

Abstract
In this study, we introduce a multilocus sequence typing (MLST) scheme, comprised of seven single-copy housekeeping genes, to genetically characterize Trichomonas vaginalis. Sixty-eight historical and recent isolates of T. vaginalis were sampled from the American Type Culture Collection and female patients at area health care facilities, respectively, to assess the usefulness of this typing method. Forty-three polymorphic nucleotide sites, 51 different alleles, and 60 sequence types were distinguished among the 68 isolates, revealing a diverse T. vaginalis population. Moreover, this discriminatory MLST scheme retains the ability to identify epidemiologically linked isolates such as those collected from sexual partners. Population genetic and phylogenetic analyses determined that T. vaginalis population structure is strongly influenced by recombination and is composed of two separate populations that may be nonclonal. MLST is useful for investigating the epidemiology, genetic diversity, and population structure of T. vaginalis.
INTRODUCTION
Trichomonas vaginalis is a protozoan parasite that infects the urogenital tract of men and women, causing trichomoniasis. An estimated 153 million new cases of trichomoniasis occur worldwide each year, making it the most common nonviral, sexually transmitted disease (STD) in the world (52). The annual incidence of trichomoniasis in the United States is estimated to be 3 to 5 million, with a 3.1% prevalence rate among reproductive-aged women (14 to 49 years of age) (61). However, the prevalence of T. vaginalis infection is significantly underestimated because of the high frequency of asymptomatic infections. In women, symptoms of T. vaginalis infection include vaginitis and cervicitis (23, 66). Trichomoniasis is associated with a higher risk for other sexually transmitted infections, as well as adverse pregnancy outcomes such as preterm birth, premature rupture of placental membranes, and low-birth-weight infants (16, 40). Trichomoniasis increases the risk of human immunodeficiency virus (HIV) acquisition and the Centers for Disease Control and Prevention estimates that as much as 20% of HIV transmission in the African American population is attributable to T. vaginalis infection (38, 54, 59). T. vaginalis infection also increases the risk of cervical neoplasia (68). The primary treatment of T. vaginalis infection, metronidazole, a heterocyclic 5-nitroimidazole, is usually very effective and well tolerated.
The growing awareness and appreciation of the serious health sequelae that are associated with T. vaginalis infection, and the widespread prevalence of trichomoniasis worldwide, have brought about the need to understand the genetic diversity of this species. Currently, there is no “gold standard” method for genotyping isolates of T. vaginalis. A number of methods have been used in previous attempts to characterize the genetic diversity and population structure of T. vaginalis including isoenzyme analysis, repetitive sequence hybridization, random amplified polymorphic DNA, restriction fragment length polymorphism (RFLP), sequence polymorphisms in rRNA genes and intergenic regions, pulsed-field gel electrophoresis, and antigenic characterization (3, 17, 26, 43, 49, 50, 58, 60, 64, 65). These methods have variable levels of reproducibility and precision in determining the genetic relatedness of isolates.
Multilocus genotyping has been successfully applied to study populations of bacterial and eukaryotic pathogens (8, 34, 63). Microsatellite markers have been used to characterize the genetic diversity and population structure of several protozoan pathogens, including Leishmania tropica, Trypanosoma cruzi, Trypanosoma brucei gambiense, Plasmodium species, and Toxoplasma gondii (2, 6, 8, 12, 29, 30, 44, 53, 55). Recently, the usefulness of microsatellite makers to assess the genetic diversity and transmission of T. vaginalis has also been demonstrated (10, 11, 46). However, the generally high mutation rate of microsatellite markers can cause homoplasy, which may distort the inferred evolutionary history of a species (19). Homoplasy is less likely to occur when single nucleotide polymorphisms (SNPs) are used. In one study, six single-copy genes, including three surface proteins and three housekeeping genes, were fully sequenced for SNPs and used in population genetics analysis of seven isolates, which demonstrated the feasibility of this approach for T. vaginalis (10).
Multilocus sequence typing (MLST) unambiguously characterizes isolates of microorganisms using internal sequence fragments of approximately seven housekeeping genes (33, 34), which are amplified by PCR and sequenced. Alleles are identified directly from the nucleotide sequences. PCR amplification is sensitive to contaminating DNA, and sequence diversity may lead to the failure of particular primers to amplify gene fragments. Nevertheless, MLST allows for high levels of discrimination between isolates because many alleles can be precisely and directly identified in a single locus (33, 34). Additional advantages of using MLST as a genotyping method are that the DNA sequences can be determined using automated technology and require minimal subjective interpretation of data (34). Moreover, the portability of MLST data allows results from different laboratories to be compared (33, 34). MLST was first utilized to genetically characterize pathogenic bacteria but has now been expanded for use in genetic and population studies of fungi and protozoa (48, 67).
In the present study, we report the development of an MLST scheme to genetically characterize T. vaginalis. We have selected seven housekeeping genes as markers to demonstrate the genetic relatedness of 68 T. vaginalis isolates from several different sampling regimens. We believe that the adoption of MLST as a genotyping method for T. vaginalis could provide the gateway to better understanding of the epidemiology, genetic diversity, and population structure of T. vaginalis.
MATERIALS AND METHODS
T. vaginalis isolates.
Sixteen reference strains of T. vaginalis were obtained from the American Type Culture Collection (ATCC; Manassas, VA). The reference strains were from nine different states in the United States, the United Kingdom, and Austria, and the dates of sample collection range from 1939 to 1986 (see Table 2). Fifty-two isolates were obtained from patients presenting to the STD clinic at the Mississippi State Department of Health (MSDH) or the Emergency Department at the University of Mississippi Medical Center (UMMC) in Jackson, Mississippi, from 2001 to 2010 (Table 2). This set included isolates from African-American women who have had sex with women and participated in a study of STD prevalence (42) at the MSDH STD clinic (W isolates, n = 28), a study of prevalence of HIV coinfection with other STDs among African American HIV-positive, African-American HIV-negative, and Caucasian HIV-negative women (T isolates, n = 21), a metronidazole-allergic patient with persistent trichomoniasis (PVC01B, n = 1), and random culture of urine specimens, regardless of patient symptoms, submitted to the UMMC Emergency Department (E isolates, n = 2). All isolates were from women with the exception of ATCC isolates 30092, 50147, and 50148, whose annotations do not indicate gender. For the 52 female subjects who provided clinical specimens, informed consent was obtained when required by the UMMC Institutional Review Board (IRB) for Human Investigation and the MSDH IRB. Personal identifiers were removed, and the source of the isolates was kept anonymous to protect patient confidentiality.
Table 2.
T. vaginalis isolates, with geographic origin and date of original culture
| Isolate | Common designation | Location | Yr |
|---|---|---|---|
| 30001 | C-1:NIH | MD | 1956 |
| 30092 | 11769 | Denver, CO | 1958 |
| 30186 | 123413 | Nashville, TN | 1964 |
| 30187 | 165307–1 | Denver, CO | 1962 |
| 30188 | RP | United Kingdom | Unknown |
| 30235 | JH 30A#4 | Baltimore, MD | 1963 |
| 30245 | TVC | Northampton, MA | 1966 |
| 30246 | TVC1 | Bethesda, MD | 1956 |
| 30247 | TV 1 | IA | 1939 |
| 50138 | IR 78 | Vienna, Austria | 1978 |
| 50141 | RU 382 | CT | 1983 |
| 50144 | CDC 337 | Columbus, OH | 1983 |
| 50147 | NYH 272 | New York, NY | 1977 |
| 50148 | NYH 286 | New York, NY | 1977 |
| 50167 | B7RC2 | Greenville, NC | 1986 |
| Pra98 | G3 | Beckenham, United Kingdom | 1973 |
| PVC01B | Jackson, MS | 2010 | |
| E-series (n = 2) | Jackson, MS | 2001 | |
| W-series (n = 28) | Jackson, MS | 2009–2010 | |
| T-series (n = 21) ` | Jackson, MS | 2010 |
Initial isolation of clinical specimens from female subjects was performed by vaginal swab and inoculation of the swab into a T. vaginalis InPouch (Biomed Diagnostics, White City, OR) culture. After confirmation of T. vaginalis growth in the InPouch media, T. vaginalis trophozoites were transferred and cultured in modified Trypticase-yeast-maltose (TYM) medium supplemented with 10% heat-inactivated horse serum and antibiotics (50 μg of gentamicin/ml, 40 μg of ciprofloxacin/ml, and 50 μg of miconazole/ml). T. vaginalis trophozoites were grown in TYM medium at 37°C and subcultured at 48 h. T. vaginalis trophozoites were grown to mid-logarithmic phase, ∼106 cells/ml, and harvested by centrifugation at 500 × g for 15 min at 4°C. Axenic parasites at mid-log phase were archived as frozen isolates in 10% dimethyl sulfoxide at −80°C.
DNA extraction.
T. vaginalis trophozoites, at an inoculating concentration of 106 cells/ml, were incubated for 48 h in TYM medium at 37°C and harvested by centrifugation at 500 × g for 15 min at 4°C. Pelleted cells were washed twice with phosphate-buffered saline prior to DNA isolation. Genomic DNA was isolated using the diethyl pyrocarbonate–Triton X-100 method of Riley and Krieger with two additional chloroform extractions to reduce nuclease contamination or a Promega Wizard Genomic DNA purification kit (Promega, Madison, WI) (47).
Choice of loci for MLST.
A publicly available T. vaginalis genome sequence (version 1.2, http://trichdb.org) was screened to identify single-copy housekeeping genes. Genes were verified as single-copy by performing an all-against-all BLASTn comparison of nucleotide sequences (5). A total of 35 loci were considered and 16 loci were further investigated (see Table S1 in the supplemental material). The final MLST scheme contains seven housekeeping loci, shown in Table 1, which were selected based on their ability to be readily amplified and sequenced on both DNA strands and based on their diversity. The assembled genome of T. vaginalis contains many contigs due to its high degree of repetitiveness. Therefore, the absolute chromosomal location of each MLST gene is unknown. Each of these seven loci were located on different contigs of the sequenced genome. The average size of the contigs that contain these seven loci is ∼90 kb, with a range of 2.8 to 164.8 kb.
Table 1.
Oligonucleotide primers used for T. vaginalis MLST
| Locus (abbreviation) | Primer |
Size (bp) |
||
|---|---|---|---|---|
| Orientation | Sequences (5′–3′) | Gene | Amplicon | |
| Tryptophanase (P1)a | Forward | CGTCAACATCGGTGGCTTCA | 1,451 | 489 |
| Reverse | GCGACAGCGACGACATTCAT | |||
| Glutaminase (P3) | Forward | GTGCCATTACAACAGCATCG | 886 | 451 |
| Reverse | CCAAGTATAGCTCCGCTGAC | |||
| Family T2 aparaginase-like threonine peptidase (P6) | Forward | GAACAGGAGCACCAGCAGAA | 990 | 412 |
| Reverse | TCTCTAGCAACGCAGCCAAC | |||
| Alanyl tRNA synthetase (P8) | Forward | TCTGTCCAGGATGGTGTCTT | 3,075 | 494 |
| Reverse | ACGCCTTCCTCCTTCATCTT | |||
| DNA mismatch repair protein (P13)b | Forward | TCATCGGCCAATGGAACCAA | 1,758 | 491 |
| Reverse | TCCGTGCGGACAATTCCAAG | |||
| Serine hydroxymethyltransferase (P14) | Forward | GCTGAGTGACGGTGGACATT | 1,356 | 449 |
| Reverse | GAAGATGAGGTCCTCCTTGA | |||
| Mannose 6-phosphate isomerase (P16) | Forward | AGCCAGTTGGCTTCTGAGTT | 1,128 | 459 |
| Reverse | AACAATTCCGCAAGCTGGAG | |||
PCR amplification and nucleotide sequence determination.
Primers for MLST loci were designed to amplify gene fragments of 450 to 500 bp and are detailed in Table 1 (Primer Design 4, v4.20; Science and Educational Software, Cary, NC). All genes were amplified as follows: 50-μl PCRs contained 0.2 μM concentrations of forward primer, 0.2 μM concentrations of reverse primer, 1× Mg-free, GoTaq Flexi buffer (Promega), 0.2 mM concentrations of each deoxynucleoside triphosphate (Promega), 2.5 mM MgCl2, 1.25 U of GoTaq DNA polymerase, and 0.5 μg of template DNA. Amplifications were performed using the Bio-Rad MyCycler thermal cycler (Bio-Rad Laboratories, Hercules, CA). The thermocycler program conditions were the same for all loci as follows: 95°C for 5 min; followed by 40 cycles of 95°C for 1 min, 55° for 1 min, and 72°C for 1.5 min; followed in turn by 72°C for 10 min. Then, 2 μl of each reaction was visualized on a 1% agarose gel to verify amplification. The remaining 48 μl of PCR product was purified by precipitation in a solution of 2.5 M NaCl and 20% polyethylene glycol 8000, washed with chilled 70% ethanol, air dried, and resuspended in 15 μl of distilled H2O. Both strands of the purified gene fragments were sequenced by SeqWright DNA technology services (Houston, TX), and the sequence data were assembled and edited with DNAStar software (DNAStar, Inc., Madison, WI). Assembled sequences were determined to have highly accurate base calls (Phred-like values >Q30 on average).
Population genetic and phylogenetic analyses.
Allele numbers and sequence types (STs) were assigned to isolates as previously described (34). The discriminatory power of the MLST scheme was measured with Simpson's index of diversity (25), using an online resource (http://darwin.phylovis.net/ComparingPartitions). This index estimates the probability that two randomly selected isolates have different STs. Likewise, the nucleotide diversity of sequences at individual loci was measured with Tajima's estimator (θπ) (62), using DnaSP v5 software (32). The association between alleles at different loci was measured with the standardized index of association (IAS) (27), using LIinkage ANalysis (LIAN) v3.5. Linkage equilibrium is defined as the random association of alleles at different loci, which is an indicator of frequent recombination, whereas linkage disequilibrium is the nonrandom association of alleles at different loci, which can indicate asexual (clonal) propagation, as well as other processes (57). When linkage equilibrium exists, IAS is 0, and when linkage disequilibrium exists, IAS differs significantly from 0 based on statistical testing.
Phylogenetic relationships between STs were initially inferred from an unrooted neighbor-joining tree, constructed with PAUP*4.0b10 (Sinauer Associates, Sunderland, MA) from a matrix of p-distances between the concatenated nucleotide sequences. Statistical support for nodes within the tree was estimated by bootstrapping with 1,000 resampled data sets. The global optimal eBURST algorithm (goeBURST, http://goeburst.phyloviz.net) was used to identify groups of closely related STs and to infer a founding ST within each group (24). Bayesian population assignments were performed using STRUCTURE v2.3 (21) and BAPS v5.4 (14). Both programs perform model-based analyses to assign isolates to populations, and both programs can make use of MLST data. For STRUCTURE, the admixture model with correlated and uncorrelated allele frequencies was used with all 68 isolates and with single isolates of each of the 60 STs. For each number of populations assumed (K = 1 to 11), five runs of 2 × 105 iterations were done, discarding the first half as burn-in. The ΔK method (20) was used to select an optimal number of populations, based on the rate of change in the log probability of the data between consecutive K values. For BAPS, the codon linkage model was used with an upper bound of K = 11 populations. BAPS was also used to test for admixture using the mixture clustering option. Five runs each of 100 iterations were performed, with all other settings in default.
A variety of methods were used to test for recombination in the MLST data. First, a count-based method was used in which the alleles of the founding STs of groups and subgroups, identified by goeBURST, were compared to the differing alleles in any single locus variants of the founding STs. This method determines whether changes in STs are due to de novo point mutations or recombination events (22). Second, the Bayesian method implemented by ClonalFrame v1.2 (18) was used to attempt to identify mutation and recombination events, but these results are not discussed because the Markov chains did not achieve satisfactory mixing. In addition, the Recombination Detection Program (RDP3 v4.4) (37) was used to identify recombination breakpoints within the MLST gene fragments using four different methods: RDP, GENECONV, MAXCHI, and CHIMAERA (36, 45, 51, 56). Recombination events were scored if they were detected based on three of the four methods. Finally, the pairwise homoplasy index test, a test that uses the notion of phylogenetic incompatibility between informative sites to detect recombination, was performed using SplitsTree4 (28).
Nucleotide sequences.
Nucleotide sequence data of alleles are available in the GenBank database under accession numbers JX239643 to JX239693. The T. vaginalis MLST database will be housed at www.mlst.net (1).
RESULTS
Selection of MLST Loci.
T. vaginalis is a haploid species with a highly repetitive genome; over 66% of the genome is repeated, and 59 repeat families have been identified to date (9, 69). The genome sequence of reference strain G3 (ATCC Pra98 [http://trichdb.org/trichdb/]) was mined for single-copy housekeeping genes. Sixteen gene fragments were PCR amplified and sequenced for a subset of 12 isolates. The seven genes ultimately selected for MLST and the primers used to amplify fragments of these genes are shown in Table 1. A total of 68 isolates were characterized using this seven-gene MLST scheme and are listed in Table 2, along with their geographic origin and year of initial isolation. All sequences obtained for a single locus could be aligned without gaps. Moreover, amplification of the selected gene fragments resulted in single sequences, which further verified that the selected loci were present in single copy in the T. vaginalis genome.
Sequence diversity and discriminatory power of the MLST scheme.
A total of 2,542 nucleotides from seven loci were sequenced for each isolate. Forty-three polymorphic nucleotide sites (1.7%) were detected among the 68 isolates. The number of polymorphic sites for each locus ranged from 1 to 12 (Table 3). The nucleotide diversity for these loci, calculated based on pairwise differences (θπ), averages 0.0035 differences/site (Table 3). Each distinct nucleotide sequence in a locus is defined by MLST nomenclature (33, 34) as a different allele, and the alleles were assigned numbers in the order of their identification. The number of different alleles for each locus ranged from 2 to 17 (Table 3). Each isolate was assigned an allelic profile or ST based on the alleles located at each of the seven gene loci in that isolate. Sixty STs were distinguished among the 68 isolates. Of the STs represented by multiple isolates, ST34 was represented by three isolates (T132, W024, and W029), and STs 17, 32, 37, 38, 39 and 44 were each represented by two isolates (T098 and W148, W049 and W055, W032 and W052, W046 and W047, W060B and W096, and 30001 and 30246, respectively). Each of these STs represented by multiple isolates was comprised of isolates from close geographic sites: W isolates and T isolates from Jackson, MS, and ATCC isolates 30001 and 30246 from Bethesda, MD. The remaining 53 STs (88%) were represented by single isolates. Among these 53 STs, 24 pairs of single-locus variants were identified. The discriminatory power of this MLST scheme is 0.996 (95% confidence interval = 0.992 to 1.000) as defined by Simpson's index. Therefore, this scheme has sufficient ability to distinguish between isolates.
Table 3.
Characteristics of housekeeping loci used for T. vaginalis MLST
| Locus (abbreviation) | Sequence length (bp) | No. of SNPs | No. of alleles | θπa | No. of changes |
|
|---|---|---|---|---|---|---|
| Synonymous | Nonsynonymous | |||||
| Tryptophanase (P1) | 351 | 3 | 5 | 0.0017 | 2 | 1 |
| Glutaminase (P3) | 285 | 8 | 5 | 0.0093 | 3 | 5 |
| Family T2 aparaginase-like threonine peptidase (P6) | 318 | 10 | 6 | 0.0037 | 4 | 6 |
| Alanyl tRNA synthetase (P8) | 430 | 12 | 17 | 0.0042 | 5 | 7 |
| DNA mismatch repair protein (P13) | 447 | 6 | 12 | 0.0033 | 2 | 4 |
| Serine hydroxymethyltransferase (P14) | 339 | 1 | 2 | 0.0011 | 1 | 0 |
| Mannose 6-phosphate isomerase (P16) | 372 | 3 | 4 | 0.0014 | 1 | 2 |
Nucleotide diversity per site.
Molecular epidemiological uses of the MLST scheme.
Twenty-eight isolates (W series) were collected as part of a study investigating the prevalence of STDs among African-American women who have sex with women (42). Isolates from two pairs of sexual partners were typed. Isolates W029 and W032 were obtained from one couple, and isolates W046 andW047 were obtained from a second couple (41). Isolates W046 and W047 both represented ST38, and isolates W029 and W032 represented two different STs (ST34 and ST37, respectively). ST34 is identical to ST37 at five of the seven sequenced loci. These data demonstrate that, along with its high discriminatory power, this MLST scheme has the ability to genetically link isolates that are epidemiologically linked.
As described above, several other STs were represented by two to three isolates that were geographically and temporally linked with isolation occurring in Jackson, MS from 2009 to 2010. Two of these STs were represented by multiple isolates from both the W and T series (ST34 and ST17). The observation that isolates from these two studies included some of the same STs suggests that this MLST scheme may be useful for studying transmission of T. vaginalis in broader sexual networks.
Linkage disequilibrium and recombination.
The level of multilocus linkage disequilibrium was assessed using LIAN and results are shown in Table 4. For the entire population of T. vaginalis isolates, the standardized index of association (IAS) was 0.0230, which was significantly greater than zero (P < 0.05). However, when each ST was treated as a single isolate, thus removing redundancy due to indistinguishable clones, the IAS was 0.0135, which was not statistically different from zero (P > 0.05). Therefore, the signal of linkage disequilibrium is lost when redundant isolates are removed from the analysis. These data suggest that T vaginalis is not a highly clonal species.
Table 4.
Analysis of multilocus linkage disequilibrium
| Data set (n) | Variance in allelic mismatches |
IAS |
P (test that IAS = 0) |
||
|---|---|---|---|---|---|
| Observed | Expected | Monte Carlo | Parametric | ||
| All isolates (68) | 1.6795 | 1.4761 | 0.0230 | 0.022 | 0.00128 |
| STs only (60) | 1.5936 | 1.4739 | 0.0135 | 0.113 | 0.077 |
To test for recombination in this population, several recombination detection methods were utilized. A count-based method compared each of the founding STs, identified by goeBURST, with their single locus variants (SLVs) within clonal complexes (Fig. 1). This analysis indicated that the founding STs diversify 10 times more frequently by recombination than by mutation (10:1 based on 11 data points). However, the RDP3 and SplitsTree programs detected no recombination breakpoints within the MLST gene fragments. This finding is reflected by the relatively even distribution (i.e., nonclustering) of polymorphic nucleotide sites within the MLST gene fragments. When the concatenated nucleotide sequences were analyzed with the Phi test, evidence of recombination was detected (P = 0.03). Thus, recombination events in T. vaginalis may tend to break in between these MLST gene fragments and replace genomic fragments larger than these MLST gene fragments.
Fig 1.
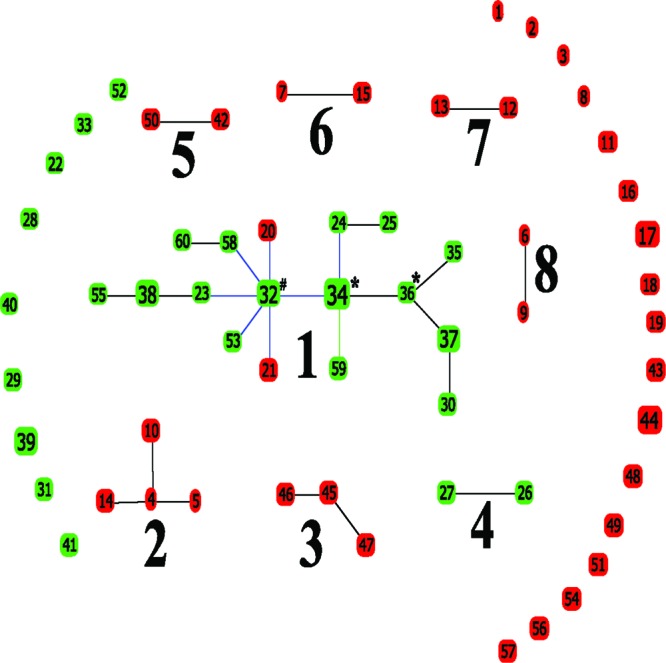
goeBURST analysis of 68 isolates representing 60 STs. goeBURST analysis revealed eight clonal complexes and 26 singleton STs within the sample population. Green ST nodes clustered to clade A in the neighbor-joining phylogram (NJ); red ST nodes clustered to clade B in the NJ phylogram. Large, bold numbers identify CCs, small numbers indicate ST nodes. Symbols: #, group founder; *, subgroup founder. Colored links: black, link drawn without recourse to tiebreak rules; blue, link drawn using tiebreak rule 1; and green, link drawn using tiebreak rule 2 (24).
Population structure.
The goeBURST analysis of all isolates revealed eight clonal complexes (CCs) of two or more single locus variants (Fig. 1). The temporal stability of T. vaginalis CCs is apparent as older ATCC strains from 1964 (30186, ST10), 1968 (30245, ST55), 1977 (50147, ST15), and 1978 (50138, ST27) are present in CCs 1, 2, 4, and 6 along with more recent isolates from 2001 to 2010. CCs 1, 2, 4, 6, and 8 each contain isolates from Jackson, MS, and isolates from elsewhere in the United States (isolates 50147 [ST15] from New York in CC6, 30245 [ST55] from Massachusetts in CC1, and 30186 [ST10] from Tennessee in CC2) and from overseas (isolates 50138 [ST27] from Austria in CC4 and 30188 [ST9] from the United Kingdom in CC8).
Trees of individual MLST gene fragments displayed two major clades in six of the MLST loci. Significant bootstrap support warranted separation of the two clades in loci P3 and P6 (98.8 and 85.7%, respectively). Modest bootstrap support was found for the two clades in loci P14 and P16 (63.5 and 63.9%, respectively). Bootstrap support for the two clades in P1, P8, and P13 was negligible (<40%). P8 contained the greatest number of alleles (17) and displayed an increased number of small groups of isolates. The two major clades identified by the individual trees had similar distributions of STs into clades with particular STs clustering together in a single clade.
A neighbor-joining tree of concatenated nucleotide sequences also displayed two major clades with modest bootstrap support (see Fig. S1 in the supplemental material). CCs 1 and 4, with the exception of two divergent isolates from CC1 (STs 20 and 21), clustered into clade A, and CCs 2, 3, 5, 6, 7, and 8 clustered into clade B of the concatenated sequence tree (Fig. 1 and see Fig. S1 in the supplemental material). Recombination at the glutaminase (P3) locus is a possible explanation for the clustering of STs 20 and 21 outside of clade A along the path that defines the two separate clades; these two STs are single-locus variants (at the same locus) of the inferred founder of CC1. We note that Pra98, the isolate used in the genomic sequencing of T. vaginalis, grouped into clade B and that both clades A and B contain local, national, and international isolates.
Population assignments were made using the model-based approaches implemented in STRUCTURE (21) and BAPS (14). Two populations (K = 2) were determined to be optimal based on analysis with STRUCTURE (see Fig. S2 in the supplemental material). STs were assigned to a population if they had at least 85% ancestry from that population. Only four STs (ST20, ST21, ST41, and ST57) were not assigned to a particular population based on this criterion (Fig. 2A, asterisks). These same four STs are along the path that defines the two clades in the neighbor-joining tree (see Fig. S1 in the supplemental material). BAPS analysis also determined that two populations (K = 2) was optimal with these MLST data. Furthermore, BAPS consistently determined that significant admixture had occurred in STs 20, 21, and 57 (Fig. 2B, asterisks). The two populations identified by STRUCTURE and BAPS are identical to clades A and B, respectively, from the neighbor-joining tree and are present at nearly equal frequencies in the sample, with 26 STs represented by 32 isolates in clade A and 34 STs represented by 36 isolates in clade B. Further subdivisions within these clades/populations could not be resolved with these MLST data. When population structure is taken into account by measuring IAS in the two separate populations, no multilocus linkage disequilibrium is evident, suggesting that either free recombination occurs within the separate populations or that the power to reject the null hypothesis of panmixia is reduced with the smaller sample sizes of the separate populations.
Fig 2.
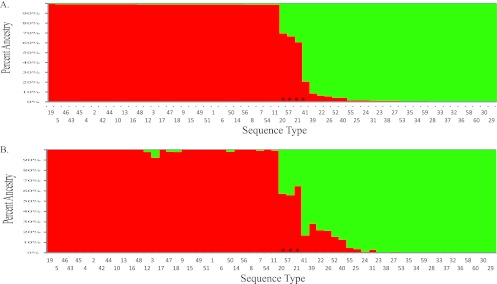
Two-cluster population structure of T. vaginalis. Assignment of STs into two populations by STRUCTURE (A) and BAPS (B) was performed. The order of STs is the same in both graphs, and they are sorted by the percent ancestry into clade B (red) based on STRUCTURE analysis. The green cluster (n = 26 STs) and red cluster (n = 34 STs) are identical to the clades identified in the neighbor-joining tree. Asterisks indicate STs unassigned to populations (in panel A) and STs with significant admixture (in panel B).
DISCUSSION
We have introduced the first MLST markers for investigating the epidemiology, genetic diversity, and population structure of Trichomonas vaginalis. A number of different typing methods, with various levels of reproducibility and precision, have been used in previous attempts to characterize the genetic diversity and population structure of T. vaginalis. Single-copy genes were identified using the publicly available draft genome sequence of T. vaginalis (9). With two-thirds of the genome comprised of repeats and transposable elements, the identification and verification of single-copy loci was imperative to ensure the usefulness of the MLST scheme (9). We performed BLASTn analysis to verify the copy number of the selected loci, analyzed the sequence diversity at each locus, and examined the discriminatory power of this MLST scheme to distinguish between isolates. Moreover, a variety of population genetic and phylogenetic analyses were performed in order to understand the diversity and the coarse-level population structure of the species. A limitation of the present study is the lack of isolates from male patients. It is important to note that isolates from men could yield different MLST results. Therefore, further MLST studies are needed to compare isolates from men and women.
In the present study, 68 recent and historical isolates of T. vaginalis were sampled from the ATCC and female patients at local health care facilities This MLST scheme revealed a high level of genetic diversity in the T. vaginalis population. These data are in agreement with other genetic typing methods used by our laboratory and others (10, 15, 39, 50, 60). Although the average nucleotide diversity of 0.0035 differences/site is lower than some pathogenic bacteria (13), it is higher than that of its human hosts (31). The ST diversity was sufficiently high; each characterized locus was comprised of more than one allele and the discriminatory power of 0.996 clearly demonstrates the ability of this MLST scheme to distinguish between isolates.
Sixty STs were distinguished among the 68 isolates. Isolates W046 and W047, representing ST38, also displayed identical EcoRI-restricted Hsp70 RFLP patterns (D. C. Cornelius, C. A. Muzny, L. A. Mena, and J. C. Meade, unpublished data). These isolates were obtained from women in a sexual partnership with each other (41), thus providing a plausible explanation for these isolates to share an ST. Likewise, two reference isolates (30001 and 30246) that are geographically and temporally linked (obtained in Maryland in 1956) represented ST44. These results indicate that shorter-term clonality is detectable with this MLST scheme. Thus, in addition to possessing high discriminatory power and demonstrating the genetic diversity of the T. vaginalis population, this MLST scheme is also able to identify epidemiologically linked isolates.
A second set of isolates, W029 and W032 were also obtained from two women in a sexual partnership with each other. However, these isolates represented two different STs, ST34 and ST37, respectively, which differed at two of the seven MLST loci but are in the same CC. The allelic differences between these two STs could be a result of in vivo recombinations with other strains, acquisition of a different strain by one of the women outside of this particular sexual relationship, or a mixed infection in which a different isolate in the infection was selected during laboratory cultivation of the samples from each woman. Mixed infections can occur in the host at a frequency of approximately 11% (11). MLST could be useful in detecting mixed infections, if the isolates are all relatively frequent in the host and one particular isolate is not selected during culture. The sequence traces (electropherograms) of a single-copy gene should indicate the presence of multiple alleles by the presence of multiple nucleotide peaks. However, there was no evidence of mixed infections in any of the isolates tested here.
Multilocus linkage disequilibrium was measured using standard methods (27, 57). Statistically significant linkage disequilibrium was detectable only when performing analysis on all 68 isolates, in which some STs were represented by more than one isolate. When each ST was analyzed as a single isolate, the signal of linkage disequilibrium disappeared. This suggests that the T. vaginalis population is not highly clonal; however, some coarse-level population structure is clearly identified with these MLST data. Analysis of multilocus linkage disequilibrium within the two separate populations did not detect linkage disequilibrium, suggesting the possibility of free recombination within the separate clusters; however, larger samples of each population are needed to test this hypothesis. Consistent with a role for recombination in T. vaginalis population structure, a count-based method that is capable of detecting recombination events indicated that the founding STs change 10 times more often by recombination than by mutation. The RDP3 and SplitsTree programs were unable to identify recombination breakpoints within the MLST gene fragments. The Phi test for recombination did detect evidence of recombination when analyzing the concatenated nucleotide sequences. In summary, these data suggest that recombination may occur between the MLST gene fragments and replace genome fragments larger than the MLST gene fragments. A recent study also detected recombination events between three single-copy genes in T. vaginalis (11). Meiotic gene homologs have been identified in T. vaginalis, indicating that the species possesses the near-universal machinery to perform meiotic recombination (35). Furthermore, evidence of horizontal gene transfer has been presented for T. vaginalis and other protist species (4, 7). The MLST data suggest that recombination makes an important contribution to the evolution of T. vaginalis. Increased knowledge of recombination in this species may improve our ability to use patterns of linkage disequilibrium to identify loci responsible for clinically relevant phenotypes.
The population genetic and phylogenetic analyses revealed that the coarse-level population structure detected with these MLST data consists of two clades/populations. A neighbor-joining tree subdivided the isolates into two distinct clades (see Fig. S1 in the supplemental material). Bayesian population assignments also determined that the MLST data are best represented by two populations (Fig. 2). Moreover, the two clades and populations identified by these different methods were identical to each other. These results are in agreement with previous results obtained in our laboratory using RFLP and with microsatellite results obtained by other laboratories (10, 11, 15, 39). The microsatellite studies indicated that the two populations were almost always present in nearly equal frequencies worldwide (11). Our MLST data demonstrate a similar pattern of distribution of the two populations within the Jackson, MS area with 26 STs (32 isolates) in population A and 34 STs (36 isolates) in population B. Recombination between the two populations may be a rare occurrence as only a few STs had enough admixture to obscure their population of origin.
The MLST scheme developed here for T. vaginalis has several applications, including the rational selection of isolates for vaccine design and a population-based approach for the identification of genetic factors that regulate the clinical manifestations of trichomoniasis. Classification of T. vaginalis isolates into distinct populations presents the opportunity for further molecular epidemiological analysis of this species. The two population clusters identified in this species may be differentially associated with various outcomes and health sequelae that accompany trichomoniasis. Furthermore, MLST studies could reveal differences in the genetic backgrounds of unusually virulent and antimicrobial-resistant clones. There is currently no “gold standard” method for genetic characterization of T. vaginalis, and the use of MLST may provide a portable, precise, and unambiguous method for this purpose.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Mississippi Institute for Improvement of Geographic Minority Health grant 1 CPI MP061018-03 from the National Institutes of Health and the Intramural Research Support Program at the University of Mississippi Medical Center.
We thank Shernica Ferguson, Xiao Luo, LaKesha Smith, Jonathan Thomas, Antrice Walker, and Liangfen Zhang for technical support.
Footnotes
Published ahead of print 1 August 2012
Supplemental material for this article may be found at http://jcm.asm.org/.
REFERENCES
- 1. Aanensen DM, Spratt BG. 2005. The multilocus sequence typing network: mlst.net. Nucleic Acids Res. 33:W728–W733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ajzenberg D, Banuls AL, Tibayrenc M, Darde ML. 2002. Microsatellite analysis of Toxoplasma gondii shows considerable polymorphism structured into two main clonal groups. Int. J. Parasitol. 32:27–38 [DOI] [PubMed] [Google Scholar]
- 3. Alderete JF, et al. 1987. Phenotypes and protein-epitope phenotypic variation among fresh isolates of Trichomonas vaginalis. Infect. Immun. 55:1037–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alsmark UC, Sicheritz-Ponten T, Foster PG, Hirt RP, Embley TM. 2009. Horizontal gene transfer in eukaryotic parasites: a case study of Entamoeba histolytica and Trichomonas vaginalis. Methods Mol. Biol. 532:489–500 [DOI] [PubMed] [Google Scholar]
- 5. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 6. Anderson TJ, et al. 2000. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol. Biol. Evol. 17:1467–1482 [DOI] [PubMed] [Google Scholar]
- 7. Andersson JO, Hirt RP, Foster PG, Roger AJ. 2006. Evolution of four gene families with patchy phylogenetic distributions: influx of genes into protist genomes. BMC Evol. Biol. 6:27 doi:10.1186/1471-218-6-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barker GC. 2002. Microsatellite DNA: a tool for population genetic analysis. Trans. R. Soc. Trop. Med. Hyg. 96(Suppl 1):S21–S4 [DOI] [PubMed] [Google Scholar]
- 9. Carlton JM, et al. 2007. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315:207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conrad M, et al. 2011. Microsatellite polymorphism in the sexually transmitted human pathogen Trichomonas vaginalis indicates a genetically diverse parasite. Mol. Biochem. Parasitol. 175:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conrad MD, et al. 2012. Extensive genetic diversity, unique population structure, and evidence of genetic exchange in the sexually transmitted parasite Trichomonas vaginalis. PLoS Negl. Trop. Dis. 6:e1573 doi:10.1371/journal.pntd.0001573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper A, et al. 2008. Genetic analysis of the human infective trypanosome Trypanosoma brucei gambiense: chromosomal segregation, crossing over, and the construction of a genetic map. Genome Biol. 9:R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cooper JE, Feil EJ. 2006. The phylogeny of Staphylococcus aureus: which genes make the best intra-species markers? Microbiology 152:1297–1305 [DOI] [PubMed] [Google Scholar]
- 14. Corander J, Marttinen P, Siren J, Tang J. 2008. Enhanced Bayesian modeling in BAPS software for learning genetic structures of populations. BMC Bioinform. 9:539 doi:10.1186/1471-2105-9-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cornelius DC, Mena L, Lushbaugh WB, Meade JC. 2010. Genetic relatedness of Trichomonas vaginalis reference and clinical isolates. Am. J. Trop. Med. Hyg. 83:1283–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cotch MF, et al. 1997. Trichomonas vaginalis associated with low birth weight and preterm delivery. Sex. Transm. Dis. 24:353–360 [DOI] [PubMed] [Google Scholar]
- 17. Crucitti T, Abdellati S, Van Dyck E, Buve A. 2008. Molecular typing of the actin gene of Trichomonas vaginalis isolates by PCR-restriction fragment length polymorphism. Clin. Microbiol. Infect. 14:844–852 [DOI] [PubMed] [Google Scholar]
- 18. Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Estoup A, Jarne P, Cornuet JM. 2002. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 11:1591–1604 [DOI] [PubMed] [Google Scholar]
- 20. Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14:2611–2620 [DOI] [PubMed] [Google Scholar]
- 21. Falush D, Stephens M, Pritchard JK. 2007. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol. Ecol. Notes. 7:574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feil EJ, Enright MC, Spratt BG. 2000. Estimating the relative contributions of mutation and recombination to clonal diversification: a comparison between Neisseria meningitidis and Streptococcus pneumoniae. Res. Microbiol. 151:465–469 [DOI] [PubMed] [Google Scholar]
- 23. Fichorova RN. 2009. Impact of Trichomonas vaginalis infection on innate immune responses and reproductive outcome. J. Reprod. Immunol. 83:185–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Francisco AP, Bugalho M, Ramirez M, Carrico JA. 2009. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 10:152 doi:10.1186/1471-2105-10-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grundmann H, Hori S, Tanner G. 2001. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J. Clin. Microbiol. 39:4190–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hampl V, Vanacova S, Kulda J, Flegr J. 2001. Concordance between genetic relatedness and phenotypic similarities of Trichomonas vaginalis strains. BMC Evol. Biol. 1:11 doi:10.1186/1471-2148-1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haubold B, Hudson RR. 2000. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage Anal. Bioinform. 16:847–848 [DOI] [PubMed] [Google Scholar]
- 28. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267 [DOI] [PubMed] [Google Scholar]
- 29. Imwong M, et al. 2007. Contrasting genetic structure in Plasmodium vivax populations from Asia and South America. Int. J. Parasitol. 37:1013–1022 [DOI] [PubMed] [Google Scholar]
- 30. Imwong M, et al. 2006. Microsatellite variation, repeat array length, and population history of Plasmodium vivax. Mol. Biol. Evol. 23:1016–1018 [DOI] [PubMed] [Google Scholar]
- 31. Li WH, Sadler LA. 1991. Low nucleotide diversity in man. Genetics 129:513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452 [DOI] [PubMed] [Google Scholar]
- 33. Maiden MC. 2006. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 60:561–588 [DOI] [PubMed] [Google Scholar]
- 34. Maiden MC, et al. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95:3140–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Malik SB, Pightling AW, Stefaniak LM, Schurko AM, Logsdon JM., Jr 2008. An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. PLoS One 3:e2879 doi:10.1371/journal.pone.0002879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin D, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16:562–563 [DOI] [PubMed] [Google Scholar]
- 37. Martin DP, et al. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McClelland RS, et al. 2007. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J. Infect. Dis. 195:698–702 [DOI] [PubMed] [Google Scholar]
- 39. Meade JC, et al. 2009. Genetic diversity of Trichomonas vaginalis clinical isolates determined by EcoRI restriction fragment length polymorphism of heat-shock protein 70 genes. Am. J. Trop. Med. Hyg. 80:245–251 [PMC free article] [PubMed] [Google Scholar]
- 40. Minkoff H, et al. 1984. Risk factors for prematurity and premature rupture of membranes: a prospective study of the vaginal flora in pregnancy. Am. J. Obstet. Gynecol. 150:965–972 [DOI] [PubMed] [Google Scholar]
- 41. Muzny CA, Rivers CA, Mena LA, Schwebke JR. Genotypic characterization of Trichomonas vaginalis isolates among women who have sex with women in sexual partnerships. Sex. Transm. Dis. 39:556–558 [DOI] [PubMed] [Google Scholar]
- 42. Muzny CA, Sunesara IR, Martin DH, Mena LA. 2011. Sexually transmitted infections and risk behaviors among African American women who have sex with women: does sex with men make a difference? Sex. Transm. Dis. 38:1118–1125 [DOI] [PubMed] [Google Scholar]
- 43. Nadler SA, Honigberg BM. 1988. Genetic differentiation and biochemical polymorphism among trichomonads. J. Parasitol. 74:797–804 [PubMed] [Google Scholar]
- 44. Oliveira RP, et al. 1998. Probing the genetic population structure of Trypanosoma cruzi with polymorphic microsatellites. Proc. Natl. Acad. Sci. U. S. A. 95:3776–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Posada D, Crandall KA. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. U. S. A. 98:13757–13762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prokopi M, Chatzitheodorou T, Ackers JP, Clark CG. 2011. A preliminary investigation of microsatellite-based genotyping in Trichomonas vaginalis. Trans. R. Soc. Trop. Med. Hyg. 105:479–481 [DOI] [PubMed] [Google Scholar]
- 47. Riley DE, Krieger JN. 1992. Rapid and practical DNA isolation from Trichomonas vaginalis and other nuclease-rich protozoa. Mol. Biochem. Parasitol. 51:161–163 [DOI] [PubMed] [Google Scholar]
- 48. Robles JC, Koreen L, Park S, Perlin DS. 2004. Multilocus sequence typing is a reliable alternative method to DNA fingerprinting for discriminating among strains of Candida albicans. J. Clin. Microbiol. 42:2480–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rojas L, Fraga J, Sariego I. 2004. Genetic variability between Trichomonas vaginalis isolates and correlation with clinical presentation. Infect. Genet. Evol. 4:53–58 [DOI] [PubMed] [Google Scholar]
- 50. Ryu JS, Min DY, Shin MH, Cho YH. 1998. Genetic variance of Trichomonas vaginalis isolates by Southern hybridization. Korean J. Parasitol. 36:207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sawyer S. 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6:526–538 [DOI] [PubMed] [Google Scholar]
- 52. Schmid G, Samuelson J, Rowley JT. 2011. Prevalence and incidence of selected sexually transmitted infections. World Health Organization, Geneva, Switzerland [Google Scholar]
- 53. Schwenkenbecher JM, et al. 2006. Microsatellite analysis reveals genetic structure of Leishmania tropica. Int. J. Parasitol. 36:237–246 [DOI] [PubMed] [Google Scholar]
- 54. Shafir SC, Sorvillo FJ, Smith L. 2009. Current issues and considerations regarding trichomoniasis and human immunodeficiency virus in African-Americans. Clin. Microbiol. Rev. 22:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simo G, et al. 2010. Population genetic structure of Central African Trypanosoma brucei gambiense isolates using microsatellite DNA markers. Infect. Genet. Evol. 10:68–76 [DOI] [PubMed] [Google Scholar]
- 56. Smith JM. 1992. Analyzing the mosaic structure of genes. J. Mol. Evol. 34:126–129 [DOI] [PubMed] [Google Scholar]
- 57. Smith JM, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 90:4384–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Snipes LJ, et al. 2000. Molecular epidemiology of metronidazole resistance in a population of Trichomonas vaginalis clinical isolates. J. Clin. Microbiol. 38:3004–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sorvillo F, Smith L, Kerndt P, Ash L. 2001. Trichomonas vaginalis, HIV, and African-Americans. Emerg. Infect. Dis. 7:927–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stiles JK, et al. 2000. Molecular typing of Trichomonas vaginalis isolates by HSP70 restriction fragment length polymorphism. Am. J. Trop. Med. Hyg. 62:441–445 [DOI] [PubMed] [Google Scholar]
- 61. Sutton M, et al. 2007. The prevalence of Trichomonas vaginalis infection among reproductive-age women in the United States, 2001–2004. Clin. Infect. Dis. 45:1319–1326 [DOI] [PubMed] [Google Scholar]
- 62. Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tibayrenc M. 2009. Multilocus enzyme electrophoresis for parasites and other pathogens. Methods Mol. Biol. 551:13–25 [DOI] [PubMed] [Google Scholar]
- 64. Upcroft JA, et al. 2006. Genotyping Trichomonas vaginalis. Int. J. Parasitol. 36:821–828 [DOI] [PubMed] [Google Scholar]
- 65. Vanacova S, Tachezy J, Kulda J, Flegr J. 1997. Characterization of trichomonad species and strains by PCR fingerprinting. J. Eukaryot. Microbiol. 44:545–552 [DOI] [PubMed] [Google Scholar]
- 66. World Health Organization 2001. Global prevalence and incidence of selected curable sexually transmitted infections: overview and estimates. World Health Organization, Geneva, Switzerland [Google Scholar]
- 67. Yeo M, et al. 2011. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 5:e1049 doi:10.1371/journal.pntd.0001049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang ZF, Begg CB. 1994. Is Trichomonas vaginalis a cause of cervical neoplasia? Results from a combined analysis of 24 studies. Int. J. Epidemiol. 23:682–690 [DOI] [PubMed] [Google Scholar]
- 69. Zubacova Z, Cimburek Z, Tachezy J. 2008. Comparative analysis of trichomonad genome sizes and karyotypes. Mol. Biochem. Parasitol. 161:49–54 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.