed to the fact that PrAMPs can enter only some cells of the immune system but not mammalian cells in general. By covalently ligating four different insect-derived PrAMPs to penetratin, we could show that even intracellular PrAMPs are only slightly toxic to human cell lines. This nicely fits the low cytotoxicity of PrAMPs and other cell-permeating proline-rich basic peptides in phagocytic mammalian immune cells (26, 28, 31, 33). In considering MIC values (Table 3), the values determined for E. coli of about 2 μmol/liter and the unaltered cell viability for penetratin-PrAMP concentrations below 25 μmol/liter show that PrAMPs can be considered nontoxic to mammalian cells, as they cannot enter most of these, but that even if transported into human cells, they are only slightly toxic. This also indicates that PrAMPs can efficiently inhibit DnaK and its orthologs in other bacteria but are only slightly cross-reactive toward human chaperones or other intracellular mammalian proteins when ligated to penetratin.
It is tempting to think that the antibacterial spectrum of PrAMPs, which are typically active against Gram-negative pathogens, could be extended by elongating their sequence with that of another cell-penetrating peptide, such as penetratin. This concept might even extend the treatment options toward bacteria that can hide inside mammalian cells, although the toxic side effects obtained toward mammalian cells might limit such applications. Interestingly, this concept of adding additional sequences to improve their cellular uptake was already invented by evolution. Whereas insect-derived PrAMPs are relatively short, mammalian PrAMPs are significantly longer. Bactenicins, for example, which are processed from their precursor cathelicidins, contain different repeat units that are responsible for the cellular uptake and the antimicrobial activity (28–30). These mammalian PrAMPs have broader antimicrobial effects against Gram-negative bacteria (e.g., E. coli), Gram-positive bacteria (e.g., Staphylococcus aureus), and fungi (e.g., Candida albicans) and are less susceptible to higher medium concentrations than short PrAMPs, which has been attributed to their high cell-permeant activities (28). Future studies will have to show if it is possible to combine insect-derived PrAMPs with short CPPs to enhance their antimicrobial activity against Gram-positive bacteria and fungi, as well as Gram-negative bacteria, in full-strength medium without being toxic toward mammalian cells. This could be accomplished, for example, by a shortened construct that is able to enter only pathogens and not regular mammalian cells. The higher level of activity of drosocin, containing three PRP motifs, against M. luteus (0.5 μmol/liter) compared to the levels of activity with two PRP motifs in oncocin (8 μmol/liter) and pyrrhocoricin (∼ 32 μmol/liter) and only one PRP motif in apidaecin 1b (16 μmol/liter) may already indicate that such an approach is promising. The broader activity spectrum of the penetratin-PrAMP constructs and their higher activities in full-strength medium could also be explained by increased net charges or charge densities, especially for M. luteus. Penetratin contains seven positively charged side chains (Lys and Arg), compared to only four (apidaecin 1b and pyrrhocoricin) or six (drosocin and oncocin) basic residues in the linked PrAMPs, potentially enhancing interactions with the negatively charged bacterial surface.
Conclusions.
The activity spectrum of insect-derived proline-rich AMPs can be extended toward Gram-positive bacteria by ligating them with a cell-penetrating peptide, such as penetratin. Such constructs are able to enter mammalian cells, where they possess only a slight toxicity. This could allow the development of peptide derivatives that optimize the activity of short, insect-derived PrAMPs against a range of pathogens and potentially prevent resistance mechanisms related to cellular uptake.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Singer for technical assistance with the thioether ligation.
Financial support from the European Fund for Regional Structure Development (EFRE, European Union and Free State Saxony) is gratefully acknowledged.
Footnotes
Published ahead of print 30 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Benincasa M, et al. 2010. The proline-rich peptide Bac7(1-35) reduces mortality from Salmonella typhimurium in a mouse model of infection. BMC Microbiol. 10:178 doi:10.1186/1471-2180-10-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boucher HW, et al. 2009. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 3. Bradford PA, et al. 2004. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 beta-lactamases in New York City. Clin. Infect. Dis. 39:55–60 [DOI] [PubMed] [Google Scholar]
- 4. Bulet P, et al. 1993. A novel inducible antibacterial peptide of Drosophila carries an O-glycosylated substitution. J. Biol. Chem. 268:14893–14897 [PubMed] [Google Scholar]
- 5. Casteels P, Ampe C, Jacobs F, Vaeck M, Tempst P. 1989. Apidaecins: antibacterial peptides from honeybees. EMBO J. 8:2387–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castle M, Nazarian A, Yi SS, Tempst P. 1999. Lethal effects of apidaecin on Escherichia coli involve sequential molecular interactions with diverse targets. J. Biol. Chem. 274:32555–32564 [DOI] [PubMed] [Google Scholar]
- 7. Cociancich S, et al. 1994. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus. Biochem. J. 300(Pt 2):567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Czihal P, Hoffmann R. 2009. Mapping of apidaecin regions relevant for antimicrobial activity and bacterial internalization. Int. J. Pept. Res. Ther. 15:157–164 [Google Scholar]
- 9. Czihal P, et al. 2012. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant Gram-negative pathogens. ACS Chem. Biol. 7:1281–1291 [DOI] [PubMed] [Google Scholar]
- 10. Derossi D, Chassaing G, Prochiantz A. 1998. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 8:84–87 [PubMed] [Google Scholar]
- 11. Fischer R, Kohler K, Fotin-Mleczek M, Brock R. 2004. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J. Biol. Chem. 279:12625–12635 [DOI] [PubMed] [Google Scholar]
- 12. Giacometti A, et al. 2003. In vitro effect on Cryptosporidium parvum of short-term exposure to cathelicidin peptides. J. Antimicrob. Chemother. 51:843–847 [DOI] [PubMed] [Google Scholar]
- 13. Hooper DC. 2001. Emerging mechanisms of fluoroquinolone resistance. Emerging Infect. Dis. 7:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones RN. 2001. Resistance patterns among nosocomial pathogens: trends over the past few years. Chest 119:397S–404S [DOI] [PubMed] [Google Scholar]
- 15. Knappe D, et al. 22 June 2012. Oncocin derivative Onc72 is highly active against Escherichia coli in a systemic septicaemia infection mouse model. J. Antimicrob. Chemother. [Epub ahead of print.] doi:10.1093/jac/dks241 [DOI] [PubMed] [Google Scholar]
- 16. Knappe D, et al. 2010. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): A novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 53:5240–5247 [DOI] [PubMed] [Google Scholar]
- 17. Knappe D, et al. 2011. Rational design of oncocin derivatives with superior protease stabilities and antibacterial activities based on the high-resolution structure of the oncocin-DnaK complex. Chembiochem 12:874–876 [DOI] [PubMed] [Google Scholar]
- 18. Kragol G, et al. 2002. Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur. J. Biochem. 269:4226–4237 [DOI] [PubMed] [Google Scholar]
- 19. Langel U. (ed). 2006. Handbook of cell-penetrating peptides, p 5–28 CRC Press, Boca Raton, FL [Google Scholar]
- 20. Liebscher M, Roujeinikova A. 2009. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J. Bacteriol. 191:1456–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Livermore DM, Brown DFJ. 2001. Detection of beta-lactamase-mediated resistance. J. Antimicrob. Chemother. 48:59–64 [DOI] [PubMed] [Google Scholar]
- 22. Mattiuzzo M, et al. 2007. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 66:151–163 [DOI] [PubMed] [Google Scholar]
- 23. Otvos L. 2002. The short proline-rich antibacterial peptide family. Cell. Mol. Life Sci. 59:1138–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Otvos L, et al. 2000. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 39:14150–14159 [DOI] [PubMed] [Google Scholar]
- 25. Otvos L, et al. 2005. Designer antibacterial peptides kill fluoroquinolone-resistant clinical isolates. J. Med. Chem. 48:5349–5359 [DOI] [PubMed] [Google Scholar]
- 26. Otvos L, Cudic M, Chua BY, Deliyannis G, Jackson DC. 2004. An insect antibacterial peptide-based drug delivery system. Mol. Pharmaceutics. 1:220–232 [DOI] [PubMed] [Google Scholar]
- 27. Pitout JDD, Laupland KB. 2008. Extended-spectrum beta-lactamase-producing enterobacteriaceae: an emerging public-health concern. Lancet Infect. Dis. 8:159–166 [DOI] [PubMed] [Google Scholar]
- 28. Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. 2002. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry 41:14150–14157 [DOI] [PubMed] [Google Scholar]
- 29. Scocchi M, Tossi A, Gennaro R. 2011. Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action. Cell. Mol. Life Sci. 68:2317–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shamova O, et al. 1999. Purification and properties of proline-rich antimicrobial peptides from sheep and goat leukocytes. Infect. Immun. 67:4106–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Splith K, Neundorf I. 2011. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 40:387–397 [DOI] [PubMed] [Google Scholar]
- 32. Szabo D, et al. 2010. The designer proline-rich antibacterial peptide A3-APO is effective against systemic Escherichia coli infections in different mouse models. Int. J. Antimicrob. Agents 35:357–361 [DOI] [PubMed] [Google Scholar]
- 33. Tomasinsig L, et al. 2006. Mechanistic and functional studies of the interaction of a proline-rich antimicrobial peptide with mammalian cells. J. Biol. Chem. 281:383–391 [DOI] [PubMed] [Google Scholar]
- 34. Zhou YS, Chen WN. 2011. iTRAQ-coupled 2-D LC-MS/MS analysis of cytoplasmic protein profile in Escherichia coli incubated with apidaecin IB. J. Proteomics 75:511–516 [DOI] [PubMed] [Google Scholar]
- 35. Zhou YS, Chen WN. 2011. iTRAQ-coupled 2-D LC-MS/MS analysis of membrane protein profile in Escherichia coli incubated with apidaecin IB. PLoS ONE. 6:e20442 doi:10.1371/journal.pone.0020442 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.