

Abstract
The abundances, compositions, and activities of microbial communities were investigated at bog and fen sites in the Glacial Lake Agassiz Peatland of northwestern Minnesota. These sites contrast in the reactivity of dissolved organic matter (DOM) and the presence or absence of groundwater inputs. Microbial community composition was characterized using pyrosequencing and clone library construction of phylogenetic marker genes. Microbial distribution patterns were linked to pH, concentrations of dissolved organic carbon and nitrogen, C/N ratios, optical properties of DOM, and activities of laccase and peroxidase enzymes. Both bacterial and archaeal richness and rRNA gene abundance were >2 times higher on average in the fen than in the bog, in agreement with a higher pH, labile DOM content, and enhanced enzyme activities in the fen. Fungi were equivalent to an average of 1.4% of total prokaryotes in gene abundance assayed by quantitative PCR. Results revealed statistically distinct spatial patterns between bacterial and fungal communities. Fungal distribution did not covary with pH and DOM optical properties and was vertically stratified, with a prevalence of Ascomycota and Basidiomycota near the surface and much higher representation of Zygomycota in the subsurface. In contrast, bacterial community composition largely varied between environments, with the bog dominated by Acidobacteria (61% of total sequences), while the Firmicutes (52%) dominated in the fen. Acetoclastic Methanosarcinales showed a much higher relative abundance in the bog, in contrast to the dominance of diverse hydrogenotrophic methanogens in the fen. This is the first quantitative and compositional analysis of three microbial domains in peatlands and demonstrates that the microbial abundance, diversity, and activity parallel with the pronounced differences in environmental variables between bog and fen sites.
INTRODUCTION
Peatland ecosystems are generally classified into ombrotrophic (rain-fed) bogs and minerotrophic (groundwater-fed) fens, depending mainly on trophic status and water source (12). These ecosystems accumulate carbon in the form of a partially decomposed peat layer. The important role of peatlands in global carbon sequestration (78) and emission of greenhouse gases (38) is well established, but the response of these ecosystems to changing climate is still uncertain due to inherent spatial and temporal heterogeneity as well as the complex interplay of hydrological and biogeochemical processes (52).
Despite the important role of peatlands in the global carbon cycle, microbial communities in these environments remain understudied. Peats have been gradually recognized as a home to diverse microbial assemblages primarily through cultivation-based studies (29). Modern molecular techniques have begun to be employed to explore microbial community structure in peatlands. However, the focus of this research was restricted largely to specific functional guilds of microorganisms, such as methanogens (7, 65), methanotrophs (32), fermenters (34), sulfate reducers (60), or microbes involved in nitrogen cycling (76). To date, cultivation-independent studies of peatland microbial diversity have provided only a limited phylogenetic resolution and depth of coverage due to practical limitations of the techniques used, e.g., terminal restriction fragment length polymorphism (T-RFLP) and clone library construction (2, 8, 15, 20, 44, 55, 58, 61, 73). Studies of fungi, for example, have suffered from the lack of a high-quality curated database for taxonomic assignment (6).
Walker et al. and others proposed that climate change, particularly warming, is likely to alter the vegetation balance in northern terrestrial biomes (74). Grasses, such as sedges, and other vascular plants (Carex, Eriophorum) will be favored and expand in biomass and areal extent, while bryophytes (mosses, such as Sphagnum) will likely decrease in areal extent and importance. These changes could have important ramifications for the carbon cycle, since plant species composition is thought to intimately drive microbial carbon turnover. For example, geochemical evidence indicates that the reactivity of organic matter, and therefore its susceptibility to microbial degradation, is dependent upon plant species composition in peatlands (11, 14). Dissolved organic matter (DOM) produced in Sphagnum woody plant-dominated bogs is significantly more recalcitrant and aromatic in character than is DOM within vascular plant (sedge)-dominated fens (70). In this study, we sought to compare the microbial ecology of a bog and fen as an analogous scenario to plant species transitions that are projected to occur during climate change. We provide a comprehensive investigation of peatland microbial communities that is closely coupled to geochemical investigations focused on the carbon cycle. Here, we show that the shift of microbial abundance, community composition, and activity parallels with the stark differences in pH and DOM reactivity observed at a bog and fen in the Glacial Lake Agassiz Peatland (GLAP) of northern Minnesota.
MATERIALS AND METHODS
Sampling and chemical characterization.
Field sampling was conducted within the Glacial Lake Agassiz region of northwestern Minnesota. Approximately 56% (or nearly 5,000 km2) of this region is covered by peatlands, which average 2 to 3 m in depth and were established approximately 5,000 years ago in response to climatic cooling (30). Four peat cores were sampled on 11 August 2009 in the Red Lake II raised bog complex, whose landform patterns are similar to those found in other large peat basins (30). The distance between the bog and fen site was approximately 5 km. The water table was at 0 m. Two cores were from a bog, which is an acidic and nutrient-deficient environment that receives water inputs primarily from precipitation and is covered by Sphagnum moss. The remaining two cores were taken from a minerotrophic fen, which is influenced by inputs of more alkaline, nutrient-rich groundwater and is dominated by sedges. Cores from each site were sectioned into surface (0 to 10 cm) and subsurface (40 to 50 cm) samples.
Fen and bog pore water samples were also collected at the same depths as the peat samples (0 to 10, 40 to 50 cm) using piezometers and a peristaltic pump. Experimental approaches for sample handling and the determination of dissolved organic carbon (DOC) and organic nitrogen were described previously (11, 71). Optical properties of chromophoric DOM (CDOM) were determined using absorption and fluorescence spectroscopy as described in reference 71. Two optical properties (E2/E3 ratio and SUVA254) were included here to indicate their link with shifts in microbial community composition. The E2/E3 ratio of absorbance at 250 to 365 nm is used to estimate DOM molecular size. E2/E3 decreases as molecular size increases, because high-molecular-weight DOM absorb at a longer wavelength (21). The SUVA254 (liters mg C−1 m−1) is calculated by normalizing the UV absorbance at 254 nm by the concentration of DOM. SUVA254 correlates with the fraction of aromatic compounds contained in CDOM (77).
Assays for polyphenolic oxidase enzyme activity followed the protocol described in reference 26, with addition of H2O2 to determine total potential activities of both polyphenolic oxidases and peroxidases. Briefly, 1 g of wet peat was suspended in 9 ml of acetate buffer (10 mM, pH 4) at 4°C and blended for 5 min to release adsorbed enzyme in suspension. Enzyme reactions began by mixing the soil suspension with 2,2′-azinobis-(−3 ethylbenzothiazoline-6-sulfononic acid) diammonium salt (ABTS; final concentration of 5 mM) in the buffer (pH 4) and incubating in the dark at 4°C for 10 min. To stop the reaction, the mixture was centrifuged at 10,000 rpm for 10 min. The absorbance of oxidized ABTS was detected with a spectrophotometer at 420 nm.
DNA extraction, PCR amplification, pyrosequencing, and qPCR.
Total genomic DNA was extracted from 0.5 g peat soil per sample (in triplicate) using a MoBio PowerSoil DNA extraction kit (MoBio, Carlsbad, CA) by following the manufacturer's protocol. The extracted DNA was pooled, amplified, and sequenced at the Emory University Genomics Facility (Atlanta, GA) using the standard pyrosequencing protocol (Roche 454, Branford, CT). PCR amplification of the bacterial small-subunit (SSU) rRNA gene and fungal internal transcribed spacer (ITS) regions of the rRNA gene was performed using the primer pairs of 27F/534R (43) and ITS1F/ITS4 (28), respectively. Targeted gene fragments were amplified in 50-μl reaction mixtures containing 5 PRIME MasterMix and 10 ng genomic DNA. Thermal cycling was performed by following the manufacturer's protocol with annealing temperature at 55°C for 35 cycles. Real-time PCR was performed to quantify the abundance of total bacterial and archaeal SSU rRNA genes according to the procedures described elsewhere (40). The original genomic DNA was diluted to 1:40 to remove inhibition effects. Fungal quantitative real-time PCR (qPCR) was performed following the protocol described in reference 62. Standard curves were created with PCR amplicons from genomic DNA of an Ascomycota isolate from GLAP using primers EukA and EukB (49).
Clone library construction.
Construction of clone libraries for archaeal SSU rRNA gene sequences was based on PCR products amplified using primers ARC344F and ARC915R (63). PCR products were purified with a MinElute PCR purification kit (Qiagen Inc., Valencia, CA) according to the manufacturer's instructions. The amplicons were cloned using the TOPO TA cloning kit (Invitrogen, Carlsbad, CA). Colonies were picked into 0.85% NaCl and sent to Sequetech for plasmid isolation and sequencing. Sequence processing and phylogenetic analysis followed the procedures described elsewhere (47).
Processing of pyrosequencing data and diversity estimation.
All 454 sequencing data were analyzed in QIIME (9). The sequencing reads were first binned and filtered to remove low-quality sequences, which included those sequences of <200 bp in length with an average quality score of <25 and containing ambiguous characters or a homopolymer run exceeding 6 nt. Sequences without the correct primer sequence were excluded. A workflow script in QIIME was used to pick operational taxonomic units (OTUs) at the 97% sequence identity level through building OTU tables. For bacterial SSU rRNA gene sequences, the PyNAST-aligned representative sequences were used to identify chimeric sequences using ChimeraSlayer in QIIME. Chimera check of fungal ITS gene sequences was done with UCHIME (23) in a de novo mode. Taxonomy was assigned to each representative sequence by using BLAST with a maximum E value of 0.001 against the Greengenes core set for bacteria and using the QIIME-compatible UNITE database for fungi (http://qiime.org/home_static/dataFiles.html). For all OTU-based analyses, the original OTU table was rarefied to depths of 44,576 bacterial sequences and 774 fungal sequences per sample, to minimize the effects of a different sampling effort in comparing alpha- and beta-diversity across the samples.
Statistical analyses.
Microbial diversity expressed as Chao1 richness and equitability was calculated in QIIME based on the OTU tables rarefied to the same sampling depths. Pairwise comparisons of microbial community structure were computed as Bray-Curtis distances and were visualized using principal coordinate analysis in PRIMER version 6. Pearson correlations between the OTU abundance matrix and the environmental matrix shown in Table 1 were calculated with PRIMER Permanova+. Environmental parameters having their Pearson correlation with the first two PCO axes larger than >0.5 were displayed as vectors in the principal coordinate analysis. Mantel tests by RELATE in PRIMER were performed to calculate the significance of fit between the community similarity matrix and distance matrices of each environmental variable with 1,000 permutations. A distance-based multivariate linear model (DistLM) was performed to select the best combination of predictor variables explaining the community variation, with significance tests by permutation.
Table 1.
Biogeochemical comparison between the bog and fen environmentsa
| Site | Depth (cm) | pH | Amt |
C/N ratio | E2/E3 | SUVA254 | Enzyme activity (μmol/g/h) | Abundance of bacteria and archaea (×108)/g | % of archaea | Abundance of fungi (×106)/g | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DOC (mM) | DON (μM) | Ammonia (μM) | ||||||||||
| Bog | 0–10 | 4.2 | 5.6 | 59.7 | 14 | 93.3 | 4.8 ± 0.1 | 4.6 ± 0.1 | 3.0 ± 0.8 | 2.5 ± 0.4 | 1.2 ± 0.4 | 4.8 ± 0.7 |
| Bog | 40–50 | 4.3 | 6.2 | 44.1 | 94.2 | 139.6 | 5.3 ± 0.1 | 4.0 ± 0.0 | 2.3 ± 0.8 | 1.9 ± 0.3 | 4.1 ± 0.8 | 2.3 ± 0.4 |
| Fen | 0–10 | 5.7 | 2.9 | 53.7 | 11.6 | 53.9 | 6.9 ± 0.3 | 3.0 ± 0.0 | 22.6 ± 3.4 | 6.9 ± 0.7 | 1.3 ± 0.9 | 14.0 ± 0.8 |
| Fen | 40–50 | 5.9 | 3.6 | 39.6 | 53.9 | 90.6 | 7.3 ± 2.5 | 3.3 ± 0.1 | 19.1 ± 1.9 | 6.0 ± 0.4 | 10.5 ± 1.3 | 3.4 ± 0.5 |
Numbers following “±” indicate standard deviations. All assays associated with peats are normalized to the wet soil weight. The last 3 columns are total prokaryotic rRNA gene abundance, percentages of archaeal rRNA gene abundance, and fungal 18S rRNA gene abundance.
Comparison of spatial patterns in bacterial and fungal distribution was assessed using a Procrustean analysis at the overall degree of association between bacterial and fungal OTU abundance matrices. Peres-Neto and Jackson demonstrated the advantages of the procrustean approach over the Mantel test for detecting matrix association (59). Procrustes analysis was carried out in QIIME, which outputs the correlation value M2 (1 − r2, where r is Pearson correlation coefficient) and P values indicating significance levels. Smaller values of M2 indicate higher concordance between two data matrices.
Nucleotide sequence accession numbers.
Archaeal sequences from this study are available in GenBank (JQ807525 to JQ807565).
RESULTS
Pore water geochemistry, enzyme activity, and rRNA gene abundance in peat soils.
The bog and fen sites showed largely contrasting geochemical conditions linked to plant species composition (Table 1). Bog pore waters were more acidic (pH of ∼4) than the fen (pH of ∼6) and accumulated approximately twice as much DOC in comparison to that of the fen. Dissolved organic nitrogen content was about 10% higher in the bog, and it decreased with depth at both sites. Ammonia concentrations increased by a factor of 5 from the surface to the subsurface at both sites and did not substantially differ between sites.
Several independent parameters indicated that DOM was more recalcitrant in bog pore waters. The C/N ratio was about 1.6 times lower in the fen than in the bog for the same depth, and the bog subsurface showed the highest C/N ratio. The difference in DOM quality between sites was further reflected in its optical properties: E2/E3 ratio and SUVA254. Lower E 2/E3 ratios in the bog are representative of a higher-molecular-weight DOM relative to the fen. In addition, the SUVA254 was higher in the bog than in the fen, suggesting that the bog DOM is more aromatic in nature than that in the fen.
The sum of phenol oxidase and peroxidase activity was about 8 times higher on average in the fen than in the bog, and the activity declined about 19% on average to the subsurface (Table 1). The spatial variation between the enzyme activity and rRNA gene abundance was correlated. The abundance of prokaryotic rRNA genes was 2.5 × 108 and 1.9 × 108 per gram of wet soil in the bog surface and subsurface, respectively. SSU rRNA gene abundance was about 3 times lower in the bog than in the fen. The proportion of archaea was >3 times higher in the subsurface than at the surface of both sites, with the lowest percentage (1.2%) in the bog surface and the highest (10.5%) in the fen subsurface. Fungal rRNA gene abundance comprised on average 1.4% of the total prokaryotic rRNA gene abundance. At the surface, fungal rRNA gene abundance was 2.9 times higher in the fen than in the bog. Fungal gene abundance declined with depth at both sites.
Microbial species richness and evenness.
Microbial diversity was determined based on the analysis of 215,928 bacterial SSU rRNA gene sequences, 10,609 fungal ITS sequences, and 181 archaeal SSU rRNA gene sequences (Table 2). Microbial richness and evenness were estimated by using Chao1 and equitability (1 = complete equitability). Bacterial and archaeal species richness was more than twice as high in the fen as in the bog site for both depths. Fungal richness was highest in the bog surface, followed by the fen site, and was lowest in the bog subsurface. Similar patterns in microbial evenness were observed for these samples, with the equitability ranging from 0.39 (fungi in the bog subsurface) to 0.93 (archaea in the fen subsurface).
Table 2.
Microbial diversity indicated by Chao1 richness and equitability evenness in the bog and fen sitesa
| Site | Depth (cm) | Bacteria |
Archaea |
Fungi |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sequence read | Chao1 | Equitability | Library size | Chao1 | Equitability | Sequence read | Chao1 | Equitability | ||
| Bog | 0–10 | 71,199 | 7,783 | 0.57 | 43 | 18 | 0.66 | 774 | 243 | 0.84 |
| Bog | 40–50 | 44,576 | 8,559 | 0.55 | 45 | 13 | 0.48 | 1,189 | 89 | 0.37 |
| Fen | 0–10 | 49,361 | 22,100 | 0.73 | 47 | 90 | 0.91 | 6,436 | 210 | 0.75 |
| Fen | 40–50 | 50,487 | 19,873 | 0.47 | 46 | 40 | 0.93 | 2,139 | 220 | 0.85 |
Calculation was based on OTU tables rarefied to the same sequencing depth, namely, the smallest one of either total pyrosequencing reads or clone library size. The sequence read is the number of sequences generated per sample. Chao1 richness is estimated from OTU number with 97% sequence identity. Library size is the number of clones picked for sequencing.
Microbial community composition.
Bacteria were diverse in the peat soils, and their OTUs were affiliated with a total of 53 phyla (Fig. 1; see also Table S1 in the supplemental material), with the most abundant 10 phyla including (in order of decreasing abundance) Acidobacteria, Firmicutes, Proteobacteria, Verrucomicrobia, Chloroflexi, Planctomycetes, Actinobacteria, Bacteroidetes, Chlorobi, and TM7 (Fig. 1). Bacterial community compositions were markedly different between bog and fen sites. The bog and fen environments were dominated by Acidobacteria and Firmicutes, respectively. The 20 most abundant bacterial OTUs were affiliated with Acidobacteria group 1 (dominant in the bog), Clostridium akagii (50% at the fen subsurface), Verrucomicrobia subdivision 3, Pelosinus sp., and uncultured Oxalobacteraceae species (see Fig. S1 and Table S2 in the supplemental material).
Fig 1.
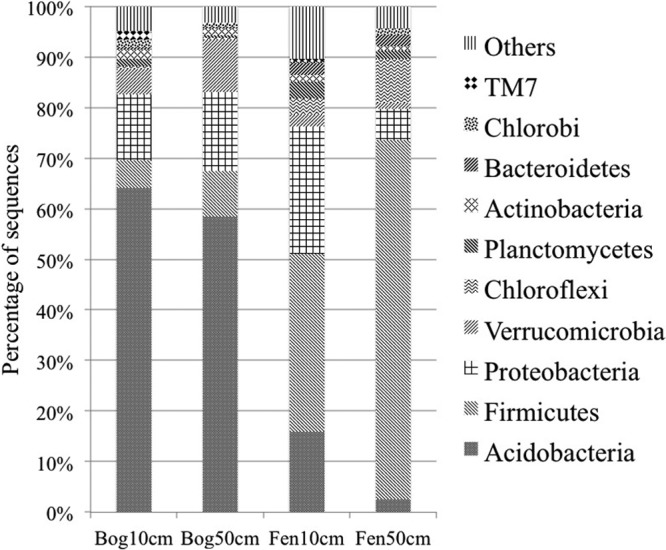
Bacterial community composition in the bog and fen sites. “Others” contains the other 43 phyla relatively rare in these sites (see Table S1 in the supplemental material).
Distribution patterns of bacterial clades associated with several functional guilds also indicated the environmental differences between the bog and fen sites and across depths (see Table S2 in the supplemental material). For example, levels of both type I (Methylococcaceae) and type II (Methylocystaceae) methanotrophs were on average 10 times higher at the surface than at the subsurface for both sites. Metal respiring bacteria (Geobacter and Shewanella) were detectable only at the fen site.
Archaeal community composition was dominated by methanogenic Euryarchaeota, including members of Methanosarcinales, Methanomicrobiales, and Methanococcales (Fig. 2; see also Fig. S2 in the supplemental material). The C2 clade was the dominant phylotype of Crenarchaeota observed in all samples. In the bog, the archaeal community was dominated by phylotypes affiliated with acetoclastic Methanosarcinales, which accounted for 65% and 91% of the total archaea in the bog surface and subsurface, respectively (Fig. 2). Samples from the fen contained more diverse groups of archaea, including both acetoclastic and hydrogenotrophic methanogens. Clone GLAP87 accounted for 55% and 75% of total archaeal sequences at the bog surface and subsurface, respectively. Interestingly, this OTU shares 98% sequence similarity with a Methanosarcina sp. isolated from a peat bog of northern England (33), but it is phylogenetically distant from well-characterized members of acetoclastic Methanosarcinales (see Fig. S2 in the supplemental material), suggesting diverse ecological adaptation strategies of different members of the Methanosarcinales.
Fig 2.

Archaeal community composition in the bog and fen sites.
Fungal community structure was distinct in the bog subsurface and dominated by sequences affiliated with the Zygomycota (90% of total sequences) (Fig. 3). A high relative abundance (23% of total) of Zygomycota sequences was also observed in the fen subsurface, in contrast to less than 5% of Zygomycota sequences observed in both surface samples. The bog surface was dominated by phylotypes affiliated with the Ascomycota. The surface bog and the two fen samples showed a similar relative abundance of Basidiomycota and Glomeromycota sequences, while sequences from the Chytridiomycota in the fen surface were about one order of magnitude higher in relative abundance than those in the other 3 samples on average. Among the 20 most abundant fungal OTUs detected in this study, only four were classified to the genus level, including Mortierella spp., Stilbella sp., Cryptococcus sp., and Rhizopus sp. (see Table S2 in the supplemental material).
Fig 3.
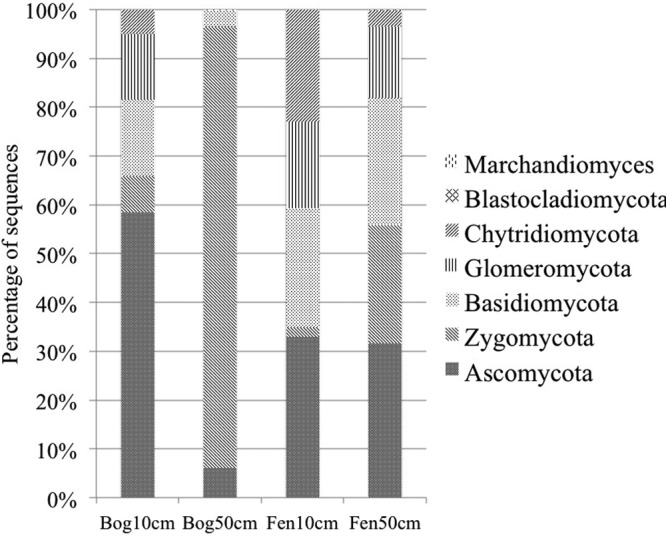
Fungal community composition in the bog and fen sites.
Community comparison across the depth and sites.
Principal coordinate analysis based on Bray-Curtis similarity separated the bog bacterial communities from those of the fen along the first principal coordinate axis (PCO1) (Fig. 4A). PCO1 explains 57.3% of total variation, and this component negatively correlates with environmental parameters, such as pH, total SSU rRNA gene abundance, and E2/E3 ratio. PCO1 positively correlates with C/N ratio, DOC concentration, and aromatic fraction (SUVA) (significance level of <5%). PCO2 explained 30.7% of total variation and separates the fen surface from the fen subsurface. Both enzyme activity and DON concentration negatively correlated with the PCO2 component, and the two vectors point to the higher values in the fen surface.
Fig 4.

Principal coordinates analyses for bacterial (A) and fungal (B) community composition. Environmental vectors with Pearson correlation of R of >0.5 are shown. The initials “B” and “F” followed with depth (10 cm or 50 cm) indicate bog and fen, respectively. Abundance represents the total SSU rRNA gene abundance.
The distribution pattern of fungal communities (Fig. 4B) was significantly different from that seen in the bacterial community (Fig. 4A) based on a matrix comparison using Procrustes analysis (M2 = 0.59, P = 0.11), highlighting a unique fungal community composition in the bog subsurface. PCO1 explained 52.8% of total variation and separated the bog subsurface sample from all others (Fig. 4B). The fungal PCO1 negatively correlates with enzyme activities, DON, and SSU gene abundance and positively correlated with C/N ratio and concentrations of DOC and ammonia. In contrast, several factors (pH, molecular size, and SUVA) that influenced bacterial distribution were less significant (significance level of >8%) for the fungal PCO1, suggesting that bacterial and fungal communities respond differently to environmental stressors (e.g., pH) and DOM properties. PCO2 explained 28.3% of total variation, and enzyme activity is still an important factor correlating with this axis.
To further explore the determinants of community structure, a distance-based multivariate linear model was used to partition community variation into environmental components. Table 3 shows that pH explained the greatest proportion (53.4%) of bacterial community variation. A combination of pH, DOC concentration, and biomass (rRNA gene abundance) predicts most (93.3%) of the bacterial community variation. In contrast, the concentrations of NH4, DON, and DOC are combined to explain 86.0% of total fungal community variation.
Table 3.
Sequential tests of predictor variables for bacterial and fungal community variation using the distance-based multivariate linear model
| Variables | Adjusted R2a | P value | Proportion of variation explained | Cumulated variation explained |
|---|---|---|---|---|
| Bacterial community | ||||
| pH | 0.457 | 0.005 | 0.534 | 0.534 |
| DOC | 0.699 | 0.013 | 0.251 | 0.785 |
| Gene abundance | 0.882 | 0.023 | 0.148 | 0.933 |
| Fungal community | ||||
| NH4 | 0.314 | 0.002 | 0.412 | 0.412 |
| DON | 0.454 | 0.049 | 0.198 | 0.610 |
| DOC | 0.755 | 0.015 | 0.250 | 0.860 |
Correlation coefficient adjusted for number of parameters.
DISCUSSION
Bacterial and archaeal community composition and their functional significance in peatlands.
Prokaryotes are major players in the mineralization of organic matter via fermentation and respiration in peats (10, 79), especially in the catotelm, where anoxia may limit the metabolism of fungi. We observed a significant correlation between bacterial community composition and pH, concentrations of DOC and DON, C/N ratios, fractions of aromatic compounds, and molecular size of DOM. In corroboration of previous work in peatlands, we observed that low pH in the moss-dominated bog selects for acid-tolerant phylotypes (e.g., Acidobacteria group 1) (20, 68), while the fen is dominated by phylotypes (e.g., Clostridia of the Firmicutes) favoring a more neutral pH condition. Bacterial community composition in peat soils of the bog was dominated by Acidobacteria, whose relative abundance was at the high end of the range reported for soil environments (45), while the relative abundance of Firmicutes in the fen was high in comparison to other soil environments and comparable to that observed in human intestinal environments with high fermentation activity (22).
Although more information is available for peatland bacterial communities than for fungi, the respective role of specific bacterial groups in the degradation of Sphagnum-derived litter is largely unknown. Acidobacteria are well adapted to acidic, nutrient-poor conditions (24, 68). Recent studies showed that 1/3 of laccase (phenol oxidase) genes in peat soils are affiliated with Acidobacteria (3), suggesting their important role in degrading aromatic compounds in peatlands. A potential role for the Acidobacteria in degrading cellulose has also been suggested based on the analysis of genome sequences (75) and laboratory tests with novel acidobacterial isolates (56, 57). In contrast, the Clostridia appear to thrive at relatively neutral pH in fen soils inhabited by more flowering plants that release labile DOM from their roots and are known to be key players in the degradation of cellulose and labile organic compounds (20, 31, 67). Interestingly, the fen subsurface contained about 50% of bacterial sequences affiliated with Clostridium akagii, an N2-fixing fermenter isolated from acidic soils (42), suggesting their adaptation to this environment. Further work is needed to link dominant bacterial groups to the degradation of specific polyphenolic and carbohydrate compounds which are abundant in plant DOM in peatlands.
Methane-oxidizing bacteria (methanotrophs) are diverse and consume a significant fraction of greenhouse gas produced in peatlands. In the GLAP bog and fen, type II (Methylocystaceae of Alphaproteobacteria) methanotrophs were the dominant methane-oxidizing bacteria and accounted for 0.8 to 1.6% (or ∼106 sequences/g) of the total community (see Table S2 in the supplemental material), comparable to that in other acidic soils (19, 39). In contrast, type I (Methylococcaceae of Gammaproteobacteria) methanotrophs were barely detectable, especially in the fen. Relative to the type I methanotrophs, type II methanotrophs generally have the ability to fix N2 and grow better at lower O2 concentrations (1). This is consistent with our observation of the dominance of type II methanotrophs in this high C/N ratio peat environment with low O2 concentrations, especially in the subsurface, although we do not exclude other environmental factors affecting the abundance and diversity of methanotrophs (46).
Surprisingly, sequences from members of the Methanosarcinales, known as acetoclastic methanogens, predominated in the bog and in the subsurface of both sites, in contrast to the previous observations that hydrogenotrophic methanogenesis is the primary pathway in the ombrotrophic bog and oligotrophic fen of GLAP (13) or in other northern peatlands in general (36, 37). The isotopic fractionation of mineralization products, CO2 and CH4, also indicates that hydrogenotrophic methanogenesis should dominate and is energetically more favorable in the bog than in the fen at GLAP (14). Although methanogens are perhaps the best studied of microbial groups in peatlands, linkages between the phylogenetic structure of archaeal communities and pathways of methanogenesis are not fully resolved (41, 65). For example, Kotsyurbenko et al. (41) reported that Methanosarcina spp. accounted for approximately half of the archaeal community in acidic peat, while members of Methanosarcinaceae were undetectable in acidic environments of Alaskan peatlands (65).
It should be recognized that members of the Methanosarcinales (e.g., Methanosarcina) exhibit the highest physiological diversity among methanogens and are capable of most major metabolic pathways for methane production (27). In addition, the most abundant Methanosarcina clone in our samples, GLAP87, is phylogenetically distant from well-characterized cultures of the Methanosarcinales (see Fig. S2 in the supplemental material) but closely related (>98% identity) to an isolate from a peat bog of northern England (33), suggesting their adaptation and potentially novel metabolism in bog environments. Methanosarcina species are believed to succeed in nature by following a generalist strategy (81). Thus, the ecological function and controls of methanogenesis in the acidic bog may need to be reevaluated.
Fungal abundance, community composition, and distribution in peatlands.
Assessing the relative contributions of fungi and bacteria to the microbial biomass and activity is of substantial interest for soil ecology, as fungi and bacteria govern most of C transformation and favor different degradation pathways (69). Our qPCR results showed that fungal SSU rRNA gene abundance was 1 to 2 orders of magnitude lower than that observed in grassland, cropland, and forest soils compared using the same technique (62). This is consistent with the observation that microbial biomass relative to soil organic carbon was much lower in peatlands than in agricultural forest soils (5.6 versus 24 mg microbial C g−1 soil organic C) (51). Moreover, aromatic and phenolic compounds released from Sphagnum litter exhibit antimicrobial properties and may impede the decomposition of carbohydrate-rich DOM (66). These properties of peatland DOM to some extent explain the lower biomass observed in the bog.
More interestingly, we found very low fungus/prokaryote ratios (<0.02), close to the lower end of the range (0.01 to 1 in soils) reported globally (25). This finding is contrary to prior cultivation-dependent studies, which suggested that surface peatlands with extensive hyphal growth are usually dominated by fungal biomass (29, 72). Our qPCR values simply represent ratios of fungal/bacterial SSU gene abundance, instead of biomass. However, our results are consistent with a more recent activity-based study demonstrating a strong bacterial dominance in a bog and fen of northern peatlands (79). Thus, our culture-independent study, together with activity-based studies, suggests a less important role for fungi in carbon cycling of peatlands relative to that in other soil environments.
Fungi, in particular, are understudied in peat soils, although these microorganisms are thought to play an important role in the carbon cycle (72). In this study, fungal communities appear to respond differently to environmental variables compared to bacteria. Statistical analysis indicates that pH is less important as a selective force in structuring fungal community composition, which can be attributed to the ability of fungi to tolerate a wider pH range for optimal growth as well as their optimal extracellular enzyme activity at low pH (5). Furthermore, our results suggest that concentrations of ammonia, DOC, and DON are more important than the molecular size and aromaticity of DOM in structuring fungal distribution patterns (Fig. 4). The gradient of ammonia with depth may be indicative of a redox condition, which is known to affect the distribution and activity of fungi in peatlands (72). Unpublished data from our group in another peatland site show a sharp decline of fungal 18S gene abundance below 50 cm, further supporting the potential importance of oxygen governing the abundance and activity of fungi. Several studies have demonstrated the relationship between fungal community distribution pattern and plant species composition in mineral soils (50, 53). However, to our knowledge, this is the first study to report the feature that fungal community pattern differentially responds to quantity, quality (e.g., C/N ratio), and reactivity of DOM in peatland ecosystems.
The spatial distribution pattern of the fungal community exhibited a vertical stratification of major phyla, reflecting their response to different environmental conditions (e.g., plant community composition, DOM quantity). Fungal taxa found in this study belong mainly to 3 phyla: Ascomycota, Basidiomycota, and Zygomycota, consistent with previous work in peat soils (2, 29, 72). The predominance of Ascomycota and Basidiomycota at the surface is consistent with their ability to degrade DOM (e.g., cellulose and polyphenolic compounds) under oxic conditions, which has been observed primarily in laboratory studies (48, 72). In contrast to previous work, members of the Zygomycota were detected in abundance in the subsurface of both the bog and fen sites, suggesting that their physiology is distinct from that of the Ascomycota and Basidiomycota. The Zygomycota produce thick-walled, resistant spores that allow the fungi to survive over long periods of dormancy (4). In this aspect, it will be important to determine if members of the Zygomycota in the subsurface are active or dormant. Additionally, members of the Zygomycota do not respond as well as the Dikarya (Ascomycota and Basidiomycota) to soluble plant degradation products (e.g., cellulose, sucrose) and instead appear to be associated with carbon substrates of animal and fungal origin, such as fungal hyphae (16, 18, 35). For example, Mortierella spp., dominant among the zygomycete phylotypes in our samples, are strong digesters of chitin (80), which is the fundamental component of fungal hyphae. Experiments show that zygomycetes degrade chitin nearly as efficiently as chitinolytic actinomycetes (18). Although the vertically stratified distribution of Zygomycota as well as Dikarya appears consistent with the gradient of carbon quantity and quality in the GLAP peatland, we can only speculate on the functional role of these fungal taxa in degrading organic carbon substrates. Our findings suggest that the Zygomycota may play a more important role in the carbon cycle than was previously perceived (72).
Our findings of both microbial community analysis and geochemical measurements were generally reproducible. Previous studies have reported high reproducibility of 454-based DNA sequencing of the same sample (54) or habitat (64). Reproducibility of microbial community analysis was also verified by a fingerprinting method, which indicated that similarity in community membership (Sorensen similarity) and structure (Bray-Curtis similarity) among triplicated DNA extractions per sample was >85% and >65%, respectively (see Table S3 in the supplemental material). In addition, the spatial variation in our geochemical measurements is also consistent with previous studies of multiple-year measurements at GLAP (17, 70). Therefore, the results reported here are robust and likely representative of peatland microbial communities and chemical environments.
In summary, this study revealed prokaryotic dominance in peatland microbial communities and demonstrated a distinct response of fungal and bacterial communities to environmental variables changing along the depth and vegetation gradients in the bog and fen environments. Ammonia (presumably correlating negatively with oxygen concentration vertically) and pH are determined to be primary environmental parameters structuring fungal and bacterial community composition, respectively. Additionally, DOM molecular size and aromaticity showed a more significant correlation, with the shift in bacterial community composition relative to that in fungi. Our study identified unique distribution patterns of specific microbial groups in the heterogeneous peatland environment, which may select for microbes with poorly recognized metabolic capability. Zygomycota prevalent at the subsurface are targets for further functional understanding in their role in peatland carbon cycling. A novel clade of Methanosarcinales dominant in the bog indicates their potential capability of both acetoclastic and hydrogenotrophic methanogenesis. A mechanistic understanding of the role of important clades in peatland carbon cycling requires both additional field experiments and ecophysiological studies in the laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Office of Biological and Environmental Research, Terrestrial Ecosystem Science Program, under U.S. DOE contract number PE-SC0007144, and the U.S. National Science Foundation (NSF-EAR-0628349).
We thank J. Delgardio, P. Chanton, and R. Poretsky for help with sample handling and discussions. We thank the personnel of the Emory University Genomics Facility for their assistance with sequencing of the SSU rRNA gene amplicons.
Footnotes
Published ahead of print 27 July 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Amaral JA, Knowles R. 1995. Growth of methanotrophs in methane and oxygen counter gradients. FEMS Microbiol. Lett. 126:215–220 [Google Scholar]
- 2. Artz RRE, et al. 2007. Changes in fungal community composition in response to vegetational succession during the natural regeneration of cutover peatlands. Microb. Ecol. 54:508–522 [DOI] [PubMed] [Google Scholar]
- 3. Ausec L, van Elsas JD, Mandic-Mulec I. 2011. Two- and three-domain bacterial laccase-like genes are present in drained peat soils. Soil Biol. Biochem. 43:975–983 [Google Scholar]
- 4. Bartnicki-Garcia S. 1987. The cell wall: a crucial structure in fungal evolution, p 389–403 In Rayner ADM, Brasier CM, Moore D. (ed), Evolutionary biology of the fungi. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 5. Beales N. 2004. Adaptation of microorganisms to cold temperatures, weak acid preservatives, low pH, and osmotic stress: a review. Compr. Rev. Food Sci. Food Safety 3:1–20 [DOI] [PubMed] [Google Scholar]
- 6. Begerow D, Nilsson H, Unterseher M, Maier W. 2010. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 87:99–108 [DOI] [PubMed] [Google Scholar]
- 7. Cadillo-Quiroz H, Yashiro E, Yavitt JB, Zinder SH. 2008. Characterization of the archaeal community in a minerotrophic fen and terminal restriction fragment length polymorphism-directed isolation of a novel hydrogenotrophic methanogen. Appl. Environ. Microbiol. 74:2059–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cadillo-Quiroz H, Yavitt JB, Zinder SH, Thies JE. 2010. Diversity and community structure of archaea inhabiting the rhizoplane of two contrasting plants from an acidic bog. Microb. Ecol. 59:757–767 [DOI] [PubMed] [Google Scholar]
- 9. Caporaso JG, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chanton JP, et al. 1995. Radiocarbon evidence for the substrates supporting methane formation within northern Minnesota peatlands. Geochim. Cosmochim. Acta 59:3663–3668 [Google Scholar]
- 11. Chanton JP, et al. 2008. Radiocarbon evidence for the importance of surface vegetation on fermentation and methanogenesis in contrasting types of boreal peatlands. Global Biogeochem. Cycles 22:GB4022 doi:10.1029/2008GB003274 [Google Scholar]
- 12. Charman D. 2002. Peatlands and environmental change. J. Wiley & Sons, London, United Kingdom [Google Scholar]
- 13. Chasar LS, Chanton JP, Glaser PH, Siegel DI. 2000. Methane concentration and stable isotope distribution as evidence of rhizospheric processes: comparison of a fen and bog in the Glacial Lake Agassiz Peatland complex. Ann. Bot. 86:655–663 [Google Scholar]
- 14. Chasar LS, Chanton JP, Glaser PH, Siegel DI, Rivers JS. 2000. Radiocarbon and stable carbon isotopic evidence for transport and transformation of dissolved organic carbon, dissolved inorganic carbon, and CH4 in a northern Minnesota peatland. Global Biogeochem Cycles 14:1095–1108 [Google Scholar]
- 15. Clare JT, Johnson D, Artz RRE. 2009. Litter type, but not plant cover, regulates initial litter decomposition and fungal community structure in a recolonising cutover peatland. Soil Biol. Biochem. 41:651–655 [Google Scholar]
- 16. Cromack K, Caldwell BA. 1992. The role of fungi in litter decomposition and nutrient cycling, p 653–668 In Carroll GC, Wicklow DT. (ed), The fungal community: its organization and role in the ecosystem, vol 2nd Marcel Dekker, New York, NY [Google Scholar]
- 17. D'Andrilli J, Chanton JP, Glaser PH, Cooper WT. 2010. Characterization of dissolved organic matter in northern peatland soil porewaters by ultra high resolution mass spectrometry. Org. Geochem. 41:791–799 [Google Scholar]
- 18. De Boer W, Gerards S, Gunnewiek PJA, Modderman R. 1999. Response of the chitinolytic microbial community to chitin amendments of dune soils. Biol. Fert. Soils 29:170–177 [Google Scholar]
- 19. Dedysh SN, Derakshani M, Liesack W. 2001. Detection and enumeration of methanotrophs in acidic Sphagnum peat by 16S rRNA fluorescence in situ hybridization, including the use of newly developed oligonucleotide probes for Methylocella palustris. Appl. Environ. Microbiol. 67:4850–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dedysh SN, Pankratov TA, Belova SE, Kulichevskaya IS, Liesack W. 2006. Phylogenetic analysis and in situ identification of bacteria community composition in an acidic Sphagnum peat bog. Appl. Environ. Microbiol. 72:2110–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Haan H, De Boer T. 1987. Applicability of light absorbance and fluorescence as measures of concentration and molecular size of dissolved organic carbon in humic Lake Tjeukemeer. Water Res. 21:731–734 [Google Scholar]
- 22. Eckburg PB, et al. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eichorst SA, Breznak JA, Schmidt TM. 2007. Isolation and characterization of soil bacteria that define Teniglobus gen. nov., in the phylum Acidobacteria. Appl. Environ. Microbiol. 73:2708–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC. 2009. Global patterns in belowground communities. Ecol. Lett. 12:1238–1249 [DOI] [PubMed] [Google Scholar]
- 26. Floch C, Alarcon-Gutierrez E, Criquet S. 2007. ABTS assay of phenol oxidase activity in soil. J. Microbiol. Meth. 71:319–324 [DOI] [PubMed] [Google Scholar]
- 27. Galagan JE, et al. 2002. The genome of M-acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12:532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gardes M, Bruns TD. 1993. Its primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol. Ecol. 2:113–118 [DOI] [PubMed] [Google Scholar]
- 29. Gilbert D, Mitchell EAD. 2006. Microbial diversity in Sphagnum peatlands, p 287–318 In Martini IP, Cortizas AM, Chesworth W. (ed), Developments in earth surface processes: peatlands—evolution and records of environmental and climate changes, vol 9 Elsevier, Oxford, United Kingdom [Google Scholar]
- 30. Glaser PH. 1992. Vegetation and water chemistry, patterned peatlands of northern Minnesota. University of Minnesota Press, Minneapolis, MN [Google Scholar]
- 31. Gossner AS, Picardal F, Tanner RS, Drake HL. 2008. Carbon metabolism of the moderately acid-tolerant acetogen Clostridium drakei isolated from peat. FEMS Microbiol. Lett. 287:236–242 [DOI] [PubMed] [Google Scholar]
- 32. Gupta V, Smemo KA, Yavitt JB, Basiliko N. 2012. Active methanotrophs in two contrasting North American peatland ecosystems revealed using DNA-SIP. Microb. Ecol. 63:438–445 [DOI] [PubMed] [Google Scholar]
- 33. Hales BA, et al. 1996. Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis. Appl. Environ. Microbiol. 62:668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamberger A, Horn MA, Dumont MG, Murrell JC, Drake HL. 2008. Anaerobic consumers of monosaccharides in a moderately acidic fen. Appl. Environ. Microbiol. 74:3112–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hobbie EA, Watrud LS, Maggard S, Shiroyama T, Rygiewicz PT. 2003. Carbohydrate use and assimilation by litter and soil fungi assessed by carbon isotopes and BIOLOG (R) assays. Soil Biol. Biochem. 35:303–311 [Google Scholar]
- 36. Horn MA, Matthies C, Kusel K, Schramm A, Drake HL. 2003. Hydrogenotrophic methanogenesis by moderately acid-tolerant methanogens of a methane-emitting acidic peat. Appl. Environ. Microbiol. 69:74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keller JK, Bridgham SD. 2007. Pathways of anaerobic carbon cycling across an ombrotrophic-minerotrophic peatland gradient. Limnol. Oceanogr. 52:96–107 [Google Scholar]
- 38. Khalil MAK. 2000. Atmospheric methane: its role in the global environment. Springer, New York, NY [Google Scholar]
- 39. Kolb S, Knief C, Dunfield PF, Conrad R. 2005. Abundance and activity of uncultured methanotrophic bacteria involved in the consumption of atmospheric methane in two forest soils. Environ. Microbiol. 7:1150–1161 [DOI] [PubMed] [Google Scholar]
- 40. Kostka JE, et al. 2011. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 77:7962–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kotsyurbenko OR, et al. 2004. Acetoclastic and hydrogenotrophic methane production and methanogenic populations in an acidic West-Siberian peat bog. Environ. Microbiol. 6:1159–1173 [DOI] [PubMed] [Google Scholar]
- 42. Kuhner CH, et al. 2000. Clostridium akagii sp. nov. and Clostridium acidisoli sp. nov.: acid-tolerant, N-2-fixing clostridia isolated from acidic forest soil and litter. Int. J. Syst. Evol. Microbiol. 50:873–881 [DOI] [PubMed] [Google Scholar]
- 43. Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow M. (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 44. Lara E, Mitchell EAD, Moreira D, Garcia PL. 2011. Highly diverse and seasonally dynamic protist community in a pristine peat bog. Protist 162:14–32 [DOI] [PubMed] [Google Scholar]
- 45. Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75:5111–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le Mer J, Roger P. 2001. Production, oxidation, emission and consumption of methane by soils: a review. Eur. J. Soil Biol. 37:25–50 [Google Scholar]
- 47. Lin XJ, Kennedy D, Fredrickson J, Bjornstad B, Konopka A. 2012. Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ. Microbiol. 14:414–425 [DOI] [PubMed] [Google Scholar]
- 48. Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66:506–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medlin L, Elwood HJ, Stickel S, Sogin ML. 1988. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71:491–499 [DOI] [PubMed] [Google Scholar]
- 50. Millard P, Singh BK. 2010. Does grassland vegetation drive soil microbial diversity? Nutr. Cycl. Agroecosyst. 88:147–158 [Google Scholar]
- 51. Moore TR, Basiliko N. 2006. Decomposition in boreal peatlands, p 125–143 In Wieder RK, Vitt DH. (ed), Boreal peatland ecosystems. Springer-Verlag, Berlin, Germany [Google Scholar]
- 52. Moore TR, Roulet NT, Waddington JM. 1998. Uncertainty in predicting the effect of climatic change on the carbon cycling of Canadian peatlands. Climat. Change 40:229–245 [Google Scholar]
- 53. Nielsen UN, Osler GHR, Campbell CD, Burslem DFRP, van der Wal R. 2010. The influence of vegetation type, soil properties and precipitation on the composition of soil mite and microbial communities at the landscape scale. J. Biogeogr. 37:1317–1328 [Google Scholar]
- 54. Oh S, et al. 2011. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl. Environ. Microbiol. 77:6000–6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Opelt K, et al. 2007. Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ. Microbiol. 9:2795–2809 [DOI] [PubMed] [Google Scholar]
- 56. Pankratov TA, Ivanova AO, Dedysh SN, Liesack W. 2011. Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ. Microbiol. 13:1800–1814 [DOI] [PubMed] [Google Scholar]
- 57. Pankratov TA, Kirsanova LA, Kaparullina EN, Kevbrin VV, Dedysh SN. 2011. Telmatobacter bradus gen. nov., sp. nov., a cellulolytic facultative anaerobe from subdivision 1 of the Acidobacteria and emended description of Acidobacterium capsulatum Kishimoto et al. 1991. Int. J. Syst. Evol. Microbiol. 62(Pt 2):430–437 [DOI] [PubMed] [Google Scholar]
- 58. Peltoniemi K, Fritze H, Laiho R. 2009. Response of fungal and actinobacterial communities to water-level drawdown in boreal peatland sites. Soil Biol. Biochem. 41:1902–1914 [Google Scholar]
- 59. Peres-Neto PR, Jackson DA. 2001. How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia 129:169–178 [DOI] [PubMed] [Google Scholar]
- 60. Pester M, Knorr K-H, Friedrich M, Wagner M, Loy A. 2012. Sulfate-reducing microorganisms in wetlands—fameless actors in carbon cycling and climate change. Front. Microbiol. 3:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Preston MD, Smemo KA, McLaughlin JW, Basiliko N. 2012. Peatland microbial communities and decomposition processes in the James Bay Low lands, Canada. Front. Microbiol. 3:70 doi:10.3389/fmicb.2012.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Prevost-Boure NC, et al. 2011. Validation and application of a PCR primer set to quantify fungal communities in the soil environment by real-time quantitative PCR. PLoS One 6:e24166 doi:10.21371/journal.pone.0024166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Raskin L, Stromley JM, Rittmann BE, Stahl DA. 1994. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 60:1232–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodriguez-Brito B, et al. 2010. Viral and microbial community dynamics in four aquatic environments. ISME J. 4:739–751 [DOI] [PubMed] [Google Scholar]
- 65. Rooney-Varga JN, Giewat MW, Duddleston KN, Chanton JP, Hines ME. 2007. Links between archaeal community structure, vegetation type and methanogenic pathway in Alaskan peatlands. FEMS Microbiol. Ecol. 60:240–251 [DOI] [PubMed] [Google Scholar]
- 66. Rudolph H, Samland J. 1985. Occurrence and metabolism of Sphagnum acid in the cell-walls of Bryophytes. Phytochemistry 24:745–749 [Google Scholar]
- 67. Rui JP, Peng JJ, Lu YH. 2009. Succession of bacterial populations during plant residue decomposition in rice field soil. Appl. Environ. Microbiol. 75:4879–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sait M, Davis KE, Janssen PH. 2006. Effect of pH on isolation and distribution of members of subdivision 1 of the phylum Acidobacteria occurring in soil. Appl. Environ. Microbiol. 72:1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Strickland MS, Rousk J. 2010. Considering fungal:bacterial dominance in soils—methods, controls, and ecosystem implications. Soil Biol. Biochem. 42:1385–1395 [Google Scholar]
- 70. Tfaily MM, Corbett JE, Chanton JP, Cooper WT. 2011. Characterization of dissolved organic matter in peatland porewaters using Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS), excitation-emission matrix fluorescence spectroscopy and parallel factor analysis (EEMS-PARAFAC), p 557 In Geological Society of America abstracts with programs, vol 43, no 5 Geological Society of America, Boulder, CO [Google Scholar]
- 71. Tfaily MM, Podgorski DC, Corbett JE, Chanton JP, Cooper WT. 2011. Influence of acidification on the optical properties and molecular composition of dissolved organic matter. Anal. Chim. Acta 706:261–267 [DOI] [PubMed] [Google Scholar]
- 72. Thormann MN. 2006. Diversity and function of fungi in peatlands: a carbon cycling perspective. Can. J. Soil Sci. 86:281–293 [Google Scholar]
- 73. Trinder CJ, Johnson D, Artz RRE. 2008. Interactions among fungal community structure, litter decomposition and depth of water table in a cutover peatland. FEMS Microbiol. Ecol. 64:433–448 [DOI] [PubMed] [Google Scholar]
- 74. Walker MD, et al. 2006. Plant community responses to experimental warming across the tundra biome. Proc. Natl. Acad. Sci. U. S. A. 103:1342–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ward NL, et al. 2009. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 75:2046–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weedon JT, et al. 2012. Summer warming accelerates sub-arctic peatland nitrogen cycling without changing enzyme pools or microbial community structure. Global Change Biol. 18:138–150 [Google Scholar]
- 77. Weishaar JL, et al. 2003. Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon. Environ. Sci. Technol. 37:4702–4708 [DOI] [PubMed] [Google Scholar]
- 78. Wigley TML, Schimel DS. 2000. The carbon cycle. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 79. Winsborough C, Basiliko N. 2010. Fungal and bacterial activity in northern peatlands. Geomicrobiol. J. 27:315–320 [Google Scholar]
- 80. Young-Ju K, Zhao Y, Oh KT, Nguyen VN, Park RD. 2008. Enzymatic deacetylation of chitin by extracellular chitin deacetylase from a newly screened Mortierella sp. DY-52. J. Microbiol. Biotechnol. 18:759–766 [PubMed] [Google Scholar]
- 81. Zinder SH. 1993. Physiological ecology of methanogens, p 128–206 In Ferry JG. (ed), Methanogenesis. Routledge, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.