

Abstract
The extracellular acid proteases of non-Saccharomyces wine yeasts may fulfill a number of roles in winemaking, which include increasing the available nitrogen sources for the growth of fermentative microbes, affecting the aroma profile of the wine, and potentially reducing protein haze formation. These proteases, however, remain poorly characterized, especially at genetic level. In this study, two extracellular aspartic protease-encoding genes were identified and sequenced, from two yeast species of enological origin: one gene from Metschnikowia pulcherrima IWBT Y1123, named MpAPr1, and the other gene from Candida apicola IWBT Y1384, named CaAPr1. In silico analysis of these two genes revealed a number of features peculiar to aspartic protease genes, and both the MpAPr1 and CaAPr1 putative proteins showed homology to proteases of yeast genera. Heterologous expression of MpAPr1 in Saccharomyces cerevisiae YHUM272 confirmed that it encodes an aspartic protease. MpAPr1 production, which was shown to be constitutive, and secretion were confirmed in the presence of bovine serum albumin (BSA), casein, and grape juice proteins. The MpAPr1 gene was found to be present in 12 other M. pulcherrima strains; however, plate assays revealed that the intensity of protease activity was strain dependent and unrelated to the gene sequence.
INTRODUCTION
Aspartic proteases (EC 3.4.23), also known as acid proteases (APs), have been isolated from a range of organisms, including retroviruses, bacteria, fungi, insects, and vertebrates. Some common examples include pepsin, cathepsin D, chymosin, and the microbial penicillopepsin, with pepsin being the most studied AP. These enzymes may be intracellular or extracellular, and they are active under acidic conditions (pH 2 to 5) and have molecular masses ranging from 35 kDa to 50 kDa and isoelectric points (pIs) of 3.9 to 4.9 (3, 12, 42). The APs have two reactive aspartic acid residues in their catalytic sites that are essential for their functioning. The two aspartic acid residues are found within two characteristic hydrophobic sequences, Asp32-Thr-Gly-Ser in the N-terminal domain and a corresponding Asp215-Thr-Gly-Ser/Thr in the C-terminal domain (according to pepsin numbering) (14). The enzymes are inhibited by pepstatin A, a hexapeptide from Streptomyces (12), and are also sensitive to diazoacetylnorleucinemethyl (DAN), and 1,2-epoxy-3-(p-nitrophenoxy)propane (EPNP) in the presence of copper ions (http:/merops.sanger.ac.uk).
These proteases are monomers and have a bilobal structure, with the active-site cleft located at the interface where the two lobes meet. Each lobe contributes an aspartic acid residue to the active site. The enzymes also have a flap region that closes over the active-site cleft. The retroviral APs are smaller homodimers formed by two identical domains (46). APs are endopeptidases that cleave peptides of at least 6 residues long with hydrophobic residues in the scissile bond. The mechanism of action is a general acid-general base mechanism, where one of the aspartic acid residues in the active site acts as an acid while the other functions as a base. A water molecule, hydrogen bonded between the two aspartic acid residues, plays a central role in the catalytic reaction (14).
The extracellular acid proteases of fungi and yeasts have been studied extensively, for example, those of Aspergillus, Penicillium, and Candida spp. They show specificity against aromatic or bulky amino acid residues on both sides of the scissile peptide bond, and their action is less stringent than that of, e.g., pepsin (39, 42). The pathogenic yeast Candida albicans has 10 secreted aspartic proteases (Saps), which are encoded by 10 genes (SAP1 to SAP10) (37). The secretory pathway of the APs secreted by C. albicans and other Candida spp. has been investigated. The process is similar to that found in Saccharomyces cerevisiae. The SAP genes are translated as preproenzymes on the rough endoplasmic reticulum (ER). The prepeptide (signal peptide) is cleaved in the ER by a signal peptidase complex, followed by glycosylation and formation of disulfide bonds. The Saps undergo further maturation during transportation via the Golgi apparatus, where processing is performed by a Kex2 protease (37, 38, 45). At the end of the secretory pathway, Saps are either incorporated to the cell wall via a glycosylphosphatidylinositol (GPI) anchor or released into the extracellular space (1).
Most commercial winemakers inoculate their grape juice with commercially available S. cerevisiae strains (inoculated at an initial concentration of 3 × 106 cells/ml) in order to obtain a uniform and predictable product and also to minimize the growth of other microbes present in the grape must (15). Recently, however, the role and contribution of the non-Saccharomyces yeasts (present mostly during the initial stages of spontaneous wine fermentation) in the final wine product have been investigated (8, 21, 26). These include yeasts from the genera Rhodotorula, Pichia, Candida, Metschnikowia, Kloeckera, and Hansenula, among others. Some of the non-Saccharomyces yeasts produce metabolites such as esters, higher alcohols, acetic acid, and acetoin, which may contribute positively or negatively to the flavor complexity of the wine (9, 34). They also secrete enzymes, e.g., pectinases, β-glucosidases, and proteases, that might be of interest to the winemaker. The proteases not only can increase the concentration of assimilable nitrogen sources for the growth of desirable (and spoilage) microbes but also can improve clarification and possibly reduce wine protein haze (21, 25, 41). Treatments such as bentonite fining and aging on total yeast lees to reduce the risk of haze formation can be expensive, among other disadvantages (51). Successful protease treatment prior to fining may reduce costs. Although many non-Saccharomyces wine yeasts have been shown to have extracellular proteolytic activity, the characteristics of these enzymes have not been studied at the genetic level.
This study describes the isolation and partial characterization of acid protease-encoding genes from two wine-associated yeasts, Metschnikowia pulcherrima IWBT Y1123 and Candida apicola IWBT Y1384. The deduced protein sequences were characterized by in silico investigations, and the gene isolated from M. pulcherrima IWBT Y1123 was expressed in S. cerevisiae YHUM272. Protease induction studies were also performed.
MATERIALS AND METHODS
Strains and culture conditions.
The strains used in this study as well as their sources are listed in Table 1. Yarrowia lipolytica UOFS Y1698 was provided by Lodewyk Kock, University of the Free State, South Africa. Metschnikowia pulcherrima FOEB L0642 was provided by Isabelle Masneuf-Pomarède, ENITA de Bordeaux, France. The yeast strains were maintained on yeast extract-peptone-dextrose (YPD) agar (Biolab Diagnostics, Wadenville, South Africa) and freshly cultured prior to use in experiments. Yeast strains were grown at 30°C in YPD broth (Biolab Diagnostics). Plasmids were constructed and grown in Escherichia coli DH5α grown at 37°C on a rotary shaker at 150 rpm in Luria-Bertani medium (Biolab Diagnostics, Wadenville, South Africa) supplemented with 100 mg/liter ampicillin (ampicillin sodium salt; Sigma-Aldrich, Johannesburg, South Africa), 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) (Sigma-Aldrich), and 80 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (Sigma-Aldrich) when appropriate. Chemically competent E. coli DH5α cells were transformed according to the Promega technical manual (TM042; Promega, Whitehead Scientific, Cape Town, South Africa). Solid media contained 20 g/liter agar. All strains were stored at −80°C in broth containing 40% (vol/vol) glycerol.
Table 1.
Strains used in this study
| Microbe | Strain | Description or genotype | Collection or reference |
|---|---|---|---|
| Saccharomyces cerevisiae | VIN13 | Wine yeast | Anchor Yeast, Cape Town, South Africa |
| Yarrowia lipolytica | Y1698 | Dairy yeast | UOFSa |
| Saccharomyces cerevisiae | YHUM272 | Σ1278b MATα ura3-52 Δtrp1::hisG Δleu2::hisG Δhis3::hisG | 47 |
| Aureobasidium pullulans | Y1008 | Wine yeast-like fungus | IWBTb |
| Candida apicola | Y1384 | Wine yeast | IWBT |
| Metschnikowia pulcherrima | L0642 | Wine yeast | FOEBc |
| Y1123 | Wine yeast | IWBT | |
| Y1072 | Wine yeast | IWBT | |
| Y1065 | Wine yeast | IWBT | |
| Y1063 | Wine yeast | IWBT | |
| Y1094 | Wine yeast | IWBT | |
| Y1325 | Wine yeast | IWBT | |
| Y1213 | Wine yeast | IWBT | |
| Y1207 | Wine yeast | IWBT | |
| Y1125 | Wine yeast | IWBT | |
| Y1120 | Wine yeast | IWBT | |
| Y1108 | Wine yeast | IWBT | |
| Y1115 | Wine yeast | IWBT | |
| Y1337 | Wine yeast | IWBT | |
| Escherichia coli | DH5α | [F− ϕ80lacZΔM15Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 thi-1 gyrA96 relA1 λ−] | GIBCO-Invitrogen Life Technologies, Mowbray, South Africa |
UFS MIRCEN Yeast Culture Collection, Bloemfontein, South Africa.
Institute for Wine Biotechnology, Stellenbosch, South Africa.
Faculté d'Oenologie de Bordeaux, Bordeaux, France.
Induction of protease secretion.
To induce protease secretion, a method described previously (25) was followed with modifications. The yeast strain M. pulcherrima IWBT Y1123 was grown in 10 ml MYGP medium (0.3% malt, 0.3% yeast extract, 0.5% peptone, and 2% glucose) for 24 h at 30°C on a rotating wheel. The cells were harvested by centrifugation at 5,000 rpm for 5 min at 4°C and washed twice with 0.9% physiological water. The cells were then added to a base medium containing 1% glucose, 0.1% Difco yeast nitrogen base (YNB) (without amino acids and ammonium sulfate), and 0.066% ammonium sulfate. The pH was adjusted to 5.5 with 1 N NaOH, and the medium was filter sterilized. The culture was grown for 24 h at 30°C with shaking at 160 rpm. After the 24-h incubation period, the cultures were spiked with different nitrogen sources, which included 0.250 mg/ml grape juice proteins, 0.250 mg/ml bovine serum albumin (BSA) (fraction V; Roche, Mannheim Germany), 0.250 mg/ml casein (Sigma-Aldrich), and 0.250 mg/ml ammonium sulfate (Merck, Wadeville, South Africa). As negative control, one flask was not spiked with any nitrogen source. The cultures were grown for a further 48 h at 30°C and 160 rpm. At the end of induction, the cultures were centrifuged at 5,000 rpm for 10 min at 4°C. The cell pellet was frozen at −80°C for RNA extraction. The crude supernatant was filtered through a 0.45-μm filter and concentrated 6 times by filtration using Amicon centrifugal filter units (Millipore, Davies Diagnostics, Randburg, South Africa) with a 10-kDa cutoff. The retentate was used as the crude protease preparation and stored at 4°C. The total protein concentration was determined using the Bio-Rad protein assay kit (Bio-Rad Labs, Hercules, CA) according to the manufacturer's protocol. Experiments were performed in triplicate.
Grape juice proteins were extracted from Chardonnay grape juice by acetone precipitation. Grape juice was obtained from the Nietvoorbij experimental cellar (Agricultural Research Council, Stellenbosch, South Africa) from grapes harvested during the 2011 harvest season. One volume of ice-cold 100% acetone (Merck, Wadeville, South Africa) was added to the grape juice and incubated overnight at −20°C. The proteins were recovered by centrifugation at 10,000 rpm for 30 min at 4°C. The protein pellet was washed twice with 4:1 acetone-water and dried overnight at −20°C. The proteins were resuspended in 0.05 M citrate phosphate buffer, pH 3.5.
Acid protease activity determination.
Acid protease activity was determined by spotting of the cells on skim milk plates at pH 3.5 as previously described (7). Pure colonies were suspended in 10 μl of Milli-Q water (Millipore) and spotted on plates. The plates were incubated at 30°C for 3 days. Enzymatic activity was visualized by a zone of clearance of at least 1 mm around the edges of the yeast colony. All assays were performed in triplicate.
Nucleic acid extraction.
Genomic DNA (gDNA) was isolated from 24-h YPD cultures grown at 30°C with shaking at 160 rpm. gDNA and RNA were extracted from yeast cultures using the methods described in references 19 and 10, respectively. Plasmid DNA was recovered from E. coli cultures using the QIAprep Spin Miniprep kit (Qiagen, Southern Cross Biotechnology, Cape Town, South Africa) according to the manufacturer's instructions. Genomic DNA and RNA concentrations were quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Products, Wilmington, DE).
In silico analyses.
Homology searches of nucleotide sequences and database searches were carried out using the Basic Local Alignment Search Tool (BLAST) service provided by the National Center for Biotechnology Information (NCBI) website (www.ncbi.nlm.nih.gov/BLAST). Multiple-sequence alignments and comparisons of DNA sequences were performed using the programs of MultAlin (http://multalin.toulouse.inra.fr/multalin) and ClustalW alignment software provided by the European Biotechnology Institute (EBI) (www.ebi.ac.uk/clustalw). The nucleotide sequences were translated into amino acid sequences and alignments were performed using Transeq and Showalign from the EMBOSS software suite (http://www.ebi.ac.uk). Degenerate primers were designed using the program http://hcgs.unh.edu/protocol/basic/pcrdegenpri.html. The secretion signal peptide of the putative protein was detected by the use of the software (http://cbs.dtu.dk/services/SignalP) (version 3.0) on the CBS website. For the calculation of the pIs and molecular weights of the putative proteins, the software of the Expasy website was utilized (http://web.expasy.org), and for prediction of N-glycosylation sites, the software at http://www.cbs.dtu.dk/services/NetNGlyc was used. To investigate and compare the conserved regions and motifs in the protein sequences with other proteins, the conserved-domain database (CDD) (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) at NCBI was used.
PCR methods.
The primers and PCR programs used in this study are listed in Table 2 and Table 3, respectively. Primer oligonucleotide sequences were synthesized by Integrated DNA Technologies (IDT, Whitehead Scientific). All PCRs were performed using the Phusion high-fidelity DNA polymerase system (Finnzymes, Vantaa, Finland) unless otherwise specified. PCR programs were run with an Applied Biosystems 2720 thermal cycler (Applied Biosystems, Foster City, CA). In order to generate sticky ends, PCR products were incubated for a further 10 min at 72°C with addition of dATP nucleotides (TaKaRa Bio Inc., Separations, Randburg, South Africa) when necessary. Fragments were purified from agarose gel using the Zymoclean Gel DNA recovery kit (Zymo Research, Irvine, CA). PCR products were cloned with pGEM-T Easy vector systems (Promega).
Table 2.
Primers used in this study
| Primer type and name | Primer sequence (5′→3′)a | Reference |
|---|---|---|
| ITS1 | TCCGTAGGTGAACCTGCGG | 52 |
| ITS4 | TCCTCCGCTTATTGATATGC | 52 |
| Degenerate primers | ||
| AXP1 forward | GAYACNGGNTCNTCNGAY | This study |
| AXP1 reverse | NGANGTNGCNGARTCNAR | This study |
| Inverse primers | ||
| Mpulch_ IPCR_ F1 | CAGATCTCGGCAAGGGCTCGTTGAT | This study |
| Mpulch_ IPCR_ R1 | TGCGCAAACTGGAAATTGGAAAGCA | This study |
| Mpulch_ IPCR_ F2 nested | GGTGAACGCCCCAGTTCT | This study |
| Mpulch_ IPCR_ R2 nested | AGCTGGAACTGTCTGGGCTA | This study |
| Capi_IPCR_F1 | CAGTGCCCAGAATGGAAGCAGTGTG | This study |
| Capi_IPCR_R1 | ACACCGGCAGTTTCGCCCTGAGTAG | This study |
| Capi_IPCR_F2 nested | TGGTAACAAGTCGAATGGTGTG | This study |
| Capi_IPCR_R2 nested | CCTTGAAGTTTGTCTGATTTGTG | This study |
| Primers for full gene | ||
| MpAPr1-F | GGATCCATGCAATTCCTCACTCTTCTTTC | This study |
| MpAPr1-R | CTCGAGTTAAGCACTTATGATGTTTGACGA | This study |
| CaAPr1-F | GGATCCATGGTACTAGCTAAGAACTATGTT CATTTA | This study |
| CaAPr1-R | CTCGAGTTAGTTGACAGATCCGGGAAT | This study |
| 5′-KPNPGK-631 | GGGGTACCCTTTATTTTGGCTTCACCC | 48 |
| NL4 | GGTCCGTGTTTCAAGACGG | 23 |
| NL1 | GCATATCAATAAGCGGAGGAAAAG | 23 |
| MpAPr1-qPCR-fw | ACACCCAAGGCGTCATACTC | This study |
| MpAPr1-qPCR-rev | ACAGGTCAATCGGGTACAGC | This study |
| ACT1-qPCR-fw | CTCCATGCCTCACGGTATTT | This study |
| ACT1-qPCR-rev | CTCCTGCTCAAAGTCCAAGG | This study |
Underlined sequences indicate restriction sites: GGATCC, BamHI; CTCGAG, XhoI; GGTACC, KpnI.
Table 3.
PCR amplification programs
| Primer pair | Initial denaturation temp (°C)/time (min) | Main cycling conditions |
Final extension temp (°C)/time (min) | |||
|---|---|---|---|---|---|---|
| No. of cycles | Temp (°C)/time (s) |
|||||
| Denaturing | Annealing | Extension | ||||
| ITS1/ITS4 | 95/5 | 40 | 95/1 | 58/1 | 72/1 | 72/7 |
| AXP1 forward/reverse | 94/7 | 45 | 94/30 | 50/60 | 72/0.6 | 72/7 |
| Mpulch_ IPCR_ F1/R1 | 94/2 | 30 | 94/20 | 68/20 | 68/5 | 68/5 |
| Capi_IPCR_F1/R1 | 94/2 | 30 | 94/20 | 60/20 | 68/5 | 68/5 |
| Mpulch_ IPCR_ F2/R2 nested | 94/2 | 30 | 94/20 | None | 68/5 | 68/5 |
| Capi_IPCR_F2/R2 nested | 94/2 | 30 | 94/20 | None | 68/5 | 68/5 |
| MpAPr1-F/R | 98/0.5 | 35 | 98/10 | 58/20 | 72/0.5 | 72/7 |
| CaAPr1-F/R | 98/0.5 | 35 | 98/10 | 58/20 | 72/0.5 | 72/7 |
| 5′-KPNPGK-631/Mpulch_ IPCR_ R2 | 96/2 | 35 | 94/30 | 58/30 | 72/0.6 | 72/10 |
| NL4/NL1 | 98/0.5 | 35 | 98/10 | 58/20 | 72/0.5 | 72/7 |
(i) Yeast identification.
In order to identify the yeasts, gDNAs were amplified with the primers ITS1 and ITS4 (Table 2) using the previously described (29, 52) PCR program (Table 3).
(ii) Degenerate PCR.
To obtain the partial gene sequences of the acid proteases of M. pulcherrima IWBT Y1123, Aureobasidium pullulans IWBT Y1008, and C. apicola IWBT Y2384, a PCR-based approach was followed. The amino acid sequence of the aspartic protease of Y. lipolytica strain CLIB122 (accession number XP_504725.1) (13) was subjected to BLAST analysis and aligned with aspartic proteases of other yeast species. The conserved sequences, which are also identified as the active-site regions of the enzymes, were used to design degenerate primers.
(iii) IPCR.
gDNA was digested separately with 5 different restriction enzymes, EcoRI, EcoRV, DraI, HpaI, and XbaI (Roche Diagnostics, Randburg, South Africa). These enzymes were selected based on the facts that they (i) do not cut within the sequence obtained from amplification with degenerate primers and (ii) would result in fragments of between 2,000 and 4,000 bp. This was determined by using the genome sequence of Candida glabrata (22), which is closely related to M. pulcherrima. In short, 200 ng genomic DNA was digested in a 50-μl reaction mix for 2 h at 37°C, followed by inactivation of the restriction enzymes at 65°C for 20 min. Self-ligation proceeded in 200-μl reaction mixtures at 16°C overnight using T4 ligase (Promega). The inverse PCR (IPCR) was performed with 2 μl of the ligation reaction mix in 50-μl reaction mixtures using Elongase (Invitrogen). After amplification, the reaction mix was diluted 100 times, and 2 μl thereof was used as the template for a nested IPCR. The fragments were cloned and sequenced as previously described. Following in silico analysis, new primers were designed based on the sequences obtained from the IPCR. These were used to amplify the full-length genes encoding the acid proteases from gDNA.
(iv) Reverse transcription-PCR.
Reverse transcription was performed using the ImPromII reverse transcription system (Promega) according to the manufacturer's instructions. The oligo(dT)15 primer of the kit was used to initiate reverse transcription of poly(A)+ mRNA molecules. PCR on the obtained cDNA was carried out as described above.
(v) qPCR.
Quantitative PCR (qPCR) was performed in triplicate on the cDNA obtained from 3 biological repeats on a 7500 real-time PCR system (Applied Biosystems, Johannesburg, South Africa). Relative quantification was determined using the 2−ΔΔCT method. The ACT1 gene (accession no. AJ389070) was used for normalization purposes. The Kapa SYBR Fast qPCR kit (Kapa Biosystems, Cape Town, South Africa) was used for the reaction mix according to the manufacturer's instructions. Primer sets MpAPr1-qPCR-fw/rev and MpACT1-qPCR-fw/rev (Table 2) were designed to optimally amplify the MpAPr1 and the ACT1 genes of Metschnikowia pulcherrima, respectively. The qPCR program consisted of an initial denaturation at 95°C for 3 min followed by 40 cycles comprising a denaturation step at 95°C for 5 s and an annealing step at 60°C for 1 min. PCR efficiency (93% and 90%, respectively, with r2 values of 0.993 and 0.999, respectively) was calculated by performing qPCR with each set of primers using a dilution series of genomic DNA concentrations as the template. The absence of nonspecific annealing was further confirmed by running dissociation curves. One hundred nanograms of cDNA was used for each qPCR.
Cloning and heterologous expression in S. cerevisiae YHUM272.
After amplification of the protease-encoding gene from gDNA, the PCR product was cloned into pJET1.2 (CloneJet PCR cloning kit; Fermentas), followed by transformation of E. coli DH5α. After plasmid extraction, the gene was excised from the pJET1.2 plasmid by restriction digestion with BamHI and XhoI (Roche Diagnostics), ligated into the corresponding sites of the pCEL13 expression vector (18) using T4 DNA ligase (Fermentas), and transformed into E. coli DH5α. In the new plasmid pCEL13-MpAPr1 (Apr URA3 PGK1P-MpAPr1-PGK1T), which possesses a 2μ origin of replication, MpAPr1 is positioned under the control of the PGK1 promoter, which therefore allows its strong constitutive expression. pCEL13-MpAPr1 was then transformed into S. cerevisiae YHUM272 using an electroporation method (48). Positive transformants were selected on minimal medium (0.17% Difco YNB without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose) plates supplemented with 40 μg/ml tryptophan, 60 μg/ml leucine, and 20 μg/ml histidine.
To confirm successful transformation of the yeast with pCEL13 containing the gene, colony PCR with positive transformants as the template was performed. The primers 5′-KPNPGK-631, which recognizes the promoter sequence on the plasmid, and Mpulch_IPCR_R2 (nested) were used.
DNA sequencing.
DNA strands were sequenced in an ABI 3130XL Genetic Analyzer at the Central Analytical Facility (Stellenbosch University) using the SP6 and T7 primers (Promega) or pJET1.2-specific primers (Fermentas, Inqaba Biotec, South Africa).
SDS-PAGE and zymography.
SDS-PAGE analysis was performed on concentrated culture supernatants as previously described (24) with 12.5% bisacrylamide gels on a Bio-Rad Mini-Protean Tetra Cell system (Bio-Rad Labs, Hercules, CA). Zymography was performed at pH 3.5 and with gelatin as a copolymerized substrate (16). To visualize protein bands, gels were stained with Coomassie blue R-250.
Protein sequencing.
Selected protein bands were excised from bisacrylamide gels and, following trypsin digestion, were sequenced by nanoscale liquid chromatography (nano-LC) and LC-tandem mass spectrometry (LC-MS/MS) at the Central Analytical Facility of Stellenbosch University (Bellville, South Africa). Experiments were performed on a Thermo Scientific EASY-nLC II instrument connected to an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, Bremen, Germany) equipped with a nano-electrospray source. For liquid chromatography, separation was performed on an Easy-Column (2 cm; inner diameter,100 μm; 5 μm; C18) precolumn followed by an Easy-column (10 cm; inner diameter, 75 μm; 3 μm; C18) column with a flow rate of 300 nl/min. The gradient used was from 5 to 40% B in 20 min, from 40 to 80% B in 5 min, and at 80% B for 10 min. Solvent A was 100% water in 0.1% formic acid, and solvent B was 100% acetonitrile in 0.1% formic acid. The mass spectrometer was operated in data-dependent mode to automatically switch between Orbitrap-MS and LTQ-MS/MS acquisition. Data were acquired using the Xcaliber software package. The precursor ion scan MS spectra (m/z 400 to 2000) were acquired in the Orbitrap with resolution R of 60,000, with the number of accumulated ions being 1 × 106. The 20 most intense ions were isolated and fragmented in the linear ion trap (number of accumulated ions, 3 × 104) using collision-induced dissociation. The lock mass option (polydimethylcyclosiloxane; m/z 445.120025) enabled accurate mass measurement in both the MS and MS/MS modes. In data-dependent LC-MS/MS experiments, dynamic exclusion was used with a 30-s exclusion duration. Mass spectrometry conditions were 1.5 kV and a capillary temperature of 200°C, with no sheath and auxiliary gas flow. The ion selection threshold was 500 counts for MS/MS, and an activation Q value of 0.25 and an activation time of 10 ms were also applied for MS/MS. Thermo Proteome Discoverer 1.2.0.208 (Thermo Scientific, Bremen, Germany) was used to identify proteins via automated database searching (Mascot; Matrix Science, London, United Kingdom) of all tandem mass spectra against the deduced protein sequence using a decoy database search with a false-discovery rate (FDR) of 0.01. Two missed tryptic cleavages were allowed. Proteins were considered positively identified when they were identified with at least 2 tryptic peptides per proteins, a Mascot score of more than a P value of <0.05, and peptides of high and medium confidences as determined by Proteome Discoverer with a false-discovery rate of 0.01.
RESULTS
Protease activity screening and strain selection.
A collection of 308 yeast strains were isolated from grape juice which was pressed from Chardonnay grapes harvested during the 2009 harvest season in Stellenbosch, South Africa. The strains were screened for extracellular protease activity by performing plate assays under acidic conditions (pH 3.5). Three strains displayed strong protease activity at pH 3.5, namely, IWBT Y1123, IWBT Y1008, and IWBT Y1384 (data not shown). These isolates were identified to the species level by PCR amplification of the internal transcribed spacers (ITS1 and ITS2) and 5.8S rRNA gene regions, sequencing of the amplicons, and performing BLAST searches. The strains were designated M. pulcherrima for IWBT Y1123, A. pullulans for IWBT Y1008, and C. apicola for IWBT Y1384, according to closest related species with identity scores of more than 98%.
Isolation and cloning of the aspartic protease-encoding genes.
Degenerate primers were designed based on amino acid sequence similarities (the conserved active-site regions) of aspartic proteases from various yeast species, mainly Y. lipolytica and some Candida spp. PCR was performed on gDNAs of Y. lipolytica UOFS-Y1698 (used as reference strain), A. pullulans IWBT Y1008, M. pulcherrima IWBT Y1123 and FOEB L0642 (of South African and French origins, respectively), and C. apicola IWBT Y1384. The amplicons generated by PCR were run on an agarose gel, and the fragments corresponding to the expected size (∼570 bp based on the length of the sequence between the active site-encoding regions of the Y. lipolytica CLIB122 gene) were excised and used as the template for a second amplification with the same primers (data not shown). The fragments were excised from the agarose gel, ligated into the pGEM-T Easy vector, and transformed into E. coli DH5α. After extraction, the fragments in the plasmids were sequenced. Homology searches were performed with the deduced protein sequences of the PCR fragments. The deduced sequences of M. pulcherrima IWBT Y1123 and C. apicola IWBT Y1384 yielded hits with known aspartic proteases. However, that of A. pullulans IWBT Y1008 displayed no homology to acid proteases. As mentioned above, the degenerate primers used were designed from available yeast protease sequences and not from sequences of fungal proteases, of which A. pullulans is an example, and therefore it is not surprising that the PCR product obtained for this species did not correspond to a protease. Experiments were thus continued with the sequences of the two positive strains. New primers specific for each strain were designed from the putative partial gene sequences to perform nested IPCR as described in Materials and Methods. The fragments obtained from amplification with the nested primers from the digestion with DraI, EcoRI, EcoRV, and HpaI for IWBT Y1123 and with DraI, EcoRI, EcoRV, and XbaI for IWBT Y1384 were cloned and sequenced as before, followed by performance of BLAST searches of the deduced amino acid sequences in order to confirm acid protease homology. Positive hits were obtained. Finally, primers were designed to amplify the full-length genes encoding putative aspartic proteases (data not shown). According to the sequences obtained from these new primers, the putative gene of M. pulcherrima IWBT Y1123 is 1,137 bp long and that of C. apicola IWBT Y1384 is 1,101 bp long. The nucleotide and deduced amino acid sequences were analyzed in silico.
In silico analysis of the putative gene and deduced protein sequences.
Alignments of the sequences obtained, nucleotide and amino acid, are illustrated in Fig. 1. The putative genes were named MpAPr1 for the gene from the M. pulcherrima strain and CaAPr1 for that from the C. apicola strain. The molecular masses of the putative preproenzymes deduced from the gene sequences are 40.9 kDa (MpAPr1) and 39.1 kDa (CaAPr1). The predicted secretion signal peptide cleavage site for the preproprotein of M. pulcherrima IWBT Y1123 is between Gly16 and Met17, with the first 16 amino acids being the secretion signal peptide. Conflicting results were obtained from different on-line databases and software programs with regard to the prediction of the signal peptide of the enzyme of CaAPr1. The protein was found not to have a signal peptide but was still considered to be a secreted protein that follows a nonclassical secretion pathway according to the CBS Secretome 2.0 Server, http://www.cbs.dtu.dk/services/SecretomeP/. Only one potential N-glycosylation site could be identified for the MpAPr1 protein, while three potential sites were predicted for the CaAPr1 protein. Both putative proteins were classified as eukaryotic aspartic proteases (A1 family, EC 3.4.23) according to the conserved catalytic motifs (DTGS/DSGT) and active-site flap regions, which are typically conserved motifs of this family of proteases.
Fig 1.

Alignment of the amino acid sequences of the putative secreted aspartic proteases of M. pulcherrima IWBT Y1123 (A) and C. apicola IWBT Y1384 (B) with those of their closest relatives (C. lusitaniae accession number XM_002615870.1 and Y. lipolytica accession number XP_500342.1, respectively). The putative secretion signal peptide is underlined. The active-site sequences and active-site flap sequence are shaded in light and dark gray, respectively. The putative N-glycosylation sites are boxed with solid lines. The peptides identified by mass spectroscopy are underlined with dotted lines. Amino acids that can differ depending on the alleles are boxed with dotted lines.
Putative identification based on homology studies.
The putative gene sequence of MpAPr1 was compared to other nucleotide sequences by performing homology searches against the NCBI database. Only one significant hit was observed: Clavispora lusitaniae ATCC 42720 hypothetical protein, mRNA (accession number XM_002615870.1), with 52% coverage and 65% identity. This is an indication of the novelty of the gene sequence. The rest of the hits were with sequences of a wide variety of different organisms that shared between 86% and 96% identity but only 2% to 3% coverage with the gene sequence. It should be noted that the hits showed coverage of the gene sequences that encode one of the active-site regions of the putative acid protease, indicating the potential acid protease nature of the MpAPr1 product. Potential cleavage sites to remove the propeptides for the proteins are at position 59 for the MpAPr1 protein sequence and position 28 for CaAPr1, representing the common KR motif. Thus, the size of the mature proteins would be 303 amino acids (378 − 16 −59) for MpAPr1 and 339 amino acids for CaAPr1.
The putative protein sequence for MpAPr1 was processed against the NCBI database. The best score was with the aspartyl protease of C. lusitaniae ATCC 42720 (100% coverage and 56% identity), the same as with the nucleotide alignment. Unlike with the nucleotide sequences, all the matches were to fungal proteins, particularly of the Candida genus, with an average identity score of 40% and average coverage scores of between 79% and 97%.
Similar results were obtained for the gene sequence of CaAPr1. Very low coverage scores (between 2% and 7%) with relatively high identity scores (80% to 90%) were observed for the region corresponding to the active-site-encoding gene sequences. The best score was given with the aspartyl proteinase (PAPA) gene of the soil fungus Trichoderma asperellum (AY611632.1). Although most of the matches were with fungal species, others included aspartic peptidases and unidentified proteins from a variety of species, such as the house mouse Mus musculus (JN950245.1). The putative protein sequence for CaAPr1 also shared similarities with only fungal proteins, with average identity scores of 37% and average coverage scores of 88%.
A phylogenetic tree of the two putative proteins with protease proteins from the NCBI database is shown in Fig. 2. It is clear that the CaAPr1 protein is more closely related to the aspartic proteases of Y. lipolytica than the MpAPr1 protein is. The MpAPr1 protein, on the other hand, is closely related to the Candida spp.
Fig 2.

Phylogenetic tree of the deduced protein sequences of MpAPr1 of M. pulcherrima IWBT Y1123 and CaAPr1 of C. apicola IWBT Y1384 with aspartic proteases from other yeasts found on the NCBI website. The phylogenetic tree was constructed by using the neighbor-joining method (PHYLIP 3.56). The scale bar represents the number of base substitutions per site.
The role of M. pulcherrima strains has been investigated in fermentation studies (20, 43), and they are known to secrete a number of enzymes (7, 21, 44) and therefore hold great biotechnological potential, especially for the wine industry. However, the proteolytic enzyme(s) of M. pulcherrima has not been studied at a genetic level. It was subsequently decided to continue only with the putative MpAPr1 gene.
Heterologous expression of the protease-encoding gene of M. pulcherrima IWBT Y1123 in S. cerevisiae YHUM272.
The MpAPr1 putative gene sequence of M. pulcherrima IWBT Y1123 was cloned into the shuttle vector pCEL13 for expression in the laboratory strain S. cerevisiae YHUM272. The yeast was also transformed with the vector not containing the gene. Successful transformation was confirmed by PCR with primers 5′-KPNPGK-631 and Mpulch_ IPCR_ R2 (nested). Colony PCR was performed on the yeast transformed with MpAPr1, the yeast transformed with the empty vector, and the untransformed yeast. The presence of the putative gene fragment was detected only in the yeast transformed with MpAPr1 and not in the other strains (Fig. 3A).
Fig 3.

Confirmation of heterologous expression of MpAPr1 in S. cerevisiae YHUM272. (A) PCR amplification with primers 5′-KPNPGK-631 and Mpulch_ IPCR_ R2. Lanes: a, colony PCR of YHUM272 transformed with MpAPr1; b, amplification of plasmid pCEL13 without the insert; c, colony PCR of untransformed YHUM272; d, amplification of pCEL13 ligated with MpAPr1 (PCR positive control); e, PCR negative control; M, molecular size marker (GeneRuler 100-bp DNA ladder Plus (Fermentas). (B) Plate assay of extracellular protease activity. 1, recombinant YHUM272 transformed with MpAPr1; 2, YHUM272 transformed with empty vector; 3, untransformed YHUM272; 4, M. pulcherrima IWBT Y1123 (positive control); 5, S. cerevisiae VIN13 (negative control).
The extracellular acid protease activities of the transformed strains were investigated by plate assays. The substrate used was skim milk, and the pH was adjusted to 3.5. Only the recombinant strain and the IWBT Y1123 strain showed extracellular protease activity as indicated by the zone of clearance around the colonies (Fig. 3B). No activity was observed from the strain that contained the empty plasmid or from the untransformed yeast. The results confirmed that the MpAPr1 gene from M. pulcherrima IWBT Y1123 indeed encodes an extracellular acid protease enzyme.
Induction of protease production by complex nitrogen sources.
The induction and substrate specificity of the MpAPr1 gene of M. pulcherrima IWBT Y1123 were studied upon exposure to different nitrogen sources by simultaneously investigating gene expression and the presence of the protease in the extracellular medium. After preculturing M. pulcherrima IWBT Y1123, cells were transferred to minimal medium and incubated for 1 day, after which cultures were spiked with different nitrogen sources and incubated for 2 more days, as explained in Materials and Methods. The different nitrogen sources included ammonium sulfate, BSA, casein, and grape juice proteins. As a control, a fifth culture received no nitrogen source addition for the final 2-day incubation. The experiments were performed in triplicate. The cells were harvested by centrifugation, and the culture supernatants were concentrated by ultrafiltration and used as the crude protease preparations. Total RNA was extracted from the harvested cells. Reverse transcription was performed on mRNA, and cDNA was used as the template for quantitative PCR (Fig. 4). The actin-encoding gene (ACT1) was used as a housekeeping gene. The expression of MpApr1 under the different conditions was compared to that in the absence of nitrogen source. The results show that MpApr1 was expressed at the same level regardless of the presence of a nitrogen source and of its nature.
Fig 4.
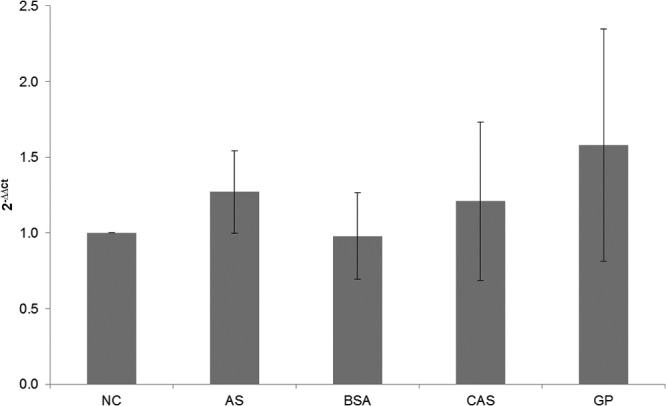
Relative expression analysis of MpAPr1 of M. pulcherrima IWBT Y1123 grown in media with different nitrogen sources using qPCR. NC, culture with no nitrogen addition (blank, used as a reference); AS, culture with ammonium sulfate addition; BSA, culture with bovine serum albumin addition; CAS, culture with casein addition; GP. culture with grape juice protein addition.
In order to assess the presence of the protease and its activity in the extracellular medium, the concentrated culture supernatants representing total extracellular proteins were analyzed by SDS-PAGE. The protease activity was visualized by zymography at pH 3.5 using gelatin as a protease substrate. The analysis of the extracellular proteins of M. pulcherrima IWBT Y1123 grown on grape juice proteins is illustrated in Fig. 5. Lane a shows the profile of the grape proteins after extraction from grape juice by acetone precipitation. The band at ∼60 kDa may be grape invertase (27, 30), and the intense band just beneath 25 kDa may be the common grape proteins thaumatin-like protein (22 kDa) and/or chitinase (25 kDa) (49, 50). These proteins are known to be very stable due to their conformation, and thus degradation due to incubation conditions was not expected (51). Lane b shows the extracellular protein profile of M. pulcherrima IWBT Y1123 grown in the presence of grape juice proteins. The band at ∼35 kDa disappeared, and those at 10 kDa, 25 kDa, and 60 kDa decreased in intensity. New faint bands could be seen at between 25 kDa and 40 kDa. Four of these bands were excised from the gel and sequenced. Very faint protein bands were observed for the ammonium sulfate-grown culture supernatant as well as for the culture that had no nitrogen source addition (data not shown). These could be proteins secreted by the yeast or released from early autolysis due to starvation of the yeast. Activity could be visualized for the culture supernatant grown with grape juice proteins (Fig. 5 lane c) but not for that grown with ammonium sulfate or the blank sample (data not shown). This indicated the presence of the protease. A clear zone was visible from the top of the lane down to a region just above 40 kDa, corresponding to the zone of migration of the protein observed at 40 kDa on the SDS-polyacrylamide gel.
Fig 5.
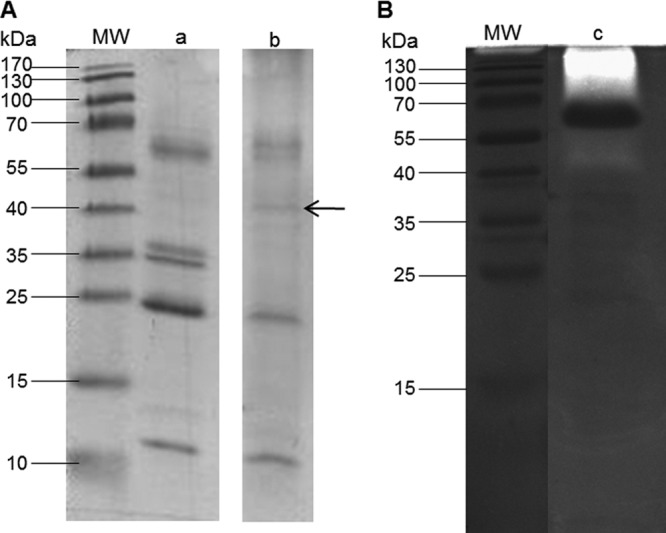
Analysis of the extracellular proteins of M. pulcherrima IWBT Y1123 cultured on grape juice proteins. (A) SDS-PAGE analysis. Lanes: a, grape juice proteins prior to culturing; b, culture supernatant. (B) Zymogram. Lane c, culture supernatant. Lanes MW, molecular mass standard proteins (PageRuler prestained ladder; Bio-Rad). The arrow points to the band excised and sequenced (estimated according to the theoretical molecular mass of 39.2 kDa).
Using casein or BSA as a source of nitrogen instead of grape proteins also showed evidence of protein degradation, with the appearance of lower-molecular-mass bands, but no clear activity was observed in the zymogram (data not shown). It is worth noting that after a 3-day incubation of casein or BSA in the medium in the absence of the yeast, no degradation of the casein was observed. This indicated that the partial degradation was not due to incubation conditions but was a result of proteolysis.
In order to identify whether the protease was indeed present in the culture supernatants, protein bands ranging from 35 kDa to 55 kDa were manually excised from lanes in SDS-polyacrylamide gels. The bands were trypsin digested, and nano-LC-MS/MS analysis was performed. The obtained peptide sequences were processed against the Mascot database and the deduced amino acid sequence for the MpAPr1 gene. One protein band from each of the two profiles, casein induced and grape protein induced, was positively identified to the deduced protein sequence for MpAPr1. For the band from the casein-induced culture, identification of the protein resulted in 21.27% sequence coverage of MpAPr1 with 6 peptides identified, and for the band from the grape protein-induced culture, identification resulted in 19.9% sequence coverage with 5 peptides identified, as shown in Fig. 1. The search against the Mascot database did not yield any other match.
A positive correlation was therefore found between the expression of the gene at the transcription level and protein production by the presence of the protease in the extracellular matrix of the yeast grown in casein and grape juice proteins. However, activity could not be visualized on the zymogram with casein. This could be because the concentration of the protease in the culture supernatant was too low.
Genetic screening of 12 M. pulcherrima strains for the presence of MpAPr1.
The identity of 12 M. pulcherrima strains previously isolated from grape juice of different harvest seasons was confirmed by amplifying and sequencing the ITS-5.8S rDNA locus and performing BLAST searches as well as by performing restriction fragment length polymorphism (RFLP) analysis on the amplicons (data not shown). RFLP analysis was performed with two restriction endonucleases, CfoI and HaeIII.
In order to evaluate the presence of the MpAPr1 gene, PCR was performed on the gDNAs of the 12 strains with the MpAPr1-amplifying primers (MpAPr1-F/MpAPr1-R). The gene appeared to be present in all the strains, as seen in Fig. 6. Extracellular acid protease activity of the 12 M. pulcherrima strains was assessed using a plate assay. Yeasts, grown to stationary growth phase in YPD broth, were spotted on skim milk plates at pH 3.5. Enzymatic activity was visualized by a clear halo of at least 1 mm around the edges of the yeast colony. Assays were performed in triplicate. Activity could be visualized in all the strains; however, strain IWBT Y1123 had a greater activity than the rest of the strains as indicated by a wider halo (Fig. 6B). S. cerevisiae VIN13, used as negative control, displayed no activity at all.
Fig 6.

Protease activity screening of M. pulcherrima strains. (A) PCR amplifications with primers MpAPr1-F/MpAPr1-R, showing the presence of the MpAPr1 protease-encoding gene. Lane M, molecular size marker (GeneRuler 100-bp DNA ladder Plus; Fermentas). (B) An example of a plate assay showing the different degrees of activity, with a positive result indicated by a clear halo around the colony. +, low activity (diameter of the halo from the edge of the colony, 2 mm); ++, medium activity (diameter of the halo from the edge of the colony, 3 mm); +++, high activity (diameter of the halo from the edge of the colony, 5 mm); −, no activity. Strain M. pulcherrima IWBT Y1123 was used as positive control (displaying high activity) and S. cerevisiae VIN13 as negative control (displaying no activity). Strains IWBT Y1072 and IWBT Y1063 have low activity, while strains IWBT Y1065 and IWBT Y1094 show medium activity.
The results showed a positive correlation between the presence of the gene, confirmed by PCR, and protease activity screening on plates. However, activity was not equal among the strains as illustrated by plate assays. The PCR fragments of the putative genes were sequenced and aligned with the sequence of IWBT Y1123 (data not shown). A number of mutation hot spots were identified in the nucleotide sequences. Single nucleotide polymorphisms indeed appear not to be random among strains but rather to be located on specific nucleotides. This analysis also revealed that all the strains possess at least two different alleles of the gene. However, only three polymorphism sites lead to different versions of the proteins, as the other mutations are silent. Depending on the allele, Val11 can appear as an isoleucine, Pro108 as a serine, and Gly235 as a valine. These amino acids are indicated in Fig. 1.
DISCUSSION
In this study, the putative gene and amino acid sequences of two aspartic proteases were retrieved and characterized by in silico analysis. The first putative gene was from M. pulcherrima IWBT Y1123 and the other from C. apicola IWBT Y1384. The putative proteins were predicted to have molecular masses of 40.8 kDa and 39.1 kDa, respectively. The molecular mass of the protein from strain IWBT Y1123 without the signal peptide (mature protease) was predicted to be 39.2 kDa. These values are in line with what has been published previously, with the sizes of aspartic proteases ranging from 35 kDa to 50 kDa (12, 42). The putative genes were named MpAPr1, isolated from IWBT Y1123, and CaAPr1, isolated from IWBT Y1384. The predicted pI values of the two putative proteins were 4.22 and 4.33, which are also in line with what has been reported in the literature (42, 46).
By performing homology searches, it was found that both gene sequences shared homology with genes from various species, including fish, insects, and humans. The coverage scores, although low, revealed high identity with the database sequences. These conserved regions correspond to the active-site-encoding regions on the putative protein sequences. It appears that the active-site-encoding regions, especially that which is located on the N-terminal domain, are highly conserved throughout different species, and this indicates how the enzymes may have evolved throughout different species but have most probably retained their function. This is expected seeing that aspartic acid proteases are found in almost all living organisms: viruses, bacteria, plants, mammals, etc. (12).
No signal peptide could be detected in the CaAPr1 protein, and it was predicted that the putative protein is also not a glycosylphosphatidylinositol (GPI)-anchored protein (data not shown). Although a signal peptide was detected for the DNA-deduced Axp aspartic protease of Y. lipolytica 148, the prepro region of the protein shared no homology with other extracellular proteins, and the secretory motif of the protein was distinct from the common motif for yeast extracellular protease processing (54). McEwen and Young (32) could not confirm whether the Axp precursor contains the signal peptide, but it was confirmed that Axp translocation occurs cotranslationally. Thus, the signal peptide could have been cleaved cotranslationally. Nonetheless the translocation process of Axp is still unknown. Because of the close homology of the CaAPr1 putative protein to the aspartic protease of Y. lipolytica, it may be suggested that the two proteins could follow a similar secretion and maturation pathway which is yet to be elucidated (4), or because of the lack of an identifiable signal peptide, the putative CaAPr1 protein may present a novel translocation process.
The putative gene MpAPr1 was ligated into pCEL13, carrying the constitutive PGK1 promoter, and expressed in S. cerevisiae YHUM272. Activity assays were performed on plates supplemented with skim milk. A zone of clearance confirmed positive activity. The activity assays confirmed that the MpAPr1 gene encodes an extracellular aspartic protease. The zone of clearance was much wider in the IWBT Y1123 strain (native host) than in the transformed YHUM272 strain, indicating weaker activity in the recombinant strain. This could be due to a number of factors. It could be caused by the differences in metabolic machinery of S. cerevisiae YHUM272 and M. pulcherrima IWBT Y1123. S. cerevisiae YHUM272 may not be systematically recognizing the (unfamiliar) secretion signal peptide of the protein, or the signal peptide may be cleaved improperly. Expression may be lower under the control of the PGK1 promoter of the plasmid. PGK1 gene expression is activated when yeast cells are grown on glucose, while PGK1 mRNA levels are low when growth is on lactate (6, 35). The skim milk medium contained lactate and had a glucose concentration of 0.8%. Due to the presence of lactate from the skim milk powder and the low glucose concentration, decreased activation of the PGK1 promoter is possible. In 1987, Mellor et al. (33) suggested that the absence of a downstream activating sequence (DAS) in expression vectors can explain in part the low yield of foreign proteins expressed under the control of the PGK1 promoter compared to the endogenous levels of 3-phosphoglycerate kinase (PGK). Codon bias may also play a role. Further investigation is needed to elucidate the decreased expression in the recombinant strain. Figure 3 shows that the colonies of the recombinant S. cerevisiae YHUM272 carrying the MpAPr1 gene and that of IWBT Y1123 were slightly larger than the colonies of the strains not carrying the gene. In order to assess the role of MpAPr1 expression in nitrogen source utilization, transformed and untransformed YHUM272 strains were spotted on skim milk medium not containing the essential amino acids tryptophan, leucine, and histidine. The cells not carrying the MpAPr1 gene did not grow at all, while the MpAPr1-transformed strain grew slightly and displayed minimal protease activity (results not shown). This indicates the important role that extracellular proteolytic activity can play in the survival of yeasts under poor carbon and/or preferred nitrogen conditions.
M. pulcherrima IWBT Y1123 was grown in media containing different nitrogen sources (casein, BSA, grape juice proteins, and ammonium sulfate) in order to evaluate induced expression of the MpAPr1 gene. The expression of the gene was investigated by qPCR. The same level of expression was observed regardless of the presence of nitrogen in the environment, indicating a constitutive expression. The presence and activity of the MpAPr1 protease were further assessed by performing SDS-PAGE analysis and zymography on cell-free concentrated supernatants. Proteolysis was observed in the SDS-PAGE analysis of the BSA-induced culture, and activity was observed in the grape protein-induced culture by zymography. Sequencing of the protein bands corresponding to the expected size of the mature enzyme from casein-induced and grape protein-induced cultures further confirmed the presence of the MpAPr1 protein in the extracellular medium. In C. albicans, protease secretion in yeasts is induced by the presence of proteins as the sole nitrogen source in the extracellular medium (11). It was not a surprise that the acid protease was able to degrade grape proteins, given that these are present in the (natural) environment from which the yeast was isolated and may be a familiar source of nitrogen for the yeast. Casein is a commonly used inducer for proteases from yeasts and fungi (2, 17), and BSA has also been used in a number of studies to induce the production and secretion of proteases (25, 45). Proteins are considered alternative or secondary nitrogen sources for yeasts, whereas amino acids, ammonium, glutamine, and urea are preferred nitrogen sources (2). Therefore, the presence of the protease was expected in the cultures containing BSA, casein, and grape juice proteins but not in those with ammonium sulfate. The genes encoding proteases are usually repressed when high concentrations of the preferred nitrogen sources are available (11), but this does not seem to be the case for MpAPr1. The fact that the MpApr1 expression pattern does not follow that of the Saps of C. albicans could be attributed to the fact that M. pulcherrima is not a pathogenic yeast and its extracellular proteases are not likely to have the same biological function. The GATA transcription factors, which control the use of alternative nitrogen sources, have been studied extensively in S. cerevisiae and C. albicans (31, 36). Investigating the presence and role of these transcription factors in M. pulcherrima would broaden our understanding of the expression of MpAPr1.
The MpAPr1 protease gene transcript was found to be of the same size as the gene (data not shown), therefore showing that MpAPr1 has no introns. Moreover, seeing that the predicted size of the mature enzyme correspond to the size (∼39 kDa) of the protein band excised from the SDS-polyacrylamide gel, it can be hypothesized that the mature protein displays a low level of N-glycosylation of the protein, resulting in the ease of migration through the gel. Only one potential N-glycosylation site was predicted for the deduced protein sequence.
In 1995, Gotoh and coworkers (17) reported on the purification of an acid protease from C. pulcherrima KSY 188-5 (teleomorph Metschnikowia) with a molecular mass of 36.5 kDa as estimated by SDS-PAGE and gel filtration and a pI of 4.7 as determined by isoelectric focusing, which is similar to what has been found in this study for the protein of M. pulcherrima IWBT Y1123 (39.2 kDa and pI 4.22, respectively). The enzyme also had a wide substrate specificity, hydrolyzing casein, BSA, hemoglobin, and collagen. Further characterization of MpAPr1 is needed to confirm whether it is identical to the enzyme described by Gotoh et al. (17). Sequencing the gene encoding the protease from C. pulcherrima KSY 188-5 would also enable one to better compare the two proteases. An acid protease gene of Metschnikowia reukaufii W6b has also been characterized (28). This gene is 1,527 bp long without any introns and encodes a protein with an estimated molecular mass of 53.5 kDa and a predicted pI of 4.2. The molecular mass of this protein is somewhat higher than the usual molecular masses of yeast aspartic proteases, but it had the same pI of the MpAPr1 protease.
The presence of MpAPr1 and extracellular protease activity were confirmed in 12 M. pulcherrima strains isolated from grape juice. As stated above, activity was not equal among the strains as illustrated by plate assays. Sequencing of the MpAPr1 genes of these 12 strains revealed that all strains possessed at least two slightly different alleles. However, only 3 amino acid polymorphisms were identified. The first substitution is located in the secretion signal, but the two amino acids, valine and isoleucine, are both aliphatic and nonpolar, and a substitution between the two is unlikely to affect the secretion process. The other two substitutions, however, may affect protein activity, as in each case the two amino acids differ in their polarity. No correlation was found between protein sequence and the degree of activity displayed on plates. Sequencing and examining the nucleotide regions upstream and downstream of the open reading frame may explain the varying degrees of activity.
Future work should include purification of the MpAPr1 enzyme (both versions) and determination of its biochemical properties, such as optimum pH and temperature for the enzyme activity. Another important aspect is to test the activity of the acid protease in wine and whether it is able to hydrolyze wine proteins and reduce protein haze formation under enological conditions, as potential inhibitors such as phenolic compounds and ethanol in wine may affect the activity of the protease. This study showed that MpAPr1 can digest some grape juice proteins and thus has potential to be tested for haze reduction abilities, its impact on aroma profile, and whether the protease activity will lead to a nonnegligible increase in available assimilable nitrogen content to be used by S. cerevisiae and lactic acid bacteria during alcoholic and malolactic fermentations. An increase in assimilable nitrogen content will certainly have an impact on the fermentation kinetics. It has been suggested that heat treatment of the wine together with protease treatment could effectively reduce white wine haze formation (41). Other potential biotechnological applications of the enzyme include cheese manufacturing (42) and beer haze chill-proofing (40).
Our work has provided insight into how some non-Saccharomyces yeasts may survive in wine by extracellular proteolytic activity (5). To our knowledge, this the first report on the extracellular aspartic protease-encoding genes of M. pulcherrima and C. apicola.
ACKNOWLEDGMENTS
We thank Winetech, the THRIP funding program of the National Research Foundation, Stellenbosch University (Subcommittee B), and the Harry Crossley Foundation for financial support.
We thank Alexis Eschstruth for technical assistance.
Footnotes
Published ahead of print 20 July 2012
REFERENCES
- 1. Albrecht A, et al. 2006. Glycosylphosphatidylinositol-anchored proteases of Candida albicans target proteins necessary for both cellular processes and host-pathogen interactions. J. Biol. Chem. 281:688–694 [DOI] [PubMed] [Google Scholar]
- 2. Banerjee A, Ganesan K, Datta A. 1991. Induction of secretory acid proteinase in Candida albicans. J. Gen. Microbiol. 137:2455–2461 [DOI] [PubMed] [Google Scholar]
- 3. Barrett AJ, Rawlings ND, O'Brien EA. 2001. The MEROPS database as a protease information system. J. Struct. Biol. 134:95–102 [DOI] [PubMed] [Google Scholar]
- 4. Beckerich J-M, Boisramé-Baudevin A, Gaillardin C. 1998. Yarrowia lipolytica: a model organism for protein secretion studies. Int. Microbiol. 1:123–130 [PubMed] [Google Scholar]
- 5. Bossi A, Bonizzato L, Zapparoli G. 2006. Acidic extracellular proteases from microrganisms of fairly acidic niche. Protein. Pept. Lett. 13:737–741 [DOI] [PubMed] [Google Scholar]
- 6. Chambers A, et al. 1989. Transcriptional control of the Saccharomyces cerevisiae PGK gene by RAPI. Mol. Cell. Biol. 9:5516–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Charoenchai C, Fleet GH, Henschke PA, Todd BENT. 1997. Screening of non-Saccharomyces wine yeasts for the presence of extracellular hydrolytic enzymes. Aust. J. Grape Wine Res. 3:2–8 [Google Scholar]
- 8. Ciani M, Comitini F. 2011. Non-Saccharomyces wine yeasts have a promising role in biotechnological approaches to winemaking. Ann. Microbiol. 61:25–32 [Google Scholar]
- 9. Ciani M, Maccarelli F. 1998. Oenological properties of non-Saccharomyces yeasts associated with wine-making. World J. Microbiol. Biotechnol. 14:199–203 [Google Scholar]
- 10. Collart MA, Oliviero S. 1993. Preparation of yeast RNA, p 13.12.1–13.12.5 In Ausubel FM, et al. (ed), Current protocols in molecular biology, vol 2.13.1 John Wiley, New York, NY: [DOI] [PubMed] [Google Scholar]
- 11. Dabas N, Morschhäuser J. 2008. A transcription factor regulatory cascade controls secreted aspartic protease expression in Candida albicans. Mol. Microbiol. 69:586–602 [DOI] [PubMed] [Google Scholar]
- 12. Davies DR. 1990. The structure and function of aspartic proteases. Annu. Rev. Biophys. Biophys. Chern. 19:189–215 [DOI] [PubMed] [Google Scholar]
- 13. Dujon B, et al. 2004. Genome evolution in yeasts. Nature 430:35–44 [DOI] [PubMed] [Google Scholar]
- 14. Dunn BM. 2002. Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem. Rev. 102:4431–4458 [DOI] [PubMed] [Google Scholar]
- 15. Fleet GH. 2003. Yeast interactions and wine flavour. Int. J. Food Microbiol. 86:11–22 [DOI] [PubMed] [Google Scholar]
- 16. Folio P, Ritt J, Alexandre H, Remize F. 2008. Characterization of EprA, a major extracellular protein of Oenococcus oeni with protease activity. Int. J. Food Microbiol. 127:26–31 [DOI] [PubMed] [Google Scholar]
- 17. Gotoh T, et al. 1995. Purification and properties of extracellular carboxyl proteinase secreted by Candida pulcherrima. Biosci. Biotechnol. Biochem. 59:367–371 [DOI] [PubMed] [Google Scholar]
- 18. Gundllapalli S, Cordero Otero R, Pretorius IS. 2006. Development of a screening method for the identification of a novel Saccharomyces cerevisiae mutant over-expressing Trichoderma reesei cellobiohydrolase II. Ann. Microbiol. 56:143–150 [Google Scholar]
- 19. Hoffman CS, Winston F. 1987. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57:267–272 [DOI] [PubMed] [Google Scholar]
- 20. Jolly NP, Augustyn OPH, Pretorius IS. 2003. The use of Candida pulcherrima in combination with Saccharomyces cerevisiae for the production of Chenin blanc wine. S. Afr. J. Enol. Vitic. 24:63–69 [Google Scholar]
- 21. Jolly NP, Augustyn OPH, Pretorius IS. 2006. The role and use of non-Saccharomyces yeasts in wine production. S. Afr. J. Enol. Vitic. 27:15–39 [Google Scholar]
- 22. Koszul R, et al. 2003. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett. 534:39–48 [DOI] [PubMed] [Google Scholar]
- 23. Kurtzman CP, Robnett CJ. 1998. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek 73:331–371 [DOI] [PubMed] [Google Scholar]
- 24. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 277:680–685 [DOI] [PubMed] [Google Scholar]
- 25. Lagace LS, Bisson LF. 1990. Survey of yeast acid proteases for effectiveness of wine haze reduction. Am. J. Enol. Vitic. 41:147–155 [Google Scholar]
- 26. Lambrechts MG, Pretorius IS. 2000. Yeast and its importance to wine aroma. S. Afr. J. Enol. Vitic. 21:97–129 [Google Scholar]
- 27. Le Bourse D, et al. 2011. Quantification of chitinase and thaumatin-like proteins in grape juices and wines. Anal. Bioanal. Chem. 401:1541–1549 [DOI] [PubMed] [Google Scholar]
- 28. Li J, et al. 2009. Cloning and characterization of a novel aspartic protease gene from marine-derived Metschnikowia reukaufii and its expression in E. coli. Appl. Biochem. Biotechnol. 159:119–132 [DOI] [PubMed] [Google Scholar]
- 29. Lott TJ, et al. 1998. Sequence analysis of the internal transcribed spacer 2 (ITS2) from yeast species within the genus Candida. Curr. Microbiol. 36:63–69 [DOI] [PubMed] [Google Scholar]
- 30. Marangon M, Van Sluyter SC, Haynes PA, Waters EJ. 2009. Grape and wine proteins: their fractionation by hydrophobic interaction chromatography and identification by chromatographic and proteomic analysis. J. Agric. Food Chem. 57:4415–4425 [DOI] [PubMed] [Google Scholar]
- 31. Marzluf GA. 1997. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McEwen RK, Young TW. 1998. Secretion and pH-dependent self-processing of the pro-form of the Yarrowia lipolytica acid extracellular protease. Yeast 14:1115–1125 [DOI] [PubMed] [Google Scholar]
- 33. Mellor J, Dobson MJ, Kingsman AJ, Kingsman SM. 1987. A transcriptional activator is located in the coding region of the yeast PGK gene. Nucleic Acids Res. 15:6243–6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendoza L, Farías ME. 2010. Improvement of wine organoleptic characteristics by non-Saccharomyces yeasts. Curr. Res. Technol. Ed. Top. Appl. Microbiol. Microb. Biotechnol. 2:908–919 [Google Scholar]
- 35. Moore PA, Sagliocco FA, Wood RMC, Brown AJP. 1991. Yeast glycolytic mRNAs are differentially regulated. Mol. Cell. Biol. 11:5330–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morschhäuser J. 2011. Nitrogen regulation of morphogenesis and protease secretion in Candida albicans. Int. J. Med. Microbiol. 301:390–394 [DOI] [PubMed] [Google Scholar]
- 37. Naglik J, Albrecht A, Bader O, Hube B. 2004. Candida albicans proteinases and host/pathogen interactions. Cell. Microbiol. 6:915–926 [DOI] [PubMed] [Google Scholar]
- 38. Newport G, Agabian N. 1997. KEX2 influences Candida albicans proteinase secretion and hyphal formation. J. Biol. Chem. 272:28954–28961 [DOI] [PubMed] [Google Scholar]
- 39. Ogrydziak DM. 1993. Yeast extracellular proteases. Crit. Rev. Biotechnol. 13:1–55 [DOI] [PubMed] [Google Scholar]
- 40. Ormrod IHL, Lalor EF, Sharpe FR. 1991. The release of yeast proteolytic enzymes into beer. J. Inst. Brew. 97:441–443 [Google Scholar]
- 41. Pocock KF, Høj PB, Adams KS, Kwiatkowski MJ, Waters EJ. 2003. Combined heat and proteolytic enzyme treatment of white wines reduces haze forming protein content without detrimental effect. Aust. J. Grape Wine Res. 9:56–63 [Google Scholar]
- 42. Rao MB, Tanksale AM, Ghatge MS, Deshpande VV. 1998. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 62:597–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodríguez ME, Lopes CA, Barbagelata RJ, Barda NB, Caballero AC. 2010. Influence of Candida pulcherrima Patagonian strain on alcoholic fermentation behaviour and wine aroma. Int. J. Food Microbiol. 138:19–25 [DOI] [PubMed] [Google Scholar]
- 44. Strauss MLA, Jolly NPNP, Lambrechts MG, Van Rensburg P. 2001. Screening for the production of extracellular hydrolytic enzymes by non-Saccharomyces wine yeasts. J. Appl. Microbiol. 91:182–190 [DOI] [PubMed] [Google Scholar]
- 45. Togni G, Sanglard D, Quadroni M, Foundling SI, Monod M. 1996. Acid proteinase secreted by Candida tropicalis: functional analysis of preproregion cleavages in C. tropicalis and Saccharomyces cerevisiae. Microbiology 142:493–503 [DOI] [PubMed] [Google Scholar]
- 46. Tyndall JDA, Nall T, Fairlie DP. 2005. Proteases universally recognize β-strands in their active sites. Chem. Rev. 105:973–1000 [DOI] [PubMed] [Google Scholar]
- 47. Van Dyk D, Pretorius IS, Bauer FF. 2005. Mss11p is a central element of the regulatory network that controls FLO11 expression and invasive growth in Saccharomyces cerevisiae. Genetics 169:91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Volschenk H, et al. 2004. Genetic engineering of an industrial strain of Saccharomyces cerevisiae for l-malic acid degradation via an efficient malo-ethanolic pathway. S. Afr. J. Enol. Vitic. 25:63–73 [Google Scholar]
- 49. Waters EJ, Wallace W, Williams PJ. 1992. Identification of heat-unstable wine proteins and their resistance to peptidases. J. Agric. Food Chem. 40:1514–1519 [Google Scholar]
- 50. Waters EJ, Shirley NJ, Williams PJ. 1996. Nuisance proteins of wine are grape pathogenesis-related proteins. J. Agric. Food Chem. 44:3–5 [Google Scholar]
- 51. Waters EJ, et al. 2005. Preventing protein haze in bottled white wine. Aust. J. Grape Wine Res. 11:215–225 [Google Scholar]
- 52. White TJ, Bruns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungi ribosomal RNA genes for phylogenetics, p 315–322 In Innis MA, Gelfand DH, Sninsky JJ, White TJ. (ed), PCR protocols. A guide to methods and applications. Academic Press, San Diego, CA [Google Scholar]
- 53. Reference deleted.
- 54. Young TW, et al. 1996. The extracellular acid protease gene of Yarrowia lipolytica: sequence and pH-regulated transcription. Microbiology 142:2913–2921 [DOI] [PubMed] [Google Scholar]