

Abstract
Visceral leishmaniasis is an emerging neglected tropical disease (NTD) caused by the protozoan Leishmania infantum in the countries bordering the Mediterranean Basin. Currently there is no effective vaccine against this disease, and the therapeutic approach is based on toxic derivatives of SbV. Therefore, the discovery of new therapeutic targets and the development of drugs designed to inhibit them comprise an extremely important approach to fighting this disease. DNA topoisomerases (Top) have been identified as promising targets for therapy against leishmaniasis. These enzymes are involved in solving topological problems generated during replication, transcription, and recombination of DNA. Being unlike that of the mammalian host, type IB DNA topoisomerase (TopIB) from Leishmania spp. is a unique bisubunit protein, which makes it very interesting as a selective drug target. In the present investigation, we studied the effect of two TopIB poisons with indenoisoquinoline structure, indotecan and AM13-55, on a murine BALB/c model of infected splenocytes with L. infantum, comparing their effectiveness with that of the clinically tested leishmanicidal drug paromomycin. Both compounds have high selectivity indexes compared with uninfected splenocytes. SDS-KCl-precipitable DNA-protein complexes in Leishmania promastigotes and in vitro cleaving assays confirmed that these drugs are Top poisons. The inhibitory potency of both indenoisoquinolines on L. infantum recombinant TopIB was assessed in vitro, with results showing that indotecan was the most active compound, preventing the relaxation of supercoiled DNA. Experimental infections in susceptible BALB/c mice treated with 2.5 mg/kg body weight/day once every other day for a total of 15 days showed that indotecan cleared more than 80% of the parasite burden of the spleen and liver, indicating promising activity against visceral leishmaniasis.
INTRODUCTION
Neglected tropical diseases (NTD) produced by vector-borne protozoa are mostly prevalent in underdeveloped and poor countries, causing an estimated 4.1 million disability-adjusted life years (DALYs) lost (16). However, in developed countries, where these diseases were unknown or had been eradicated for a long time, an unexpected new scenario has appeared. Increased tourist exchange, soldiers deployed in countries where these diseases are endemic, migratory fluxes, and eventually climate changes, along with other pandemics, are challenging the apparent safety of rich populations (26, 30). Since NTDs are prevalent in low-income countries, large pharmaceutical companies have neglected research and development of new drugs. Therefore, old compounds that are losing efficacy are still in use. These compounds have undesirable toxic effects, and their dosage schedules are complex and repetitive, which compromises patient compliance (7).
Visceral leishmaniasis is an NTD increasingly affecting European countries because of the massive flows of immigrants from North Africa (18). Its etiologic agent, Leishmania infantum, infects domestic dogs (canine leishmaniasis), which act as reservoir, and it is transmitted to humans through sandfly bites (20). For decades, the first-line treatment of this disease consisted of the old-fashioned and toxic drugs derived from pentavalent antimony (SbV) (1). Recently, safer and more effective drugs, such as amphotericin B, miltefosine, and paromomycin, are replacing SbV, despite the fact that they are not devoid of undesirable side effects and cannot be administered during pregnancy (8, 9). For these reasons, the search for new compounds against this disease is very much needed. Since the beginning of this century, DNA topoisomerase IB (TopIB) has been identified as a potential target in therapy against Leishmania and other trypanosomatids (3, 5, 6). The choice of this target is based on two main reasons: (i) the enzyme has an increased expression during the rapidly dividing cycle of the pathogen, similar to the case with tumor cells, and especially (ii) the pathogen's enzyme is structurally different from that of the host (35).
Our research group found that Leishmania donovani TopIB (LdTopIB) was atypically composed of two different subunits, encoded by different genes, that have to be assembled in the pathogen to reconstitute the active enzyme. One of the subunits contains the four amino acids of the active site, which is fully conserved from a phylogenetic point of view (35). The other subunit contains the catalytic amino acid (Tyr222), which acts by breaking one DNA strand by a specific nucleotide sequence (13). All of these features have also been found in the other protozoan-borne NTDs pathogens: Trypanosoma brucei, the agent responsible for sleeping sickness in Africa (11), and T. cruzi, responsible for Chagas' disease in South America (36). However, despite the fact that these enzymes conserve their catalytic domains unchanged, they display two nonconserved regions, one at the C-terminal end of the large protomer and the other at the N-terminal end of the small protomer, which are extremely important in sensitivity to topoisomerase poisons (12). These compounds act by stabilizing enzyme-DNA complexes, preventing the religation step and eventually producing single-strand breaks when they collide with the replication fork during DNA synthesis (22). The most studied TopIB poison comprises camptothecin (CPT) and its derivatives, including topotecan and the prodrug irinotecan, which are being used against certain tumors (10, 32). Other, non-CPT TopIB poisons are indolocarbazoles, such as the DNA-intercalating drug rebeccamycin, and indenoisoquinolines, which were initially developed as antitumor compounds with an improved ability to stabilize cleavage complexes (2, 25).
There are several promising indenoisoquinolines being studied for therapy targeting tumor processes and more recently for therapy against experimental African trypanosomiasis (4). In this article, we show for the first time the effects of two indenoisoquinolines, indotecan and AM13-55, on the viability of free-living promastigotes and amastigote-infected murine splenocytes. For this purpose, we used a modified strain of L. infantum transfected with the gene encoding the infrared protein IFP1.4 from the extremophilic bacterium Deinococcus radiodurans (31). This fluorescent pathogenic strain retains its virulence unaffected and allows studies of high performance in both free-living promastigotes and ex vivo spleen explants, where parasite growth conditions resemble a natural infection environment. The results provided in this work demonstrate the high selectivity indexes of both molecules in vitro and the promising therapeutic potential of indotecan against visceral leishmaniasis.
MATERIALS AND METHODS
Reagents and culture media.
Pyrococcus furiosus (Pfu) and Klenow polymerases and restriction enzymes were acquired from Roche (Roche Farma SA, Spain) and GE Healthcare (Spain). T4 DNA ligase was obtained from Stratagene (La Jolla, California). Cell culture media were purchased from Sigma-Aldrich (Spain). The indenoisoquinolines indotecan and AM13-55 (Fig. 1) were kindly provided by Mark Cushman, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University. Primers for PCR amplification were from Sigma Genosys (United Kingdom).
Fig 1.
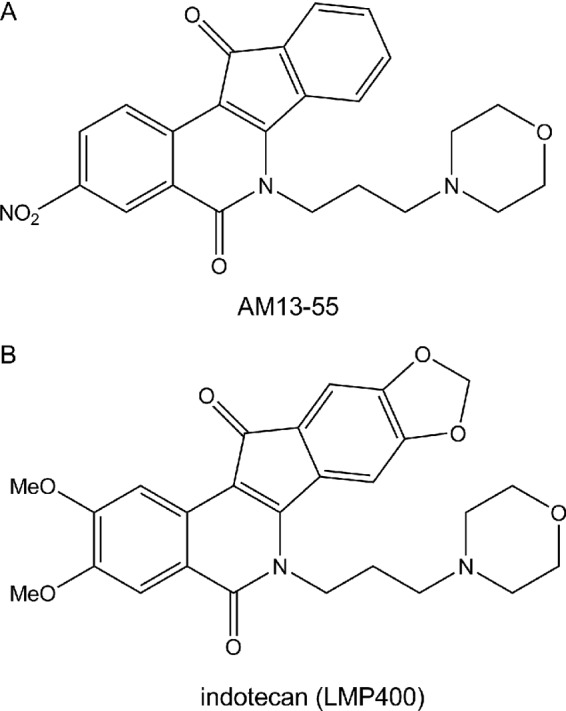
Chemical structures of the indenoisoquinolines indotecan and AM13-55, used in the present study.
Generation of an infrared fluorescent L. infantum strain.
L. infantum promastigotes (BCN150 strain) were obtained by J. M. Requena (Centro de Biología Molecular Severo Ochoa, Madrid Spain). The 987-bp IFP1.4 coding region—an improved monomeric infrared fluorescent protein derived from Deinococcus radiodurans (31)—was amplified by PCR from the IFP1.4_pcDNA3 vector, a gift from Roger Y. Tsien (Department of Pharmacology, Department of Chemistry and Biochemistry, University of California, San Diego), using the forward primer 5′CCGCTCGAGCCATGGCCACCATGGCTCGGGACCCTCTGCC3′ and the reverse primer 5′ATAAGAATGCGGCCGCTCATTTATACAGCTCGTCCATTCC3′. The amplified fragment was digested with BglII and NotI and assembled into the expression vector pLEXSY-2-HYG (Jena Bioscience GmbH, Germany), which was previously digested with the same enzymes. The resultant plasmid was named pLEXSY-IFP1.4. Parasites were electroporated with the large SwaI targeting fragment from pLEXSY-IFP1.4 and selected on semisolid media as previously described (27). Many clonal lines were obtained, and correct integration into the 18s rRNA locus was confirmed by PCR analysis. The transformant strain was routinely cultured at 26°C in M199 medium supplemented with 25 mM HEPES (pH 6.9), 10 mM glutamine, 7.6 mM hemin, 0.1 mM adenosine, 0.01 mM folic acid, 1× RPMI 1640 vitamin mix (Sigma), 10% (vol/vol) heat-inactivated fetal calf serum (FCS), and antibiotic cocktail (50 U/ml penicillin and 50 μg/ml streptomycin).
Ex vivo splenic explant cultures.
BALB/c mice were inoculated intraperitoneally with 108 L. infantum purified metacyclic promastigotes. Briefly, infective promastigotes were isolated from stationary-phase culture by negative selection with peanut agglutinin (29). Five weeks postinfection, spleens were aseptically dissected, washed in cold phosphate-buffered saline (PBS), and placed in petri dishes. Small pieces were obtained by using a scalpel. In order to obtain a single-cell suspension, tissue was incubated with 5 ml of 2 mg/ml collagenase D (Roche) prepared in buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2) for 20 min at 37°C. A cell suspension containing traces of spleen mass was gently passed through a 100-μm cell strainer to remove tissue fragments (21). Splenocytes were washed twice with PBS by centrifugation (500 × g for 7 min at 4°C) and resuspended in RPMI medium supplemented with 10% FCS, 1 mM sodium pyruvate, 1× RPMI vitamins, 10 mM HEPES, and 50 U/ml penicillin and 50 μg/ml streptomycin at 37°C with 5% CO2. Cells were counted and diluted at different cell densities. Cells were seeded until confluence, and different concentrations of the studied indenoisoquinolines were administered to the explants for 48 h. The viability of infecting amastigotes was assessed by registering the fluorescence emission at 708 nm in an Odyssey (Li-Cor) infrared imaging system. The cytotoxicities of the drugs were assessed on uninfected ex vivo explants after 48 h of incubation, using the alamarBlue staining method according to the manufacturer's recommendations.
Recombinant Leishmania infantum TopIB.
Cloning of Leishmania infantum TopIB (LiTopIB) open reading frames (ORFs) (encoding large and small subunits) and expression and purification of the enzyme were carried out as previously described for LdTopIB (35). Briefly, a Saccharomyces cerevisiae EKY3 strain deficient in TopIB activity (MATα ura3-52 his3Δ200 leu2Δ1 trp1Δ63 top1 ΔTRP1) was transformed by the lithium acetate method (15) with the bicistronic pESC-URA vector, which carries both LiTopIB ORFs. Single colonies were incubated overnight in synthetic complete (SC)-uracil medium with 2% (wt/vol) dextrose. Since dextrose traces may interfere with the expression of the protein, yeast cultures were incubated in SC-uracil medium supplemented with 2% (wt/vol) raffinose for 24 h prior to a 6-h induction with 2% (wt/vol) galactose. Yeasts were harvested, washed with cold 1× TEEG buffer (50 mM Tris–HCl [pH 7.4], 1 mM EDTA, 1 mM EGTA, and 10% glycerol) and resuspended in 15 ml of the same buffer supplemented with 0.2 M KCl and a protease inhibitor cocktail (Roche Farma SA, Spain). The cells were subjected to one freeze/thaw cycle at 80°C and lysed by vortexing in a glass bead beater at 4°C. Protein extracts were obtained by centrifugation at 15,000 × g for 45 min at 4°C. Yeast extracts were sequentially precipitated with two increasing concentrations of ammonium sulfate (35 and 75%, respectively). The second precipitate supernatant was loaded on a P-11 phosphocellulose column (Whatman International Ltd., England), which was equilibrated following the manufacturer's indications. The LiTopIB protein was eluted at 4°C with a discontinuous gradient of KCl (0.2, 0.4, 0.6, 0.8, and 1 M) in TEEG buffer supplemented with 0.1 mg/ml sodium bisulphite, 0.8 mg/ml sodium bisulfite, and the protease inhibitor cocktail. Active fractions were loaded on a phenyl-Sepharose column (Sigma-Aldrich) with a discontinuous inverse gradient of ammonium sulfate (1, 0.8, 0.6, 0.4, and 0.2 M). Elution was performed with subsequent Centricon (Millipore) concentration.
Supercoiled plasmid DNA TopIB-mediated cleavage assay.
The sensitivity of LiTopIB to indotecan, AM13-55, and CPT (used as a control drug) was assayed by DNA cleavage assays. pBluescript SK(−) phagemid DNA (pSK) was used as a nickable substrate. At least 100 U of purified LiTopIB was incubated with 0.5 μg of pSK DNA in 10 mM Tris-HCl buffer (pH 7.5), 5 mM MgCl2, 5 mM dithiothreitol (DTT), 0.1 mM EDTA, 15 μg/ml bovine serum albumin, and 50 mM KCl. Different drug concentrations in 1% (vol/vol) dimethyl sulfoxide (DMSO) (20-μl reaction volume) were also added. Following incubation for 4 min at 25°C, reactions were then stopped with up to 1% (wt/vol) SDS and incubated in the presence of 1 mg/ml proteinase K for one extra hour at 37°C. Subsequently, samples were extracted with 1 volume of phenol-chloroform and loaded on a 1% agarose gel containing ethidium bromide to a final concentration of 40 pg/μl (17). The gel was run for 16 h at 4 V/cm, and the images of cleavage products were acquired using a G-Box system (Syngene United Kingdom).
DNA relaxation assays.
TopIB activity was assayed by the relaxation of negatively supercoiled plasmid DNA. One unit of recombinant LiTopIB was incubated with the corresponding drug for 15 min at 4°C. Then, the reaction mixture, containing 0.5 μg of supercoiled pSK DNA, 10 mM Tris-HCl buffer (pH 7.5), 5 mM MgCl2, 0.1 mM EDTA, 15 μg/ml bovine serum albumin, and 50 mM KCl, in a total volume of 20 μl, was added. Reaction mixtures were incubated for 4 min at 25°C. Enzyme reactions were stopped by the addition of up to 1% (wt/vol) SDS (final concentration) and digested with 1 mg/ml proteinase K at 37°C during one extra hour to remove the protein that remained linked to DNA fragments. The extent of plasmid DNA relaxation was assessed in 1% agarose gels by electrophoresis in 0.1 M Tris borate EDTA buffer (pH 8.0) at 2 V/cm for 16 h. Gels were visualized with UV illumination after ethidium bromide (0.5 μg/ml) staining. A further electrophoresis was run in the presence of 0.1 μg/ml ethidium bromide in order to separate nicked DNA from relaxed topoisomers (33).
SDS-KCl precipitation assay.
For indenoisoquinoline-induced protein-DNA complex determination, L. infantum promastigotes, previously labeled for 24 h with 0.5 μCi/ml [2-14C]thymidine, were exposed to different concentrations of CPT and indenoisoquinolines for 30 min (3). Cells were pelleted and lysed by incubation at 60°C for 10 min in the presence of 1.25% (wt/vol) SDS, 0.4 mg/ml salmon sperm DNA, and 5 mM EDTA. After the addition of 65 mM KCl, the reaction mixture was incubated on ice for 60 min. The precipitate was harvested by filtering through glass fiber paper (GF/C; Brandel Inc., Maryland), prewetted with wash buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA, 100 mM KCl), washed (three times with 4 ml of wash buffer), and air-dried. After spotting 50 μl of the same density of labeled cell suspensions onto filter paper and precipitating nucleic acids using 5% (vol/vol) trichloroacetic acid, total incorporation of radioactivity into DNA was measured by using a liquid scintillation counter (Beckman LS600A). All assays included vehicle drug as a control. The formation of DNA fragments, as a percentage of total labeled DNA, was calculated as follows: [(dpm in SDS-KCl drug − dpm in SDS-KCl solvent)/(dpm total incorporation)] × 100.
Thymidine incorporation assay.
Leishmania infantum promastigotes (10 ml; 5 × 106 cells/ml) were incubated with [2-14C]thymidine (0.5 μCi/ml) in the presence of drug or solvent in thymidine-free complete M199 medium. Five hundred-microliter aliquots were removed after 2, 4, 8, 10, and 24 h and loaded on glass fiber paper (GF/C; Brandel Inc., MarylanD). 14C-labeled-DNA was precipitated using ice-cold 5% (wt/vol) trichloroacetic acid and sequentially washed with PBS, 90% (vol/vol) ethanol, and 70% (vol/vol) ethanol. Radioactivity was quantified by liquid scintillation. CPT was used as a positive control.
Animals.
Female BALB/c mice (ages 4 to 6 weeks) were purchased from Harlan Ibérica Laboratories (Spain). All animal procedures were approved by the University of Leon Subcommittee on Research Animal Care.
Infective-stage metacyclic L. infantum promastigotes were isolated from stationary-phase cultures (6 days old) by negative selection with peanut agglutinin (29). Mice were infected with 107 metacyclic parasites intravenously through the tail vein. Fourteen days later, once every 2 days for 15 days, mice were administered a solution of indotecan or AM13-55 indenoisoquinolines diluted in DMSO (equivalent to a dose of 2.5 mg/kg of body weight per injection) intraperitoneally, for a total of eight doses. Five days after the last treatment, the mice were euthanized, organs were removed, and parasite loads were determined by limiting-dilution assay (34). Briefly, a suspension of 20 mg of either spleen or liver was prepared by grinding the tissue in 1 ml of Schneider's medium (Gibco BRL) containing 20% (vol/vol) heat-inactivated FCS in the presence of streptomycin and penicillin. This suspension was further diluted to reach a final concentration of 1 mg/ml. The parasite burden was determined by the limiting-dilution method after a 10-day period of parasite growth. The parasite burden was expressed as the number of free-living promastigotes per gram of wet tissue and compared to that for the nontreated group.
RESULTS AND DISCUSSION
Recent studies showed that the CPT derivatives topotecan, SN38, and especially gimatecan are powerful leishmanicide agents. In vitro assays demonstrated that they act by trapping both DNA and LiTopIB in reversible ternary complexes, producing precipitable SDS-KCl material when free-living promastigotes are incubated in the presence of these drugs (Prada et al., in press [23]). These compounds had good selectivity indexes when their cytotoxicity on mammalian cells (uninfected splenocytes) was compared to that from isolated ex vivo splenic explants of BALB/c mice infected with L. infantum. However, none of these compounds were more selective than miltefosine, the only effective oral treatment for visceral and cutaneous leishmaniasis (9). The use of a model of mouse splenocytes infected with an L. infantum strain expressing IFP1.4 permits high-throughput screening of compounds under conditions that resemble those found in the animal, including the complete range of immune host cells, infected macrophages, and fibroblasts (28).
Screening of indenoisoquinolinic compounds.
The initial screening of new indenoisoquinolinic compounds was performed on free-living promastigotes: the infrared fluorescent protein 1.4 (IRF1.4)-L. infantum (BCN150) strain. To quantify cytotoxicities of the test compounds, we utilized peritoneal BALB/c mouse macrophages. We first excluded compounds, that fell below an arbitrary cytotoxicity threshold in the peritoneal macrophage cultures (50% inhibitory concentration [IC50] of <10 μM) (data not shown), and after exclusion of nine indenoisoquinolinic compounds (see Fig. S1 in the supplemental material), only two new drugs were kept for further validation studies (Fig. 1).
For these two compounds, antileishmanial activity (50% effective concentration [EC50]) was determined in the ex vivo splenic explant system. Comparison of these EC50s with cytotoxicity (IC50) values in the uninfected ex vivo splenic explant allowed determination of an in vitro selectivity index (SI) (IC50/EC50) (Table 1). As a positive control, we have used both paromomycin and CPT. Paromomycin has demonstrated a good selectivity index in the same system (Prada et al., in press [23]).
Table 1.
IC50 calculation after 48-h period of exposure to selected compounds of L. infantum promastigotes, ex vivo-infected splenocytes, and uninfected splenocytesa
| Drug (reference) | IC50 forb: |
SI48hc | ||
|---|---|---|---|---|
| L. infantum promastigotes | Amastigotes | Uninfected splenocyte culture | ||
| Paromomycin | 42.41 ± 1.65 μM | 9.20 ± 0.01 μM | 15.70 ± 3.54 mM | 1,706.5 |
| Camptothecin (22) | 1.12 ± 0.13 μM | 0.03 ± 0.01 μM | 0.62 ± 0.13 μM | 20.7 |
| Indotecan | 0.10 ± 0.08 μM | 0.10 ± 0.05 μM | 57.16 ± 6.01 μM | 571.6 |
| AM13-55 | 1.02 ± 0.09 μM | 0.10 ± 0.37 μM | 48.37 ± 3.68 μM | 483.7 |
IC50 at 48 h of the compounds selected in freshly uninfected splenocyte culture and SI48h values between this cell line and infected splenocytes with L. infantum amastigotes ex vivo were calculated from the dose-response curves determined in triplicate in separate experiments after performing a nonlinear fitting with the SigmaPlot software program.
Mean ± SD.
Selectivity index; SI48h, IC50 for amastigotes/IC50 for infected splenocytes.
CPT and both indenoisoquinolines (indotecan and AM13-55) were much more effective in preventing the proliferation of promastigotes and development of infection in splenocytes than paromomycin, an aminoglycoside antibiotic used in clinical practice against human leishmaniasis. The IC50 of paromomycin was estimated to be 9.20 μM for mouse ex vivo splenic explants infected with L. infantum, whereas the IC50 was 0.1 μM for indotecan and AM13-55. Only CPT was more effective (IC50 = 0.03 μM) at killing the parasites. To estimate the cytotoxicity on mammalian cells and therefore determine selectivity indexes of the studied compounds, we prepared noninfected ex vivo splenic explant. These cells were exposed to different concentration ranges of the drugs for 48 h, and the viability was determined by the alamarBlue method. Dose-response curves were fitted by nonlinear analysis using the Sigma-Plot statistical software package, showing that the least toxic compound was paromomycin (IC50 = 15.70 mM), followed by indotecan (IC50 = 57.16 μM) and AM13-55 (IC50 = 48.37 μM). Selectivity indexes of each compound were calculated as the ratio between the IC50s with the uninfected explants system versus the IC50s with infected ex vivo splenic explants. Both indenoisoquinolines have very high selectivity indexes, 571.6 for indotecan and 483.7 for AM13-55, which are very much higher than those of CPT-related compounds and miltefosine (23) but less than that of paromomycin (the safest compound tested, with a selectivity index of 1,706.5).
Indotecan and AM13-55 induce TopI-DNA convalent complexes.
Figure 2A shows the induction of DNA cleavage complexes by indotecan and AM13-55 in the presence of TopI as tested in supercoiled plasmid DNA (pSK). Both indotecan and AM13-55 induce DNA cleavage complexes in a pattern similar to that of CPT but with differences in their relative intensities. The indotecan cleavage complex showed the same strong intensity as CPT at 100 μM, although the cleavage complex intensity is almost the same in the range of 0.1 to 10 μM. On the other hand, the AM13-55 cleavage complex has lower intensities than those of CPT, but the pattern resembles that of CPT, being proportional to the drug concentration. We observed a dose-dependent increase in nicked DNA for the three compounds, thus confirming the TopIB-poisoning nature of both indenoisoquinolines (2, 4).
Fig 2.

Indotecan and AM13-55 induce TopI-DNA covalent complexes and inhibit supercoiled DNA relaxation. (A) Supercoiled plasmid DNA TopIB-mediated cleavage assay showing the displacement toward cleavage complexes mediated by both indenoisoquinolines and CPT on LiTopIB. Samples were run on a 1% agarose gel containing ethidium bromide to a final concentration of 40 pg/μl in order to separate supercoiled and relaxed DNA. The results are representative of three independent trials. (B) Inhibition of supercoiled DNA relaxation by indotecan (top gel) and AM13-55 (bottom gel) mediated by human (left lanes)or leishmanial (right lanes) TopIB. Reaction mixtures were incubated at 37°C in a final concentration of 150 μM KCl and then stopped with SDS up to a final concentration of 1% of reaction volume. Products were resolved in a 1% agarose gel and visualized by ethidium bromide staining. The results are representative of three independent trials.
We also studied the potency for inhibiting DNA relaxation activity, comparing the human and leishmanial TopIB enzymes. The LiTopIB enzyme was more sensitive to indotecan and AM13-55 than the human enzyme. Figure 2B (top panel) showed that Leishmania TopIB was already inhibited at 80 nM indotecan. However, 2.5 μM was required to achieve the same effect on the human enzyme. AM13-55 (Fig. 2B, lower panel) was a little bit less efficient, inhibiting Leishmania TopIB at 0.15 μM, while the human enzyme was not inhibited until 2.5 μM.
These results suggest that the indenoisoquinolines indenotecan and AM13-55 at pharmacologically relevant doses are primarily TopI poisons, with DNA cleavage patterns exhibiting similarities and differences from those of CPT, but they are much more effective for Leishmania than for human enzyme.
Induction of TopI-DNA complexes by indotecan and AM13-55 in L. infantum cultures.
To determine whether indotecan and AM13-55 induce TopI cleavage complexes in drug-treated cells, we used the SDS-KCl precipitation assay. IFP1.4-L. infantum (BCN150) promastigotes were exposed for 30 min to different concentrations of CPT (used as a positive control), indotecan, and AM13-55 in a concentration range from 0.1 to 90 μM. After this time, stabilized protein-DNA complexes were quantified by the SDS-KCl precipitation method. Figure 3A shows that CPT produced an increasing amount of SDS-KCl-precipitable complexes that were dependent on the concentration of drug that was added up to a value close to 80% of total labeled DNA. None of the indenoisoquinolines studied had such a potent effect within the same concentration range. Indotecan and AM13-55 clearly showed an increase in the amount of SDS-KCl-precipitable complexes, but unlike CPT, indotecan produced up to 20% labeled DNA at 10 μM. AM13-55 reached up to 40% at the same concentration. These results are higher than those obtained by Bakshi and coworkers with three sets of indenoisoquinolines against trypomastigotes of T. brucei (4). In that case, the indenoisoquinolines tested induced only up to 12% of cleavage complexes from the total labeled DNA.
Fig 3.

Induction of TopI-DNA complexes by indotecan and AM13-55 in L. infantum cultures and DNA synthesis inhibition. SDS-KCl-precipitable enzyme-DNA complexes at increasing concentrations of the drugs under study. (A) CPT, indotecan, and AM13-55 in promastigotes of L. infantum after 30 min of growth in the presence of different concentrations of drugs. Notice that for CPT the x scale is up to 10 μM, whereas in the panels for indotecan and AM13-55, concentrations reach 90 μM. Results are expressed as means ± SE for at least three different experiments carried out in duplicate. (B) Indotecan and AM13-55 increase CPT-induced cleavable complex formation. Leishmania promastigotes were treated with 5 μM indotecan, 5 μM AM13-55, or 10 μM CPT for 30 min or with 5 μM indotecan or AM13-55 for 5 min prior to addition of 10 μM CPT, followed by an additional 25-min incubation; *, P < 0.01; * *, P < 0.05 (paired Student t test). (C) Incorporation of [2-14C]thymidine into DNA of growing promastigotes in the presence of 1 μM CPT (empty dots), 1 μM AM13-55 (empty triangles), 1 μM indotecan (solid triangles), or solvent (solid dots). The amount of labeled DNA is expressed as total cpm per assay at different time points (2, 4, 6, 8, 10, and 24 h). Results are expressed as means ± SE for at least three different experiments carried out in duplicate.
Since indotecan and AM13-55 inhibit DNA relaxation and induce stabilization of DNA cleavage complexes, we performed a competition study between CPT and both indenoisoquinolines in order to evaluate the primary mediator of cell killing in Leishmania. Figure 3B shows that 5 μM concentrations of both tested compounds were not able to prevent CPT-mediated TopIB-DNA stabilization, thus pointing to the poisonous nature of both drugs rather than interaction with DNA.
In order to assess the arresting effect of indotecan and AM13-55 on DNA synthesis, 5 × 106 exponentially growing L. infantum promastigotes were pulsed with 0.5 μCi [2-14C]thymidine in the presence of 1 μM CPT, 1 μM AM13-55, 1 μM indotecan, or solvent (control). After a time lapse of 2, 4, 6, 8, 10, and 24 h, labeled DNA was determined by scintillation counting. All of the TopI poisons arrested DNA synthesis at all time points more than 90% with no significant differences among them (Fig. 3C). These results correlate well with those found by Cushman et al. in 2011 (19). They showed that indenoisoquilones with a large primary amine side chain, like indotecan and AM13-55, do not intercalate into free DNA and do not suppress cleavage complex formation at high concentrations, unlike those with a small primary amine (4, 19).
Indotecan and AM13-55 are used as therapeutic agents in a visceral leishmaniasis murine model of infection with L. infantum.
To evaluate the therapeutic potential of indotecan and AM13-55 in vivo, we performed an experimental infection with a susceptible BALB/c mouse strain with L. infantum promastigotes (wild-type strain). Seven million infective-stage metacyclic promastigotes were administered intravenously by the caudal vein to 15 healthy mice that were 4 to 6 weeks old. By day 15 post-parasite inoculation, mice were divided into three groups of five animals each. Mice were treated with 0.5-ml solutions of indotecan or AM13-55 in DMSO-saline, equivalent to 2.5 mg/kg body weight per injection, intraperitoneally every 2 days for 15 days (total, eight doses). As a control, indenoisoquinoline vehicle was administered.
Five days after the last administration, animals were killed by cervical dislocation and spleens and livers were aseptically dissected to determine the parasite load. This was determined by the limiting-dilution assay. Each treatment group was compared to the control, which had received only the vehicle in which the drug had been dissolved.
Figure 4A shows the parasite burden in the group of mice treated with 2.5 mg/kg of indotecan in spleen (upper panel) and liver (lower panel). After the administration schedule was completed, a drastic reduction (P < 0.001) of the number of transforming amastigotes recovered from the target organs of drug-treated animals was observed. On the other hand, mice treated with the same dose of AM13-55 (Fig. 4B) showed that only the spleen was efficiently cleared of infecting parasites, unlike the liver, which retained the pathogen load at levels similar to those for the untreated group (Fig. 4B). In a subsequent trial, 5.0 mg/kg of body weight of AM13-55 was administered under the same conditions as in previous experiments (data not shown). The parasite load was reduced more than 90% in the spleen but not in the liver, where the load remained unchanged. The resistance of liver macrophages to killing the parasites may be due to metabolic transformations of the parent compound in the hepatic parenchyma on TopIB-inactive by-products.
Fig 4.

Indenoisoquinolines cleared tissue parasitic burdens in BALB/c mice experimentally infected with L. infantum. Three groups of five animals each were challenged with 107 metacyclic promastigotes administered via the caudal vein. Fifteen days after infection, animals were injected intraperitoneally every 2 days for a total of 15 days (eight doses total) with a dose of 2.5 mg/kg body weight per injection of indotecan (A), AM13-55 (B), or the corresponding vehicle (a solution of DMSO in sterilized saline solution). Mice were killed 5 days after the last treatment, and the spleens and livers were aseptically removed, weighted, and homogenized in medium supplemented with 20% (vol/vol) FBS. After 10 days, transforming promastigotes were counted, and the limiting dilution was considered to determine the parasitic burden of each organ. The results are representative of two independent trials; statistical differences were observed between groups; *, P < 0.001 using the paired Student t test.
Despite the fact that no definitive cure was achieved with indotecan, our results are very similar to those found with 20 mg/kg body weight of paromomycin alone or in combination with 200-mg/kg-body-weight glucantime in experimental infections of BALB/c visceral leishmaniasis (14). Furthermore, both indotecan and AM13-55 were more effective than CPT (2.5 mg/kg body weight), both free and liposome encapsulated, in a murine model of L. donovani leishmaniasis. The authors of that study found that parasite loads in livers and spleens were reduced by just 55% on average when infected mice were treated with this TopI poison (24).
In conclusion, we have proven that the two indenoisoquinolines analyzed have strong leishmanicidal activities both in vitro (ex vivo splenic explant cultures) and in vivo (visceral leishmaniosis murine model). These compounds act by stabilizing the DNA-LiTopIB cleavage complexes and inhibiting the intrinsic relaxation activity of human and leishmanial TopIB enzymes. Indotecan has a very high selectivity index with respect to host cells and depleted the parasitic burden of the spleen and liver. These facts make this compound an exceptional candidate for its development as a new leishmanicidal drug with a better therapeutic profile than others currently in use.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by Ministerio de Ciencia y Tecnología (grants AGL2009-11935/GAN and AGL2010-16078/GAN), Instituto de Salud Carlos III (grant PI09/0448 and the Network of Tropical Diseases RICET RD06/0021/1004), and Junta de Castilla y León (grant Gr238). R.A.-V., C.F.P., and E.C.-A. are predoctoral fellows through RICET (ISCIII), Junta de Castilla y León (ESF; European Social Founding), and the University of León, respectively. This work was also made possible by the National Institutes of Health (NIH) through support with research grant U01 CA89566.
Footnotes
Published ahead of print 30 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Aït-Oudhia K, Gazanion E, Vergnes B, Oury B, Sereno D. 2011. Leishmania antimony resistance: what we know what we can learn from the field. Parasitol. Res. 109:1225–1232 [DOI] [PubMed] [Google Scholar]
- 2. Antony S, et al. 2005. Cellular topoisomerase I inhibition and antiproliferative activity by MJ-III-65 (NSC 706744), an indenoisoquinoline topoisomerase I poison. Mol. Pharmacol. 67:523–530 [DOI] [PubMed] [Google Scholar]
- 3. Bakshi RP, Shapiro TA. 2003. DNA topoisomerases as targets for antiprotozoal therapy. Mini Rev. Med. Chem. 3:597–608 [DOI] [PubMed] [Google Scholar]
- 4. Bakshi RP, Sang D, Morrell A, Cushman M, Shapiro TA. 2009. Activity of indenoisoquinolines against African trypanosomes. Antimicrob. Agents Chemother. 53:123–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balaña-Fouce R, Garcia-Estrada C, Perez-Pertejo Y, Reguera RM. 2008. Gene disruption of the DNA topoisomerase IB small subunit induces a non-viable phenotype in the hemoflagellate Leishmania major. BMC Microbiol. 8:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balaña-Fouce R, Redondo CM, Pérez-Pertejo Y, Díaz-González R, Reguera RM. 2006. Targeting atypical trypanosomatid DNA topoisomerase I. Drug Discov. Today 11:733–740 [DOI] [PubMed] [Google Scholar]
- 7. Balaña-Fouce R, Reguera RM, Cubría JC, Ordoñez D. 1998. The pharmacology of leishmaniasis. Gen. Pharmacol. 30:435–443 [DOI] [PubMed] [Google Scholar]
- 8. Bhattacharya SK, et al. 2007. Phase 4 trial of miltefosine for the treatment of Indian visceral leishmaniasis. J. Infect. Dis. 196:591–598 [DOI] [PubMed] [Google Scholar]
- 9. Berman J. 2005. Miltefosine to treat leishmaniasis. Expert Opin. Pharmacother. 6:1381–1388 [DOI] [PubMed] [Google Scholar]
- 10. Bodley AL, Shapiro TA. 1995. Molecular and cytotoxic effects of camptothecin, a topoisomerase I inhibitor, on trypanosomes and Leishmania. Proc. Natl. Acad. Sci. U. S. A. 92:3726–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bodley AL, Chakraborty AK, Xie S, Burri C, Shapiro TA. 2003. An unusual type IB topoisomerase from African trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 100:7539–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Díaz-González R, Pérez-Pertejo Y, Ordóñez D, Balaña-Fouce R, Reguera RM. 2007. Deletion study of DNA topoisomerase IB from Leishmania donovani: searching for a minimal functional heterodimer. PLoS One 2:e1177 doi:10.1371/journal.pone.0001177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Díaz-González R, Pérez-Pertejo Y, Pommier Y, Balaña-Fouce R, Reguera RM. 2008. Mutational study of the “catalytic tetrad” of DNA topoisomerase IB from the hemoflagellate Leishmania donovani: role of Asp-353 and Asn-221 in camptothecin resistance. Biochem. Pharmacol. 76:608–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gangneux JP, Sulahian A, Garin YJ, Derouin F. 1997. Efficacy of aminosidine administered alone or in combination with meglumine antimoniate for the treatment of experimental visceral leishmaniasis caused by Leishmania infantum. J. Antimicrob. Chemother. 40:287–289 [DOI] [PubMed] [Google Scholar]
- 15. Gietz RD, Schiestl RH. 1991. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast 7:253–263 [DOI] [PubMed] [Google Scholar]
- 16. Hotez PJ, et al. 2006. Incorporating a rapid-impact package for neglected tropical diseases with programs for HIV/AIDS, tuberculosis, and malaria. PLoS Med. 3:e102 doi:10.1371/journal.pmed.0030102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsiang YH, Hertzberg R, Hecht S, Liu LF. 1985. Camptothecin induces protein-linked DNA break via mammalian DNA topoisomerase I. J. Biol. Chem. 260:14873–14878 [PubMed] [Google Scholar]
- 18. Kimutai A, Ngure K, Kiprotich Tonui W, Muita Gicheru M, Bonareri Nyamwamu L. 2009. Leishmaniasis in Northern and Western Africa: a review. Afr. J. Infect. Dis. 3:14–25 [Google Scholar]
- 19. Kiselev E, et al. 2011. 7-Azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents. J. Med. Chem. 54:6106–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miró G, Cardoso L, Pennisi MG, Oliva G, Baneth G. 2008. Canine leishmaniosis—new concepts and insights on an expanding zoonosis: part two. Trends Parasitol. 24:371–377 [DOI] [PubMed] [Google Scholar]
- 21. Ossorio V, Travi BL, Rensio AR, Peniche AG, Melby PC. 2011. Identification of small molecule lead compounds for visceral leishmaniasis using novel ex vivo splenic explant model system. Plos Negl. Trop. Dis. 5:e962 doi:10.1371/journal.pntd.0000962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pommier Y. 2006. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6:789–802 [DOI] [PubMed] [Google Scholar]
- 23. Prada CF, et al. Gimatecan and other camptothecin derivatives poison Leishmania DNA-topoisomerase IB leading to a strong leishmanicidal effect. J. Antimicrob. Chemother., in press [DOI] [PubMed] [Google Scholar]
- 24. Proulx ME, Desormeaux A, Marquis JF, Olivier M, Bergeron MG. 2001. Treatment of visceral leishmaniasis with sterically stabilized liposomes containing camptothecin. Antimicrob. Agents Chemother. 45:2623–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prudhomme M. 2003. Rebeccamycin analogues as anti-cancer agents. Eur. J. Med. Chem. 38:123–140 [DOI] [PubMed] [Google Scholar]
- 26. Ready PD. 2010. Leishmaniasis emergence in Europe. Euro Surveill. 15:19505. [PubMed] [Google Scholar]
- 27. Robinson KA, Beverley SM. 2003. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 128:217–228 [DOI] [PubMed] [Google Scholar]
- 28. Sacks D, Anderson C. 2004. Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunol. Rev. 301:225–238 [DOI] [PubMed] [Google Scholar]
- 29. Sacks DL, Perkins PV. 1984. Identification of an infective stage of Leishmania promastigotes. Science 223:1417–1419 [DOI] [PubMed] [Google Scholar]
- 30. Schwartz E, Hatz C, Blum J. 2006. New world cutaneous leishmaniasis in travelers. Lancet Infect. Dis. 6:342–349 [DOI] [PubMed] [Google Scholar]
- 31. Shu X, et al. 2009. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 324:804–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teicher AB. 2008. Next generation topoisomerase I inhibitors: rationale and biomarker strategies. Biochem. Pharm. 75:1262–1271 [DOI] [PubMed] [Google Scholar]
- 33. Thrash C, Voelkel K, DiNardo S, Sternglanz R. 1984. Identification of Saccharomyces cerevisiae mutants deficient in DNA topoisomerase I activity. J. Biol. Chem. 259:1375–1377 [PubMed] [Google Scholar]
- 34. Titus RG, Marchand M, Boon T, Louis JA. 1985. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol. 7:545–555 [DOI] [PubMed] [Google Scholar]
- 35. Villa H, et al. 2003. A novel active DNA topoisomerase I in Leishmania donovani. J. Biol. Chem. 278:3521–3526 [DOI] [PubMed] [Google Scholar]
- 36. Zuma AA, Cavalcanti DP, Maia MC, de Souza W, Motta MC. 2011. Effect of topoisomerase inhibitors and DNA-binding drugs on the cell proliferation and ultrastructure of Trypanosoma cruzi. Int. J. Antimicrob. Agents. 37:449–456 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.