

Abstract
To determine if there is a core microbial community in the microbial populations of different wastewater treatment plants (WWTPs) and to investigate the effects of wastewater characteristics, operational parameters, and geographic locations on microbial communities, activated sludge samples were collected from 14 wastewater treatment systems located in 4 cities in China. High-throughput pyrosequencing was used to examine the 16S rRNA genes of bacteria in the wastewater treatment systems. Our results showed that there were 60 genera of bacterial populations commonly shared by all 14 samples, including Ferruginibacter, Prosthecobacter, Zoogloea, Subdivision 3 genera incertae sedis, Gp4, Gp6, etc., indicating that there is a core microbial community in the microbial populations of WWTPs at different geographic locations. The canonical correspondence analysis (CCA) results showed that the bacterial community variance correlated most strongly with water temperature, conductivity, pH, and dissolved oxygen (DO) content. Variance partitioning analyses suggested that wastewater characteristics had the greatest contribution to the bacterial community variance, explaining 25.7% of the variance of bacterial communities independently, followed by operational parameters (23.9%) and geographic location (14.7%). Results of this study provided insights into the bacterial community structure and diversity in geographically distributed WWTPs and discerned the relationships between bacterial community and environmental variables in WWTPs.
INTRODUCTION
Biological wastewater treatment is a multibillion dollar industry, the largest application of biotechnology in the world. By harnessing and concentrating microorganisms in bioreactors, the beneficial activities of naturally occurring microorganisms are accelerated, enabling removal of oxygen-depleting organics, toxins, and nutrients and preventing the discharge of pathogens. Despite the environmental and economic importance of these processes, the knowledge of the microbial communities within biological wastewater treatment is incomplete, primarily because of a lack of suitable tools for their analysis. Culture-dependent methods are biased by the selection of species which obviously do not represent the real dominance structure (4, 6). The applications of conventional molecular techniques in the last 2 decades, such as fluorescence in situ hybridization (FISH), denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP), quantitative PCR, 16S rRNA clone libraries, etc., have provided new insights into microbial community structure. However, these methods generally have difficulty detecting most of the low-abundance organisms in a wastewater microbial community, thus providing incomplete information about the microbial diversity and community within the biological WWTPs (30).
Recently developed pyrosequencing technology can generate hundreds of thousands of short sequences (14) and significantly improve researchers' ability to investigate the low-abundance microorganisms (24). This new approach has been extended to environmental systems in recent years (1, 9, 15, 19, 21). However, to our knowledge, there are few reports using pyrosequencing technology to compare bacterial communities between various WWTPs operated at different geographic locations. More recently, Zhang et al. (33) and Xia et al. (30) have observed that some core populations of bacteria were shared by multiple activated sludge samples from different wastewater treatment plants. However, they did not examine the relationships between microbial community structures and environmental variables, thus it is not clear how wastewater characteristics, operational parameters, and geographic locations influence the bacterial community structures.
In this study, we collected activated sludge samples from 14 municipal wastewater treatment systems located in different geographic regions of China and analyzed these samples via pyrosequencing technology to obtain the bacterial community structures and diversities. The objective was to address the following two questions. (i) Is there a core microbial community in the microbial populations of municipal WWTPs at different geographic locations? (ii) How do wastewater characteristics, operational parameters, and geographic locations affect bacterial community structures in WWTPs?
MATERIALS AND METHODS
Mixed-liquid samples.
Activated sludge samples were collected from the aeration tanks of 14 full-scale wastewater treatment systems located in different cities of China: Shenzhen, Wuxi, Beijing, and Harbin. Details of the locations, treatment processes, influents, and operational parameters of the 14 systems are listed in Table 1.
Table 1.
Characteristics of 14 geographically distributed wastewater treatment systems
| WWTP | Location |
Scale | Processa | Environmental variable |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| City | Latitude | Longitude | COD (mg/liter) | TN (mg/liter) | TP (mg/liter) | pH | Conductivity (μs/cm) | DO (mg/liter) | Temp (°C) | SRT (days) | MLSS (mg/liter) | |||
| A1 | Shenzhen | 22.55 | 114.15 | Plant | Oxidation ditch | 317 | 33 | 4.3 | 6.84 | 645 | 2.99 | 21.2 | 20 | 4,700 |
| A2 | Shenzhen | 22.55 | 114.15 | Plant | AB | 328 | 33 | 4.3 | 6.42 | 700 | 4.49 | 21.2 | 18 | 4,800 |
| B | Shenzhen | 22.54 | 114.10 | Plant | MUCT | 343 | 77.9 | 5.1 | 6.41 | 740 | 2.6 | 21.6 | 15 | 4,600 |
| C | Shenzhen | 22.56 | 114.25 | Plant | MSBR | 319 | 45 | 5 | 6.5 | 730 | 2.7 | 21.7 | 15 | 4,900 |
| D1 | Wuxi | 31.59 | 120.32 | Plant | Oxidation ditch | 375 | 45 | 7 | 7.06 | 1,291 | 5.6 | 18.2 | 20 | 7,989 |
| D2 | Wuxi | 31.59 | 120.32 | Plant | A/A/O + MBR | 375 | 45 | 7 | 6.9 | 1,341 | 1.7 | 18.2 | 18 | 8,386 |
| E1 | Wuxi | 31.51 | 120.33 | Plant | A/A/O | 325 | 39 | 6 | 6.88 | 950 | 2.3 | 18.1 | 15 | 4,960 |
| E2 | Wuxi | 31.51 | 120.33 | Plant | Oxidation ditch | 325 | 39 | 6 | 6.7 | 991 | 1.5 | 18.1 | 20 | 5,272 |
| F | Beijing | 39.93 | 116.11 | Plant | A/A/O + MBR | 427 | 48 | 6 | 7.1 | 1,018 | 2 | 16.9 | 18 | 5,047 |
| G1 | Beijing | 40.02 | 116.29 | Bench | A/A/O | 284.6 | 53.9 | 6.1 | 7.1 | 928 | 2.94 | 16.7 | 15 | 4,136 |
| G2 | Beijing | 40.02 | 116.29 | Bench | A/A/O | 284.6 | 53.9 | 6.1 | 7.29 | 930 | 2.83 | 16.7 | 15 | 4,083 |
| H | Beijing | 40.32 | 116.63 | Plant | A/A/O + MBR | 412 | 39.3 | 4.2 | 7.1 | 1,029 | 5 | 16.5 | 20 | 4,200 |
| I | Beijing | 39.91 | 116.53 | Pilot | A/A/O | 452.8 | 52 | 6 | 6.97 | 1,018 | 1.39 | 16.8 | 15 | 4,017 |
| J | Haerbin | 45.80 | 126.69 | Plant | A/O | 140.6 | 55.5 | 7.5 | 7.24 | 693 | 6.97 | 14.1 | 15 | 4,329 |
AB, absorption biodegradation; MUCT, modified University of Cape Town; MSBR, modified sequencing batch reactor; A/A/O, anaerobic/anoxic/aerobic; MBR, membrane bioreactor.
Among these wastewater treatment systems, G1 and G2 are laboratory-scale systems, I is a pilot-scale system, and the others are full-scale wastewater treatment systems. Systems A1, A2, D1, D2, E1, and E2 are located in plants A, D, and E, respectively. The 14 systems were operated using different treatment processes: oxidation ditch, anaerobic/anoxic/aerobic (A2O), anoxic/aerobic (AO), membrane bioreactor (MBR), absorption biodegradation (AB), modified University of Cape Town (MUCT), and modified sequencing batch reactor (MSBR) processes (Table 1).
Activated sludge samples were collected from the aeration tanks of 14 wastewater treatment systems in the winter of 2010. For archiving, each 40-ml sample was dispensed into a 50-ml sterile Eppendorf tube and centrifuged at 14,000 × g for 10 min. The supernatant was decanted, and the pellet was stored at −80°C prior to analysis.
DNA extraction.
Microbial genomic DNA was extracted by freeze-mechanical lysis as previously described (34). DNA quality was assessed by the 260/280-nm and 260/230-nm absorption ratios, measured by an ND-2000 spectrophotometer (Nanodrop Inc., Wilmington, DE) and agarose gel electrophoresis.
PCR amplification and purification.
The extracted DNA samples were amplified with a set of primers targeting the hypervariable V4 region of the 16S rRNA gene. The forward primer is 5′-AYTGGGYDTAAAGNG-3′, and the reverse primers are an equal-portion mixture of four primers, i.e., 5′-TACCRGGGTHTCTAATCC-3′, 5′-TACCAGAGTATCTAATTC-3′, 5′-CTACDSRGGTMTCTAATC-3′, and 5′-TACNVGGGTATCTAATCC-3′ (31). Barcodes that allow sample multiplexing during pyrosequencing were incorporated between the 454 adapter and the forward primers.
Pyrosequencing and sequence analysis.
The composition of the PCR products of the V4 region of 16S rRNA genes was determined by pyrosequencing using a Roche 454 FLX titanium sequencer (Roche, Nutley, NJ). Samples in this study were individually barcoded to enable multiplex sequencing. After pyrosequencing, Python scripts were written to remove sequences containing more than one ambiguous base (N) and sequences shorter than 150 bp. RDP Classifier (version 2.2) was used to assign all effective sequences to taxonomic ranks with a set confidence threshold of 50%.
Data analysis.
Shannon-Weaver index (H), evenness index (E), Jaccard index, and detrended correspondence analysis (DCA) were performed using R (v.2.13.1; http://www.r-project.org/) with a 3% cutoff in nucleic acid sequences. The contributions of wastewater characteristics (W), operational parameters (O), and geographic locations (G) to the variances of bacterial communities were assessed with variance partitioning analysis using CCA by R (v.2.13.1). All wastewater characteristics and operational parameter data were log2(x + 1) transformed for standardization. Spatial variables measured as latitude-longitude coordinates were converted into projected coordinates and are represented by a cubic trend-surface polynomial to capture broad-scale spatial trends (10).
RESULTS AND DISCUSSION
Composition of bacterial community.
By using 454 pyrosequencing, 7,422 to 11,151 effective sequence tags were yielded for each sample, resulting in 139,951 sequences in total from all 14 samples. RDP Classifier was used to assign these sequence tags to different operational taxonomic units (OTUs) with a 3% nucleotide cutoff. A total of 21,944 OTUs were recovered from the 14 samples. Individual samples contained much smaller numbers of OTUs, from 2,176 to 4,123 (Table 2). Thus, individual samples contained between 10 and 19% of the total OTUs. With a 5% nucleotide cutoff, a total of 11,628 OTUs were obtained. Each sample contained 1,338 to 2,573 OTUs, accounting for 12 to 22% of the total OTUs. Plots of OTU number versus sequence number, that is, the rarefaction curves, are shown in Fig. S1 in the supplemental material.
Table 2.
Diversity indices from 14 samples
| WWTP | No. of sequencesa | Richnessb | Hc | Evennessd |
|---|---|---|---|---|
| A1 | 11,056 | 4,123 | 7.26 | 0.87 |
| A2 | 10,886 | 4,119 | 7.36 | 0.88 |
| B | 11,151 | 3,860 | 7.16 | 0.87 |
| C | 10,901 | 3,758 | 7.15 | 0.87 |
| D1 | 11,051 | 2,970 | 6.96 | 0.87 |
| D2 | 11,061 | 3,477 | 6.26 | 0.77 |
| E1 | 9,700 | 3,620 | 7.24 | 0.88 |
| E2 | 10,601 | 3,210 | 7.12 | 0.88 |
| F | 7,422 | 2,355 | 6.73 | 0.87 |
| G1 | 7,807 | 3,027 | 7.35 | 0.92 |
| G2 | 11,001 | 2,990 | 7.07 | 0.88 |
| H | 11,086 | 2,932 | 6.9 | 0.86 |
| I | 8,551 | 2,880 | 7.11 | 0.89 |
| J | 7,677 | 2,176 | 6.63 | 0.86 |
Detected sequence number.
Detected OTU number.
Shannon-Weiner index; higher numbers represent higher levels of diversity.
Evenness index.
To assess the internal (within-sample) complexity of individual microbial populations, the Shannon-Weaver index (H) and evenness were calculated. The values of H ranged from 6.2 to 8 (Table 2) across the 14 samples. These H values are typical for diverse microbial populations without a few strongly dominant taxa (30). The evenness of all samples showed few differences, ranging from 0.77 to 0.92.
Pairwise community similarity between the samples was calculated based on the presence and absence of each OTU using a Jaccard index (see Table S1 in the supplemental material). The Jaccard values ranged from 0.21 to 0.55. Each pair of samples from the same plant (such as A1 and A2) exhibited high similarity.
DCA was performed to examine the overall variation among bacterial communities of these 14 samples (see Fig. S2 in the supplemental material). The results showed that most of the samples from the same plants (such as A1 and A2, E1 and E2, and G1 and G2) were clustered together, while the samples from plants using the same treatment process (such as E1 and I) were not clustered together. This indicated that bacterial community structures were not significantly affected by the treatment process in this study.
RDP Classifier was used to assign these sequence tags to different phylogenetic bacterial taxa. Figure 1a summarizes the relative bacterial community abundance at the phylum level for each sample. Proteobacteria was the predominant phylum, constituting between 21 and 53% of all detected OTUs. Bacteroidetes, Acidobacteria, and Chloroflexi were the subdominant groups, comprising 11 to 64%, 1 to 27%, and 1 to 17% of the detections, respectively. These four phyla represented approximately 77 to 97% of bacteria detected within the 14 samples.
Fig 1.

Relative abundance of bacterial community composition in 14 samples. (a) The relative abundance of total bacteria grouped by phyla. (b) Relative abundance of the phylum Proteobacteria. (c) Relative abundance of the Betaproteobacteria.
Within Proteobacteria, the β subdivision was the predominant group (21 to 52%), followed by Alphaproteobacteria (7 to 48%), Gammaproteobacteria (8 to 34%), and Deltaproteobacteria (2 to 18%) (Fig. 1b). Within the Betaproteobacteria, seven taxa were identified (Fig. 1c). Rhodocyclales is the dominant group within a range of 29 to 71% of all 14 samples, followed by Burkholderiales and Gallionellales, representing 16 to 56% and 9 to 28% of each population, respectively. The other four detected groups (Hydrogenophilales, Nitrosomonadales, Neisseriales, and Methylophilales) had fewer detections in all samples and constituted less than 10% of Betaproteobacteria. The abundance of Betaproteobacteria observed in this study agrees with the results revealed by the conventional molecular techniques, such as clone library sequencing (27, 28) and pyrosequencing (33).
Shared and unique orders/families.
By using RDP Classifier, a total of 75 orders were obtained (see Table S2 in the supplemental material). Among them, 38 orders were commonly shared by all activated sludge samples, including Sphingobacteriales, Anaerolineales, Rhodocyclales, Burkholderiales, Rhizobiales, Xanthomonadales, Verrucomicrobiales, Clostridiales, Planctomycetales, Myxococcales, etc. (see Fig. S3). They accounted for 96 to 99% of the classified sequences. There were three rare orders that only appeared in one sample, i.e., Caldisericales in plant C and Rubrobacterales and Natranaerobiales in plant A1, which made up <0.1% of the classified sequences in each sample.
At the family level, a total of 179 families were obtained (see Table S3 in the supplemental material). Forty-six families (accounting for 86 to 96% of the classified sequences), including Anaerolineaceae, Chitinophagaceae, Rhodocyclaceae, Verrucomicrobiaceae, Xanthomonadaceae, Planctomycetaceae, Saprospiraceae, Hyphomicrobiaceae, Comamonadaceae, Sphingomonadaceae, Rhodobacteraceae, Burkholderiales incertae sedis, Moraxellaceae, Bradyrhizobiaceae, Pasteuriaceae, etc., were commonly shared by all samples (see Fig. S4). There were 20 rare families that appeared in only one sample, such as Caldisericaceae, Rubrobacteraceae, Pseudonocardiaceae, Micrococcaceae, Beutenbergiaceae, Sanguibacteraceae, Sporichthyaceae, Geodermatophilaceae, Nakamurellaceae, Micromonosporaceae, etc., which occupied only <1% of the classified sequences (see Table S4).
Core and unique genera.
The abundance of all 543 genera acquired from all samples was summarized in Table S5 in the supplemental material. Among the 543 assigned genera, 60 were shared by all 14 samples and accounted for 62 to 86% of the classified sequences. A total of 156 genera were commonly shared by more than 10 samples, accounting for 85 to 98% of all classified sequences. There were 131 rare genera that were observed in only one sample, and they accounted for <0.5% of total classified sequences in each sample (see Table S6).
The 10 most abundant genera in each sample were selected (a total of 44 genera for all 14 samples), and their abundances were compared to those in other samples (Fig. 2). Six genera were abundant (>1%) in at least 10 samples, including Ferruginibacter, Prosthecobacter, Zoogloea, Subdivision 3 genera incertae sedis, Gp4, and Gp6. Most of them were also found to be core genera and were shared by multiple activated samples from WWTPs according to Zhang et al. (32). These results suggested that these core genera play crucial roles in wastewater treatment regardless of the treatment process and geographic locations. Members of Ferruginibacter, such as Ferruginibacter alkalilentus and Ferruginibacter lapsinanis, are capable of hydrolyzing some organic matter (11). Members of Zoogloea, such as Zoogloea ramigera, have long been considered the typical activated sludge bacteria responsible for the formation of activated sludge flocculation and to improve the purification process (11). Prosthecobacter are heterotrophic organisms which are often referred to as oligotrophic bacteria, meaning that they thrive in low-nutrient environments (25). This does not seem to apply to the Prosthecobacter organisms found here, as the influent biochemical oxygen demand (BOD) concentrations of the WWTPs were above 284.6 mg/liter. The effects of nutrients on members within this genus needs further study to better understand the implications of the presence of Prosthecobacter. The genera of Subdivision 3 genera incertae sedis, Gp4, and Gp6 are not well described, and the knowledge and roles of them in biological wastewater treatment systems are limited. This result is in accordance with the findings of Xia et al. (30), who showed that, based on the examination of bacterial community structures in wastewater treatment bioreactors via PhyloChip, five different wastewater treatment reactors share a large proportion of core bacterial population, indicating almost identical compositions in different reactors.
Fig 2.

Heat map of the 10 most abundant genera in each sample. The 10 most abundant genera in each sample were selected (a total of 44 genera for all 14 samples), and their abundances were compared to those in other samples. The color intensity in each cell shows the percentage of a genus in a sample.
Correlations of environmental data and bacterial communities.
To discern the possible relationship between microbial community structure and environmental parameters, CCA was performed (Fig. 3). Based on variance inflation factors (VIF) with 999 Monte Carlo permutations, eight significant environmental variables, chemical oxygen demand (COD), total nitrogen (TN), total phosphorus (TP), pH, dissolve oxygen (DO), conductivity, temperature, and solid retention time (SRT), were selected in the CCA biplot. The length of an environmental parameter arrow in the ordination plot indicates the strength of the relationship of that parameter to community composition.
Fig 3.
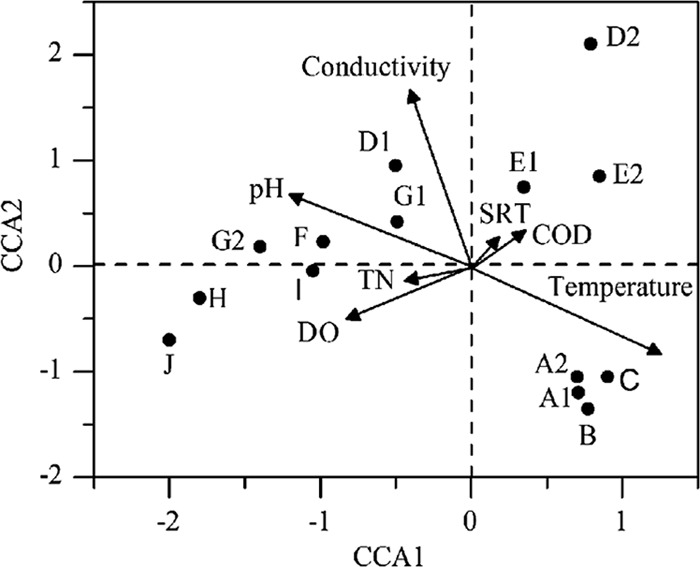
Canonical correspondence analysis (CCA) of pyrosequencing data and measurable variables in the 14 samples. Arrows indicate the direction and magnitude of measurable variables associated with bacterial community structures. Each circle represents a different bacterial community structure from a specific wastewater treatment plant.
As such, temperature appears to be the most important environmental parameter. This agreed with the findings of Siggins et al. (22), who suggested, based on the survey of bacteria community variance via DGGE in expanded granular sludge bed (EGSB) bioreactors, that water temperature is one of the most influential factors in bacterial community variance. Similar results have also been obtained in a full-scale WWTP by Wells et al. (29).
Conductivity is an index measured by the number of ions relative to salinity, and it was strongly and significantly linked to bacterial community variance in CCA. Previous studies have found salinity to be an important factor regulating bacterial composition and diversity across a variety of habitats (12, 13, 16, 26).
In addition to temperature and conductivity, bacterial community variance was also significantly linked to pH in the WWTPs in CCA. The intracellular pH of most microorganisms is usually within 1 pH unit of neutral, and any significant deviation in environmental pH should impose stress on single-cell organisms (3). Some previous studies have demonstrated that the stress of pH has a significant effect on the overall diversity and composition of microbial communities in a range of terrestrial and aquatic environments (3, 7).
DO was also strongly and significantly linked to bacterial community variance in CCA. DO is well recognized as a critical process parameter in biological wastewater treatment processes due to its impact on bacterial activity and the high operational costs of aeration, but little is known about the specific selection of distinct bacterial lineages by DO concentration. The results of this study showed that DO had a significant effect in shaping the microbial community structure in wastewater treatment systems. Park et al. (18) have also found that DO concentration was an important structuring factor based on the T-RFLP analysis of bacterial community structures in two laboratory-scale bioreactors operated with high and low DO concentrations.
Variance partitioning analyses (VPA) were further performed to assess the contributions of wastewater characteristics (COD, TN, TP, pH, and conductivity), operational parameters (DO, temperature, SRT, and mixed-liquid-suspended solids [MLSS]), and geographic location to the microbial community variance. Figure 4 indicated that 69.6% of the variance could be explained by these three components. Wastewater characteristics, operational parameters, and geographic location could independently explain 25.7, 23.9, and 14.7% of the variation of bacterial communities, respectively. Interactions among the three major components seemed to have less influence than did individual components and were only observed between wastewater characteristics and geographic locations (4.8%) and between operational parameters and geographic locations (1.8%). It should be noted that the sampling strategy and the number of samples analyzed in this study may limit our ability to fully separate the effects of environmental variables and geographic locations. More replicated samples from each plant should be collected to examine the bacterial community structures in WWTP across different geographic locations.
Fig 4.
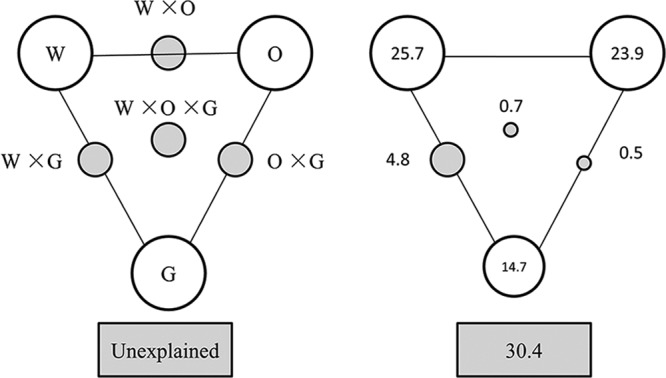
Variation partitioning analysis of microbial community explained by wastewater characteristics (W), operational parameters (O), and geographic location (G). (a) General outline; (b) bacterial communities. Each diagram represents the biological variation partitioned into the relative effects of each factor or a combination of factors, in which geometric areas were proportional to the respective percentages of explained variation. The edges of the triangle represents the variation explained by each factor alone. The sides of the triangles represent interactions of any two factors, and the middle of the triangles represent interactions of all three factors.
More than 30% of the community variance could not be explained by these three components. It is reasonable to expect that some additional factors, such as stochastic dispersal and immigration, predation, and some unmonitored inhibitory chemicals, play an influential role in mediating bacterial community structures in WWTPs. Ofiteru et al. (17) demonstrated that, in a full-scale WWTP, the variation in bacterial community was consistent with neutral community assembly, where chance and random immigration played an important and predictable role in shaping the communities. Other studies also showed that stochastic dispersal and immigration processes (2, 23), protozoan grazing (20), phage predation (8), and chaotic behavior (5) played dominant roles in structuring the microbial communities. The relative influence of deterministic environmental and stochastic factors in structuring microbial communities within bioreactors warrants further investigation.
In summary, high-throughput pyrosequencing was employed to examine the bacterial communities of activated sludge samples from 14 wastewater treatment systems located in four cities in China. In addition to certain unique genera of bacterial populations in each sample, 60 genera, including genera of Ferruginibacter, Prosthecobacter, Zoogloea, Subdivision 3 genera incertae sedis, Gp4, and Gp6, etc., were commonly shared by all samples. This demonstrated that there was a core microbial community in the microbial populations of WWTPs at different geographic locations. The CCA results showed that water temperature, conductivity, pH, and DO were correlated most strongly to the variance of bacterial communities. Variance partitioning analyses suggested that wastewater characteristics had the greatest contribution to the bacterial community variance, explaining 25.7% of the variance of bacterial communities independently, followed by operational parameters (23.9%) and geographic locations (14.7%). These findings provided insight into the bacterial community structure and diversity in geographically distributed WWTPs and identify the main factors shaping microbial community structures in WWTPs.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the NSFC (grants 51078207′ and 51178239) and research funding from Tsinghua University (20101081834).
We thank Tong Zhang at The University of Hong Kong for providing us with the primers for PCR amplification.
Footnotes
Published ahead of print 27 July 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Bibby K, Viau E, Peccia J. 2010. Pyrosequencing of the 16S rRNA gene to reveal bacterial pathogen diversity in biosolids. Water Res. 44:4252–4260 [DOI] [PubMed] [Google Scholar]
- 2. Curtis TP, Sloan WT. 2006. Towards the design of diversity: stochastic models for community assembly in wastewater treatment plants. Water Sci. Technol. 54:227–236 [DOI] [PubMed] [Google Scholar]
- 3. Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U. S. A. 103:626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilbride KA, Lee DY, Beaudette LA. 2006. Molecular techniques in wastewater: understanding microbial communities, detecting pathogens, and real-time process control. J. Microbiol. Methods 66:1–20 [DOI] [PubMed] [Google Scholar]
- 5. Graham DW, et al. 2007. Experimental demonstration of chaotic instability in biological nitrification. ISME J. 1:385–393 [DOI] [PubMed] [Google Scholar]
- 6. He Z, Van Nostrand JD, Deng Y, Zhou J. 2011. Development and applications of functional gene microarrays in the analysis of the functional diversity, composition, and structure of microbial communities. Front. Environ. Sci. Eng. China 5:1–20 [Google Scholar]
- 7. Hornstrom E. 2002. Phytoplankton in 63 limed lakes in comparison with the distribution in 500 untreated lakes with varying pH. Hydrobiologia 470:115–126 [Google Scholar]
- 8. Kunin V, et al. 2008. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 18:293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee TK, et al. 2010. Discovery of commonly existing anode biofilm microbes in two different wastewater treatment MFCs using FLX titanium pyrosequencing. Appl. Microbiol. Biotechnol. 87:2335–2343 [DOI] [PubMed] [Google Scholar]
- 10. Liang Y, et al. 2011. Functional gene diversity of soil microbial communities from five oil-contaminated fields in China. ISME J. 5:403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim JH, Baek SH, Lee ST. 2009. Ferruginibacter alkalilentus gen. nov., sp. nov. and Ferruginibacter lapsinanis sp. nov., novel members of the family ‘Chitinophagaceae’ in the phylum Bacteroidetes, isolated from freshwater sediment. Int. J. Syst. Evol. Microbiol. 59:2394–2399 [DOI] [PubMed] [Google Scholar]
- 12. Liu ST, Yang FL, Gong Z, Su ZC. 2008. Assessment of the positive effect of salinity on the nitrogen removal performance and microbial composition during the start-up of CANON process. Appl. Microbiol. Biotechnol. 80:339–348 [DOI] [PubMed] [Google Scholar]
- 13. Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U. S. A. 104:11436–11440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Margulies M, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. 2010. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 12:378–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohamed DJ, Martiny JBH. 2011. Patterns of fungal diversity and composition along a salinity gradient. ISME J. 5:379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ofiteru ID, et al. 2010. Combined niche and neutral effects in a microbial wastewater treatment community. Proc. Natl. Acad. Sci. U. S. A. 107:15345–15350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park HD, Noguera DR. 2004. Evaluating the effect of dissolved oxygen on ammonia-oxidizing bacterial communities in activated sludge. Water Res. 38:3275–3286 [DOI] [PubMed] [Google Scholar]
- 19. Petropoulos P, Gilbride KA. 2005. Nitrification in activated sludge batch reactors is linked to protozoan grazing of the bacterial population. Can. J. Microbiol. 51:791–799 [DOI] [PubMed] [Google Scholar]
- 20. Qian PY, et al. 2011. Vertical stratification of microbial communities in the Red Sea revealed by 16S rDNA pyrosequencing. ISME J. 5:507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanapareddy N, et al. 2009. Molecular diversity of a North Carolina wastewater treatment plant as revealed by pyrosequencing. Appl. Environ. Microbiol. 75:1688–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siggins A, Enright AM, O'Flaherty V. 2011. Temperature dependent (37–15°C) anaerobic digestion of a trichloroethylene-contaminated wastewater. Bioresource Technol. 102:7645–7656 [DOI] [PubMed] [Google Scholar]
- 23. Sloan WT, et al. 2006. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8:732–740 [DOI] [PubMed] [Google Scholar]
- 24. Sogin ML, et al. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takeda M, et al. 2008. Prosthecobacter fluviatilis sp. nov., which lacks the bacterial tubulin btubA and btubB genes. Int. J. Syst. Evol. Microbiol. 58:1561–1565 [DOI] [PubMed] [Google Scholar]
- 26. Wakelin SA, et al. 2012. Bacterial communities associated with a mineral weathering profile at a sulphidic mine tailings dump in arid western Australia. FEMS Microbiol. Ecol. 79:298–311 [DOI] [PubMed] [Google Scholar]
- 27. Wang X, et al. 2010. Bacterial community dynamics in two full-scale wastewater treatment systems with functional stability. J. Appl. Microbiol. 109:1218–1226 [DOI] [PubMed] [Google Scholar]
- 28. Wang XH, et al. 2011. Bacterial community dynamics in a functionally stable pilot-scale wastewater treatment plant. Bioresource Technol. 102:2352–2357 [DOI] [PubMed] [Google Scholar]
- 29. Wells GF, et al. 2009. Ammonia-oxidizing communities in a highly aerated full-scale activated sludge bioreactor: betaproteobacterial dynamics and low relative abundance of Crenarchaea. Environ. Microbiol. 11:2310–2328 [DOI] [PubMed] [Google Scholar]
- 30. Xia SQ, et al. 2010. Bacterial community structure in geographically distributed biological wastewater treatment reactors. Environ. Sci. Technol. 44:7391–7396 [DOI] [PubMed] [Google Scholar]
- 31. Ye L, Shao MF, Zhang T, Tong AHY, Lok S. 2011. Analysis of the bacterial community in a laboratory-scale nitrification reactor and a wastewater treatment plant by 454-pyrosequencing. Water Res. 45:4390–4398 [DOI] [PubMed] [Google Scholar]
- 32. Zhang G, et al. 2012. Efficient electricity generation from sewage sludge using biocathode microbial fuel cell. Water Res. 46:43–52 [DOI] [PubMed] [Google Scholar]
- 33. Zhang T, Shao MF, Ye L. 2012. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 6:1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou JZ, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.