

Abstract
A large polypeptide encoded in the genome of the thermophilic bacterium Caldicellulosiruptor bescii was determined to consist of two glycoside hydrolase (GH) modules separated by two carbohydrate-binding modules (CBMs). Based on the detection of mannanase and endoglucanase activities in the N-terminal GH5 and the C-terminal GH44 module, respectively, the protein was designated CbMan5B/Cel44A. A GH5 module with >99% identity from the same organism was characterized previously (X. Su, R. I. Mackie, and I. K. Cann, Appl. Environ. Microbiol. 78:2230-2240, 2012); therefore, attention was focused on CbMan5A/Cel44A-TM2 (or TM2), which harbors the GH44 module and the two CBMs. On cellulosic substrates, TM2 had an optimal temperature and pH of 85°C and 5.0, respectively. Although the amino acid sequence of the GH44 module of TM2 was similar to those of other GH44 modules that hydrolyzed cello-oligosaccharides, cellulose, lichenan, and xyloglucan, it was unique that TM2 also displayed modest activity on mannose-configured substrates and xylan. The TM2 protein also degraded Avicel with higher specific activity than activities reported for its homologs. The GH44 catalytic module is composed of a TIM-like domain and a β-sandwich domain, which consists of one β-sheet at the N terminus and nine β-sheets at the C terminus. Deletion of one or more β-sheets from the β-sandwich domain resulted in insoluble proteins, suggesting that the β-sandwich domain is essential for proper folding of the polypeptide. Combining TM2 with three other endoglucanases from C. bescii led to modest synergistic activities during degradation of cellulose, and based on our results, we propose a model for cellulose hydrolysis and utilization by C. bescii.
INTRODUCTION
Polysaccharides in the plant cell wall represent the most abundant, renewable biomass on earth and therefore are a promising feedstock for the emerging biofuel industry. However, efficient and cost-effective depolymerization of cell wall polysaccharides into fermentable sugars remains a fundamental challenge (30). Cellulases play a critical role in decomposing cellulose into simple sugars by cleaving the β-1,4-glycosidic linkages. At present, glycoside hydrolases (GHs) are classified into 130 families (http://www.cazy.org/Glycoside-Hydrolases.html), and 15 families, including the GH44 family, have members that hydrolyze cellulose and/or cello-oligosaccharides (5, 34). More than 30 GH44 members have been identified, and the GH44 module may be coupled with GH modules of different families (38). For example, in the Cel44C/Man26A of Paenibacillus polymyxa GS01, the GH44 module is coupled with a GH26 module that hydrolyzes mannosidic linkages (7). Previous studies have shown that GH44 modules from P. polymyxa (7), Ruminococcus flavefaciens (37), Clostridium acetobutylicum (38), and Caldicellulosiruptor saccharolyticus (14, 16) cleaved primarily the β-1,4-glucosidic linkage and the linkage between glucosyl and xylosyl residues and, to a lesser extent, the β-1,4-xylosidic linkage (1, 6). However, xylan was not hydrolyzed by the GH44 catalytic module from Paenibacillus lautus (17). GH44 modules are generally endoglucanases and rarely display β-glucosidase activity when tested on cellobiose or the synthetic compound 4-nitrophenyl-β-d-glucopyranoside as the substrate (1, 17, 38).
Caldicellulosiruptor bescii (40), originally classified as Anaerocellum thermophilum (41), is a thermophilic bacterium that utilizes various polysaccharides and grows on untreated high-lignin grasses and hardwood at an optimum temperature of about 80°C (9). Due to its potential in the biofuel industry, the complete genome was sequenced, and an array of genes predicted to encode glycoside hydrolases that may be involved in cellulose or hemicellulose degradation were identified. Three open reading frames—ORF1946, ORF1952, and ORF1953—encoding putative endoglucanases designated CbMan5B/Cel44A (this study), CbCel9B/Man5A (35), and CbMan5C/Cel5A (J. Zhang, X. Su, R. I. Mackie, and I. K. O. Cann, unpublished data), respectively, were found in a single gene cluster. Amino acid sequence alignments of the predicted proteins revealed that ORF1946 encodes an N-terminal GH5 module and a C-terminal GH44 catalytic module that are linked by two family 3 carbohydrate-binding modules (CBM3).
C. bescii is a close relative of C. saccharolyticus and grows well on crystalline cellulose, such as Avicel (4). The CbMan5B/Cel44A polypeptide sequence shares 89% amino acid sequence identity with a multidomain mannanase from C. saccharolyticus (GenBank accession no. L01257). The C. saccharolyticus multidomain mannanase was reported to lack enzymatic activity on insoluble cellulose (14). However, glycoside hydrolases may be different in their substrate specificities and activities due to subtle amino acid substitutions in their active sites. In order to determine whether the CbMan5B/Cel44A enzyme plays a role in the reported cellulose utilization by C. bescii, we expressed the wild-type gene and its truncated derivatives to analyze their polypeptides for enzymatic activities. Here we present the enzymatic activities of CbMan5B/Cel44A and its different truncated derivatives on different polysaccharides. Our attention was focused on the catalytic module that exhibited β-1,4 glucosidic linkage cleaving activity. A polypeptide composed of this catalytic module and two CBMs was analyzed for synergistic activity with other C. bescii polypeptides demonstrated to possess β-1,4 glucosidic activity, and based on our results, we propose a model for capture of nutrients from cellulose by this bacterium.
MATERIALS AND METHODS
Materials.
The nucleotide sequence (accession number CP001393) for the whole genome of C. bescii was acquired from NCBI's GenBank database. The primers used in this study were purchased from Integrated DNA Technologies (Coralville, IA), and PrimeSTAR DNA polymerase was purchased from TaKaRa Bio Inc. (Shiga, Japan). The Escherichia coli strains XL-10 (Stratagene, La Jolla, CA) and DH5α were used for plasmid propagation. The PicoMaxx high-fidelity PCR system was also purchased from Stratagene (La Jolla, CA). The pET-46b Ek/LIC cloning kit and BL21-CodonPlus (DE3) RIPL competent cells (Novagen, San Diego, CA) were used for gene expression. Talon metal affinity resin was purchased from Clontech (Valencia, CA). Spin-X UF 20-ml centrifugal concentrators with molecular mass cutoffs of 30 kDa and 50 kDa were obtained from Corning (Lowell, MA).
Manno-oligosaccharides (mannobiose [M2], mannotriose [M3], mannotetraose [M4], mannopentaose [M5], and mannohexaose [M6]), cello-oligosaccharides (cellotriose [G3], cellotetraose [G4], cellopentaose [G5], and cellohexaose [G6], and konjac glucomannan [KGM]), lichenan, birchwood xylan, xyloglycan, and 1,4-β-mannan were purchased from Megazyme (Bray, Ireland). Cellobiose (G2) was purchased from Sigma-Aldrich (St. Louis, MO). Sodium carboxymethyl cellulose (CMC) was purchased from Acros Organics (Geel, Belgium). Protein markers for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were obtained from Bio-Rad (Hercules, CA). All other reagents were of analytical grade and were from Sigma-Aldrich (St. Louis, MO).
Cloning, expression, and purification of Cb1946WT and its truncated mutants.
The genome of C. bescii was uploaded on the Rapid Annotation using Subsystem Technology (RAST) server to facilitate analyses (3). The gene encoding CbMan5B/Cel44A was amplified from the genomic DNA of C. bescii DSM6725T by PCR using PrimeSTAR DNA polymerase. Primers were designed to amplify ORF1946 without its predicted signal peptide-encoding sequence. The resulting polypeptide was designated wild-type protein (CbMan5B/Cel44A-WT). The PCR primers were designed to amplify sequences encoding the GH5 module together with the two CBMs (CbMan5B/Cel44A-TM1), the two CBMs and the GH44 module (CbMan5B/Cel44A-TM2), one CBM and the GH44 module (CbMan5B/Cel44A-TM3), and the GH44 module (CbMan5B/Cel44A-TM4). The primers used for the PCR amplifications are listed in Table S1 in the supplemental material. The PCR products for the complete gene and its truncated derivatives were each analyzed by electrophoresis on a 1% agarose gel. The bands corresponding to the expected DNA sizes were excised, and the DNA were extracted using a QIAquick gel extraction kit (Qiagen, Valencia, CA). The purified PCR products were treated with the exonuclease activity of T4 DNA polymerase (Novagen, San Diego, CA) and annealed to the pET-46 Ek/LIC vector (Novagen, San Diego, CA) according to the manufacturer's instructions. The ligation mixture was used to transform NovaBlue GigaSingles competent cells (Novagen, San Diego, CA), which were then spread on lysogeny broth (LB) plates containing ampicillin at 100 μg/ml. For each transformation, a single colony was picked from the plate and cultured in 4 ml of LB medium supplemented with 100 μg/ml ampicillin. The recombinant plasmid was extracted from the culture with a Qiagen plasmid miniprep kit (Qiagen, Valencia, CA), and the DNA insert was sequenced to confirm the integrity of the gene (W. M. Keck Center for Comparative and Functional Genomics at University of Illinois).
The plasmids harboring the DNA fragments of interest were transformed into E. coli BL-21(DE3) CodonPlus RIPL cells and grown overnight at 37°C on LB agar plates containing 100 μg/ml ampicillin and 50 μg/ml chloramphenicol. A single colony from each transformation was used to inoculate fresh LB medium (10 ml) supplemented with the same concentration of both antibiotics and cultured with aeration for 6 h at 37°C. Each culture was then transferred into 1 liter LB medium supplemented with both antibiotics and incubated at 37°C with vigorous shaking (200 rpm). When the optical density at 600 nm reached 0.3, isopropyl β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 100 μM, the temperature was decreased to 16°C, and the culture was incubated for an additional 16 h. The E. coli cells were harvested by centrifugation (4,651 × g for 30 min at 4°C). The cell pellets were resuspended in a binding buffer (50 mM Tris-HCl, 300 mM NaCl [pH 7.5]) and ruptured by passage through an EmulsiFlex C-3 homogenizer (Avestin, Ottawa, Canada). Cell lysates were centrifuged at 12,857 × g for 20 min at 4°C, and the recombinant proteins were purified from the supernatant using Talon metal affinity resin according to the supplier's protocol (Clontech). Briefly, the affinity resin was equilibrated in the binding buffer, and the clarified lysate in the same buffer was applied to the resin. After several washes, the bound protein was eluted with an elution buffer (50 mM Tris-HCl, 300 mM NaCl, 250 mM imidazole [pH 7.5]). The recombinant proteins were loaded onto a size exclusion column (HiLoad 16 × 60, Superdex 200; GE Healthcare), and the chromatogram was developed with a buffer composed of 50 mM Tris-HCl, 150 mM NaCl [pH 7.5]. The fractions containing the recombinant proteins were further purified by anion-exchange chromatography (HiTrap Q column; GE Healthcare) with a binding buffer composed of 50 mM Tris-HCl, pH 7.5, and an elution buffer composed of the binding buffer supplemented with 1 M NaCl. The proteins in the eluted fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to Laemmli's method (24). Protein concentrations were measured by absorbance spectroscopy at 280 nm using a NanoDrop 1000 from Thermo Scientific (Waltham, MA) with the extinction coefficients 314,700 M−1 cm−1, 161,350 M−1 cm−1, 247,070 M−1 cm−1, 177,170 M−1 cm−1, and 126,750 M−1 cm−1 for CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, and CbMan5B/Cel44A-TM4, respectively.
Determination of optimal pH, optimal temperature, and thermostability.
The optimal pH of CbMan5B/Cel44A-TM2 was determined by using two buffers for two pH ranges, i.e., 50 mM citrate-HCl–150 mM NaCl for pH 3.5 to 6.0 and 50 mM Na2HPO4-NaH2PO4–150 mM NaCl for pH 6.5 to 8.0. At each pH, CbMan5B/Cel44A-TM2 (2 μM enzyme) was reacted with 2.5 mg/ml phosphoric acid-swollen cellulose (PASC) in the different buffers for 30 min at 70°C. The optimal temperature was determined by incubation of CbMan5B/Cel44A-TM2 (2 μM) with 2.5 mg/ml PASC in a pH 5.0 citrate-HCl buffer at temperatures ranging from 55°C to 95°C in increments of 5°C. The reducing sugars released by the enzyme were measured using the p-hydroxybenzoic acid hydrazide (pHBAH; Sigma-Aldrich, St. Louis, MO) assay described by Lever (26). The thermostability of CbMan5B/Cel44A-TM2 was determined by incubation of the enzyme (2 μM) in a buffer composed of 50 mM Tris-HCl and 150 mM NaCl, pH 5.0, and heated at 70°C, 75°C, 80°C, and 85°C. Samples of heat-treated protein were removed at 0 min, 30 min, 1 h, 2 h, 4 h, 6 h, and 10 h, and the residual activities were measured in the same buffers in a total reaction volume of 300 μl with PASC at 2.5 mg/ml as the substrate. All analyses for residual activities were carried out at a temperature of 70°C with a final enzyme concentration of 1 μM.
Specific activities for CbMan5B/Cel44A wild-type, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 with polysaccharides as substrates.
The wild-type protein and the truncated mutants (Fig. 1) were incubated with various polysaccharide substrates (PASC, CMC, Avicel, KGM, lichenan, guar gum, locust bean gum, mannan, birchwood xylan, and xyloglycan). CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 (0.5 μM final concentration) were incubated with each substrate (final concentration, 5 mg/ml) in citrate buffer (50 mM citrate-HCl, 150 mM NaCl [pH 5.0]) at 70°C for 16 h. The final volume of the reaction mixture was 1 ml. The release of reducing ends was measured using the pHBAH assay as described previously, with glucose as the standard (26).
Fig 1.

Schematic representation of CbMan5B/Cel44A, CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, and CbMan5B/Cel44A-TM4 of C. bescii. The putative functional domains were assigned by using the Pfam search tool (http://pfam.sanger.ac.uk/search/sequence). GH5, glycoside hydrolase family 5 catalytic module; GH44, glycoside hydrolase family 44 catalytic module; CBM3, carbohydrate-binding module family 3.
Hydrolysis of cello- and manno-oligosaccharides by CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2.
CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 were each incubated (final concentration, 0.5 μM) with manno-oligosaccharides (5 mg/ml) or cello-oligosaccharides (5 mg/ml) for 16 h in citrate buffer (50 mM citrate-HCl, 150 mM NaCl [pH 5.0]) in a total volume of 40 μl. After the reaction, 1 μl of each sample was spotted on a silica gel 60 F254 thin-layer chromatography (TLC) plate (Merck, Whitehouse Station, NJ). The TLC method was similar to that reported in our previous publications (20, 31). Standards, including mannose (M1), glucose (G1), manno-oligosaccharides (M2 to M6), and cello-oligosaccharides (G2 to G6) were also spotted (1 μl of 5 mg/ml solutions) onto the TLC plate. The hydrolysis products were resolved by three ascents with a mobile phase consisting of n-butanol, acetic acid, and H2O in a volumetric ratio of 10:5:1. The sugars were then visualized by spraying the plate with a 1:1 (vol/vol) mixture of methanolic orcinol (0.1%[wt/vol]) and sulfuric acid (10% [vol/vol]), followed by heating at 80°C for 15 min.
Determination of the specific activities and kinetic parameters of CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, and CbMan5B/Cel44A-TM4.
The specific activities of CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, and CbMan5B/Cel44A-TM4 on two cellulosic substrates (Avicel and filter paper) were investigated using the method described by Su et al. (35). Briefly, five discs of filter paper (Whatman no. 1; 0.6 cm in diameter) or 2% (wt/vol) Avicel was incubated with each recombinant enzyme (final concentration, 1 μM) in a total volume of 1 ml for 10 min by end-over-end rotation at 70°C in a 50 mM citrate buffer (pH 5.0). The enzymes were inactivated by immediately transferring the reaction mixture to 100°C for 10 min. The reaction solutions were centrifuged for 10 min at 15,871 × g, and the reducing sugars, representing end products of hydrolysis from insoluble substrates, were quantified from the supernatant by the pHBAH method (26). The specific activities on insoluble substrates were expressed as μmol reducing ends/min/μmol protein. For the kinetic parameters of the enzymes on soluble substrates, PASC, KGM, guar gum, and lichenan were dissolved in citrate buffer (pH 5.0) at eight stock concentrations. For the reactions, equal volumes (25 μl) of enzymes at an appropriate concentration and substrates were mixed in a citrate buffer (pH 5.0). Both substrate and enzyme were incubated at 70°C for 10 min before mixing. At 10, 20, 30, and 40 min, the reactions were terminated by heating at 100°C for 10 min to inactivate the enzymes. The reducing sugars were quantified by the pHBAH assay (26). The software GraphPad Prism 5.01 was used to estimate the Vmax and Km using nonlinear regression analysis according to the Michaelis-Menten equation (GraphPad, San Diego, CA). The turnover number (kcat) was determined by the equation Vmax = (kcat)([E]), where Vmax represents the maximum initial velocity, kcat represents the turnover number, and [E] represents the final enzyme concentration.
Investigation of the binding subsites for the GH44 catalytic module of CbMan5B/Cel44A.
Five mg/ml of cellopentaose in the citrate buffer (pH 5.0) was incubated at 70°C for 10 min, and then 0.5 μM CbMan5B/Cel44A-TM2 was added. The total reaction volume was 1.5 ml. At different time points, 200 μl of the reaction mixture was removed, and the enzyme was inactivated immediately by incubation at 100°C for 10 min. Each of the terminated reaction mixtures was diluted 50-fold with water and centrifuged at 15,871 × g for 5 min, and the supernatants were analyzed for end products by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) as described in our previous report (11).
Truncation of polypeptide to determine the role of the β-sandwich domain.
The primers used to amplify DNA fragments for the expression of truncated mutants CbMan5B/Cel44A-TM4Δ1β, CbMan5B/Cel44A-TM4Δ2β, CbMan5B/Cel44A-TM4Δ3β, and CbMan5B/Cel44A-TM4Δ9β are listed in Table S1 in the supplemental material. The procedures for cloning and expressing these mutant proteins were the same as those for CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, or CbMan5B/Cel44A-TM4. The expression of the recombinant proteins was determined by SDS-PAGE and Western blot analysis. Ten μl of protein samples were mixed with 10 μl of SDS-PAGE loading buffer and boiled for 5 min prior to separation by SDS-PAGE. After SDS-PAGE, the protein bands were transferred to a Hybond-P polyvinylidene difluoride (PVDF) membrane (GE Healthcare, NJ) by electroblotting at 100 V for 1 h at 4°C. The PVDF membrane was then submerged in a blocking solution (25 mM Tris, 125 mM NaCl, 0.3% Tween 20, 0.05% NaN3 [pH 7.5], 3% skim milk) at 4°C and incubated overnight. The membrane was rinsed through two changes of washing buffer (25 mM Tris, 125 mM NaCl, 0.3% Tween 20, 0.05% NaN3 [pH 7.5]). The membrane was then incubated with a His probe (mouse monoclonal IgG diluted 2,000-fold in blocking solution) (Santa Cruz, CA) solution for 2 h at room temperature on a rotary shaker and rinsed twice with washing buffer. The membrane was further incubated in diluted (1:5,000) anti-mouse IgG (whole molecule)-alkaline phosphatase (Sigma-Aldrich, St. Louis, MO) for 2 h at room temperature on a rotary shaker and then rinsed twice with washing buffer and twice with distilled water. Finally the protein bands were visualized using Western blue stabilized substrate for alkaline phosphatase (Promega, Madison, WI) according to the manufacturer's protocol.
Synergistic activities of CbMan5B/Cel44A-TM2 with other cellulases from C. bescii.
Truncated mutants encoding three endoglucanases—CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1 (see Fig. 6)—from C. bescii were cloned, expressed, and purified from E. coli. Two of the genes encoding the three endoglucanases are located in the same gene cluster as the gene encoding CbMan5B/Cel44A. Separate solutions of PASC and the enzymes were made in citrate buffer (pH 5.0) and incubated at 70°C for 10 min. Combinations of the enzymes (final concentration of 1 μM for each enzyme) with CbMan5B/Cel44A-TM2 were then mixed with the substrate and incubated for 16 h. After termination of the reaction, the mixtures were centrifuged at 15,871 × g for 5 min, and the reducing ends released were estimated by the pHBAH assay (26). The degree of synergy (DOS) was calculated as described in our previous report (20). The amounts of the individual mono- and oligosaccharides (G1, G2, G3, and G4) released were quantified by the HPAEC-PAD method described above.
Fig 6.

Synergistic effects of CbMan5B/Cel44A-TM2 with the endoglucanases CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1 from C. bescii. (A) Schematic representation of CbMan5B/Cel44A-TM2, CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1. (B) PASC (5 mg/ml) was incubated with individual enzymes (1 μM) or a combination of enzymes (1 μM each) in a citrate buffer (pH 5.5) at 70°C for 16 h. The end products were analyzed by HPAEC-PAD with G1 to G6 as standards. In panel B, values above the bars indicate degree of synergy (DOS). n.d., no synergism detected.
The hydrolysis of an oligosaccharide substrate by the cellulases was assessed by adding each enzyme at a final concentration of 0.5 μM to cellopentaose (5 mg/ml in citrate buffer, pH 5.5), and the products were detected by the HPAEC-PAD method. The hydrolytic pattern with insoluble long-chain substrates was investigated by incubating enzymes at a final concentration of 2 μM with 5 mg/ml Avicel in a citrate buffer (pH 5.5) at 70°C. At different time points, 200-μl samples were removed from the reaction mixture, and the enzyme was inactivated by transferring to 100°C for 10 min. After enzyme inactivation, the insoluble products were pelleted by centrifugation at 15,871 × g for 15 min. The precipitate was washed twice with 1 ml 6 M guanidine-HCl, soaked in 6 M guanidine-HCl for 20 min, and washed four times with 1 ml distilled water and twice with 1 ml 50 mM sodium acetate buffer (pH 5.5). The washed insoluble sample was resuspended in 200 μl of sodium acetate buffer (pH 5.5), and reducing ends were measured by the bicinchoninic acid (BCA) method using glucose as the standard (12).
Amino acid sequence alignment and structural modeling.
The amino acid sequences of the GH44 catalytic modules from homologous proteins in C. saccharolyticus (GenBank accession number L01257) (16), C. acetobutylicum (GenBank accession number AAK78891) (38), and Clostridium thermocellum (GenBank accession number BAA12070) (23) were retrieved from the Carbohydrate-Active enZymes database (http://www.cazy.org) and the GenBank database (http://www.ncbi.nlm.nih.gov/protein/). An amino acid sequence alignment was performed with ClustalX (http://www.clustal.org/clustal2/). The aligned sequences were analyzed using BOXSHADE 3.21 (http://www.ch.embnet.org/software/BOX_form.html) with a default setting of the fraction of sequences parameter as 0.5. Structural modeling was carried out with the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera).
RESULTS AND DISCUSSION
Identification of C. bescii ORF1946 and its GH44 module as a cellulase.
The Rapid Annotation by Subsystem Technology (RAST) tool (3) was used to identify several putative glycoside hydrolases from the genome of C. bescii. One of several large genes in a cluster was designated open reading frame 1946 (ORF1946) and encoded a putative glycoside hydrolase (designated CbMan5B/Cel44A) with a predicted length of 1,294 amino acid residues. Analysis of the polypeptide sequence indicated that it consists of a signal peptide, a GH5 catalytic module at its N-terminal region, a GH44 catalytic module at the C-terminal region, and two carbohydrate-binding modules of family 3 positioned between the two catalytic modules (Fig. 1). The presence of a signal peptide in the polypeptide suggested that the protein functions extracellularly, and a search of the publicly available databases (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) showed that the polypeptides encoded by ORF0851 from Caldicellulosiruptor kronotskyensis (GenBank accession no. ADQ45728; 95% identity and 96% similarity) and ORF1077 from C. saccharolyticus (GenBank accession no. ABP66691; 89% identity and 93% similarity) are homologous to CbMan5B/Cel44A. The GH5 module in CbMan5B/Cel44A exhibited >99% identity to that of CbCel9B/Man5A. We have already demonstrated that the GH5 module of CbCel9B/Man5A functions as an endo-1,4-β-mannanase (35), and the catalytic residues identified for GH5 are conserved in the GH5 module in CbMan5B/Cel44A (see Fig. S1A in the supplemental material). Therefore, it was anticipated that the CbMan5B/Cel44A polypeptide would contain a mannanase activity and another catalytic activity in the GH44 module.
Cloning, expression, and purification of CbMan5B/Cel44A and its truncated mutants.
Several truncated mutants of the polypeptide were made to determine the catalytic activity present in the GH44 module of CbMan5B/Cel44A (Fig. 1). Each truncated mutant was fused to the N-terminal hexahistidine tag encoded by pET-46 Ek/LIC to facilitate purification of the recombinant proteins by immobilized metal affinity chromatography (IMAC). The predicted molecular mass of the polypeptide designated CbMan5B/Cel44A-WT (the wild-type protein lacking its signal peptide) was 139.8 kDa. We designated this polypeptide wild-type by reasoning that in the bacterium the signal peptide would be cleaved from the polypeptide after translocation out of the cytoplasm and that the functional enzyme would lack the signal peptide. The predicted molecular masses of the truncated mutants CbMan5B/Cel44A-TM1, CbMan5B/Cel44A-TM2, CbMan5B/Cel44A-TM3, and CbMan5B/Cel44A-TM4 were 71.3, 111.0, 82.3, and 65.9 kDa, respectively. The purified wild-type protein and its truncated mutants were resolved by SDS-PAGE, and their molecular masses estimated by this method were consistent with the predicted values from the polypeptide sequence (data not shown). For ease of reference, we use WT (for “wild type”), TM1, TM2, TM3, and TM4 to refer to the wild-type protein and its various truncated mutants presented in Fig. 1.
Screening of CbMan5B/Cel44A-WT and its truncated mutants for activity against polysaccharides.
The catalytic activity of the WT protein and two truncated mutants, TM1 and TM2, were assayed with a panel of polysaccharide substrates (Fig. 2A) to gain insights into the enzymatic activities in CbMan5B/Cel44A. These substrates included glucose-configured substrates (PASC, CMC, Avicel, and lichenan), mannose-configured or backbone substrates (mannan, guar gum, and locust bean gum), xylose-configured or backbone substrates (oat spelt xylan and birchwood xylan), and KGM, which is composed of mixed sugars (glucose and mannose). As shown in Fig. 2A, the WT protein degraded both glucose- and mannose-configured substrates. As stated earlier, the GH5 module in the polypeptide shares >99% identity with a C. bescii GH5 module known to function as a mannanase (35), and this activity was confirmed in the truncated protein containing the GH5 (i.e., TM1 in Fig. 1). All three polypeptides were able to hydrolyze the mixed linkage polysaccharide lichenin, but only the polypeptides with the GH44 module possessed the β-1,4-glucosidic linkage-cleaving activity (PASC and CMC). In contrast to the results observed for the C. saccharolyticus mannanase (14), the GH44 module in CbMan5B/Cel44A exhibited some catalytic activity on Avicel (Fig. 2A), and this was confirmed by HPLC analysis (Fig. 2B). However, this activity was lower than hydrolysis of other substrates. The truncated protein containing the GH44 module (TM2) also possessed the birchwood xylan- and xyloglucan-hydrolyzing activities observed in the WT polypeptide. Except for the hydrolysis of the model crystalline cellulose (Avicel), the other activities were similar to those observed for the C. saccharolyticus mannanase (14). A xyloglucan-cleaving activity was also observed in a GH44 protein from C. thermocellum (1), and xylan-cleaving activity was observed in a GH44 module from C. saccharolyticus (14).
Fig 2.

(A) Activities of CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 against polysaccharide substrates. CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 (0.5 μM) were incubated with each substrate (final concentration, 5 mg/ml) for 16 h at 70°C. The concentration of reducing ends was measured using the pHBAH assay. (B) Time course of hydrolysis of Avicel by CbMan5B/Cel44A-TM2. Avicel (10 mg/ml) was incubated with CbMan5B/Cel44A-TM2 (2 μM). At different time points, reaction mixtures were sampled, quenched, and diluted 10-fold in water and subjected to HPAEC-PAD analysis.
Determination of pH and temperature optima and thermostability of CbMan5B/Cel44A-TM2.
The optimal pH and temperature for TM2, with PASC as the substrate, were determined to be 5.0 (Fig. 3A) and 85°C (Fig. 3B), respectively, under the conditions described in Materials and Methods. The thermostability of TM2 was determined at four temperatures: 70°C, 75°C, 80°C, and 85°C (Fig. 3C). After 24 h, the residual activities of TM2 at 70°C, 75°C, 80°C, and 85°C were 93%, 76%, 39.8%, and 20.2%, respectively. The GH44 cellulase activity of TM2 appeared to have a higher optimal temperature than that of the GH44 endoglucanase reported from the thermophilic bacterium C. thermocellum (70°C) (1). Although these enzymes were not assayed under identical conditions, it seems unlikely that the discrepancies in experimental conditions could account for such a large difference in the optimal temperatures for the enzymes. The optimum temperatures for growth of C. thermocellum and C. bescii are 60 to 64°C (33) and 75°C (4), respectively.
Fig 3.
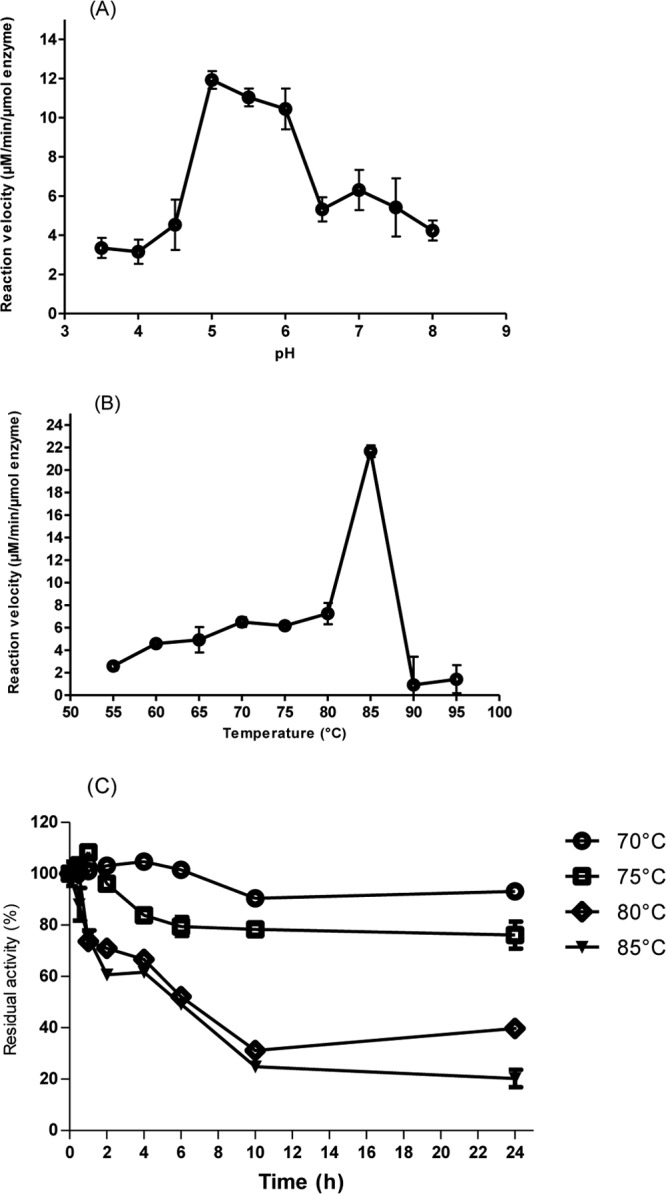
Optimal pH, optimal temperature, and thermostability assays of CbMan5B/Cel44A-TM2. (A) Optimal pH determination of CbMan5B/Cel44A-TM2. The CbMan5B/Cel44A-TM2 enzyme (2 μM) was incubated with 2.5 mg/ml phosphoric acid-swollen cellulose (PASC) in buffers differing in pH at 70°C. The released reducing ends were measured by the pHBAH assay. (B) Optimal temperature determination of CbMan5B/Cel44A-TM2. The CbMan5B/Cel44A-TM2 enzyme (2 μM) was incubated for 20 min with 2.5 mg/ml PASC in a citrate-HCl buffer (pH 5.0) at different temperatures ranging from 55 to 95°C, and the rates of end product release were determined. (C) Thermostability assay of CbMan5B/Cel44ATM2. The CbMan5B/Cel44ATM2 enzyme was incubated at 70°C, 75°C, 80°C, and 85°C. At different times, samples of the heated enzymes were taken and incubated with 2.5 mg/ml PASC dissolved in citrate-HCl buffer (pH 5.0) and reacted for 30 min at 70°C. After heat inactivation, the released reducing ends were measured as the residual activity using the pHBAH method.
Hydrolysis of PASC and cello- and manno-oligosaccharides.
Hydrolysis of PASC and cello- and manno-oligosaccharides by the WT, TM1, and TM2 proteins was performed to further clarify the enzymatic activities in the catalytic modules. As shown in Fig. 4A, cellopentaose (G5) and cellohexaose (G6) were degraded by the WT protein and TM2 to G1, G2, G3, and G4 after 16 h of incubation at 70°C. On G6, the products were mostly G2 and G4. A small amount of cellotetraose (G4) was also digested by the two polypeptides. However, none of the proteins hydrolyzed G2 or G3. On the manno-oligosaccharides, the WT protein and TM1 (Fig. 4B) hydrolyzed M3, M4, M5, and M6. Interestingly, TM2 exhibited some activity on the larger manno-oligosaccharides (M5 and M6). The catalytic activity of a GH44 module on mannose-configured substrates has not been documented previously. However, the mannanase activity of the GH44 module was weak compared with that of the GH5 module, which likely constitutes most of the mannanase activity in the WT protein. The presence of the mannanase activity in the C. bescii GH44 module may be due to subtle spatial differences between its catalytic cleft and those of hitherto-tested or -investigated GH44 module-containing polypeptides or enzymes. PASC was also incubated with the three polypeptides, and the end products were analyzed by HPAEC-PAD. As shown in Fig. 4C, the WT protein and TM2 released G1, G2, G3, and G4 from PASC, but none of the end products was observed after 16 h hydrolysis of PASC with TM1. Cellotetraose was the dominant product of the WT and the TM2 enzymes. These findings demonstrated that the cellulase activity in the WT protein is present in the GH44 catalytic module, while the majority of the mannanase activity is in the GH5 catalytic module.
Fig 4.

Hydrolysis of cello-oligosaccharides (A), manno-oligosaccharides (B), and phosphoric acid-swollen cellulose (C) by CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2. (A) Thin-layer chromatography (TLC) analysis of the reaction end products of CbMan5B/Cel44A-WT, TM1, and TM2 with glucose and cello-oligosaccharides. The reactions were carried out by incubating a 0.5 μM concentration of each protein with 5 mg/ml glucose or cello-oligosaccharide at 70°C for 16 h. At the end of the reaction, 1 μl of each reaction mixture was analyzed for end product release by TLC. G1, glucose; G2, cellobiose; G3, cellotriose; G4, cellotetraose; G5, cellopentaose; G6, cellohexaose. (B) TLC analysis of the reaction products of CbMan5B/Cel44A-WT, CbMan5B/Cel44A-TM1, and CbMan5B/Cel44A-TM2 with mannose and manno-oligosaccharides. The reactions were carried out as described for panel A. The standards are as follows: M1, mannose; M2, mannobiose; M3, mannotriose; M4, mannotetraose; M5, mannopentaose; M6, mannohexaose. (C) Hydrolysis of PASC. The analyses were as described for panel A, except that the substrate was PASC at 10 mg/ml. At the end of the reaction, samples were analyzed by high performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD). The standards for end product identification were G1 to G6.
Determination of kinetic parameters of CbMan5B/Cel44A-TM2 and its truncated mutants.
The specific activities with Avicel and filter paper as substrates were determined for the WT protein and the truncated mutants TM2, TM3, and TM4. The recombinant enzymes lacking one or two carbohydrate-binding modules retained only 53.6% (TM3) and 6.0% (TM4), respectively, of the specific activity displayed by the TM2 mutant (Table 1). Similar results were observed when filter paper was used as the substrate, indicating that the family 3 carbohydrate-binding modules in the polypeptide play a critical role in the hydrolysis of insoluble substrates by TM2 and likely the wild-type protein. PASC was used as the substrate to estimate the kinetic parameters of these truncated proteins (Table 1). The kcat values were 3.7 ± 0.5 s−1, 4.3 ± 0.6 s−1, 2.4 ± 0.3 s−1, and 0.8 ± 0.1 s−1 for WT, TM2, TM3, and TM4, respectively, demonstrating that the turnover numbers were negatively affected by loss of the carbohydrate-binding modules. These kinetic parameters strengthen our earlier observation that the family 3 CBMs in C. bescii multimodular cellulases are important in insoluble cellulose degradation (35).
Table 1.
Specific activities and kinetic parameters of CbMan5B/Cel44A-WT and the GH44-module-containing truncated mutants with cellulose substratesa
| Protein | Activity (μmol reducing ends/min/μmol enzyme) on: |
PASC |
|||
|---|---|---|---|---|---|
| Avicel | Filter paper | kcat (s−1) | Km (mg/ml) | kcat/Km (s−1 ml/mg) | |
| WT | 5.3 ± 0.8 | 12.7 ± 0.2 | 3.7 ± 0.5 | 1.2 ± 0.5 | 3.1 |
| TM2 | 5.8 ± 0.7 | 15.2 ± 0.5 | 4.3 ± 0.6 | 1.3 ± 0.5 | 3.3 |
| TM3 | 3.1 ± 0.2 | 5.4 ± 1.5 | 2.4 ± 0.3 | 1.1 ± 0.4 | 2.2 |
| TM4 | 0.4 ± 0.1 | 0.8 ± 0.5 | 0.8 ± 0.0 | 0.9 ± 0.1 | 0.9 |
Reactions were carried out at 70°C.
The kinetic parameters of the WT protein and its truncated mutants on the polysaccharide substrates KGM, guar gum, and lichenan were also determined (Table 2). The kcat values of TM2 on KGM, guar gum, and lichenan were 38.7 ± 3.3 s−1, 52.9 ± 8.8 s−1, and 927.8 ± 117.1 s−1. Compared to the WT protein (Table 2), the kcat values of TM2 obtained using mannose-configured substrates (KGM and guar gum) were sharply decreased, confirming that the mannan-hydrolyzing activity was located primarily in the GH5 catalytic module. Due to increases in both the Km and kcat of TM2, TM3, and TM4, the catalytic efficiencies of these truncated proteins against lichenan were similar to that of the WT protein (Table 2). Interestingly, only small differences in catalytic efficiencies were observed between TM2 and each of the mutants with deletions of CBMs (TM3 and TM4) on all soluble substrates (KGM, guar gum, and lichenan) suggesting that for this GH44 module, the carbohydrate-binding modules did not influence enzymatic activity against soluble substrates. The observation that TM3 displayed less than half of the catalytic efficiency of TM2 and TM4 on guar gum (Table 2) may merit further investigation. Our results also suggested that TM2 had higher activity on insoluble cellulosic substrate, particularly Avicel, than another thermostable bacterial enzyme from C. thermocellum that contains the GH44 module (1). When TM2 was incubated with Avicel at 70°C (pH 5.0), it hydrolyzed Avicel more rapidly (4.14 U/mg protein during the first hour and 0.41 U/mg protein in 24 h) than a GH44 catalytic module containing enzyme from C. thermocellum (0.13 U/mg protein in the first hour, and 0.0078 U/mg protein in 24 h) (1, 2).
Table 2.
Kinetic parameters of CbMan5B/Cel44A-WT and the GH44-module-containing truncated mutants on polysaccharides
| Protein | Konjac glucomannan |
Guar gum |
Lichenan |
||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat (s−1) | Km (mg/ml) | kcat/Km (s−1 ml/mg) | kcat (s−1) | Km (mg/ml) | kcat/Km (s−1 ml/mg) | kcat (s−1) | Km (mg/ml) | kcat/Km (s−1 ml/mg) | |
| WT | 510.2 ± 63.0 | 11.4 ± 2.7 | 50.0 | 934.3 ± 16.6 | 2.8 ± 1.6 | 333.6 | 722.9 ± 84.2 | 4.2 ± 1.4 | 172.1 |
| TM2 | 38.7.4 ± 3.3 | 1.7 ± 0.5 | 22.8 | 52.9 ± 8.8 | 1.4 ± 0.7 | 37.8 | 927.8 ± 117.1 | 5.2 ± 1.8 | 178.4 |
| TM3 | 46.4 ± 3.2 | 2.0 ± 0.5 | 23.2 | 33.1 ± 8.0 | 2.6 ± 0.7 | 12.7 | 839.5 ± 114.6 | 6.1 ± 2.1 | 137.6 |
| TM4 | 46.2 ± 4.0 | 1.67 ± 0.4 | 27.6 | 50.3 ± 9.1 | 1.4 ± 0.8 | 35.9 | 1,005.0 ± 115.4 | 8.3 ± 2.2 | 121.1 |
Investigation of sugar-binding subsites for the GH44 catalytic module.
To gain insight into the sugar-binding subsites of the GH44 module in TM2, we applied the “−n to +n” subsites nomenclature originally described by Davies and coworkers, where −n represents the nonreducing end and +n represents the reducing end, with cleavage of the glycosidic linkage taking place between the −1 and +1 subsites (10). The binding subsites of the GH44 catalytic module of CbMan5B/Cel44A were initially predicted through structural modeling (see Fig. S2 in the supplemental material). An amino acid sequence alignment then revealed that the GH44 catalytic module of C. bescii may be similar to that of the C. thermocellum Cel44A, which has −4 to + 5 subsites in its binding cleft.
In addition, based on amino acid sequence alignment of the GH44 module of CbMan5B/Cel44A with its homologs (see Fig. S1B in the supplemental material) and also by modeling of its three dimensional structure, the residues in the GH44 catalytic module in CbMan5B/Cel44A that could play key roles in hydrolysis of cellulosic substrates were revealed. Thus, the predicted catalytic proton donor/acceptor residue was Glu669 (see Fig. S1B), the predicted catalytic nucleophile residue was Glu853 (see Fig. S1B), the predicted residues forming hydrophobic platforms were Trp548, Tyr555, Trp821, Trp825, and Trp886, and the predicted residues forming hydrogen bonds with the substrate were Asn530 and Arg531. The glucopyranoses located at the −1, −3, and −4 subsites could probably interact with Trp886, Tyr555, and Trp548, respectively, of the GH44 catalytic module of CbMan5B/Cel44A. Glu669 and Glu853 were predicted to interact with the glucopyranose ring at the −1 subsite in the GH44 in a C. thermocellum endoglucanase (23), while the amino acids surrounding subsites +1 and +2 seem unlikely to interact with the pyranose ring. These predictions, while based on modeling and sequence alignment, suggested that the amino acids surrounding subsites −4 to −1 might be able to hold the chain as the substrate approaches the catalytic cleft and thus help to position the substrate to initiate the hydrolytic reaction. Therefore, we reasoned that cellotetraose would be the primary product when the GH44 catalytic module hydrolyzed glucose-configured substrates. A time course hydrolysis of cellopentaose and PASC by TM2 was performed to test this hypothesis (Fig. 5; also, see Table S2 in the supplemental material). After 2 min of hydrolysis, cellotetraose was found to be the primary product from both substrates. This pattern continued for the following 4 h, implying that the binding subsites for GH44 are indeed from −4 to +n. Cellotriose also accumulated at the beginning of the reaction, indicating that −3 to +n binding and hydrolysis also occurred to some extent. TM2 did not hydrolyze the shorter oligosaccharides cellobiose and cellotriose, which is consistent with −4 to +n binding subsites.
Fig 5.

Time course of hydrolysis of cellopentaose and PASC by CbMan5B/Cel44A-TM2. (A) Time course of hydrolysis of cellopentaose by CbMan5B/Cel44A-TM2. Cellopentaose (5 mg/ml) was incubated with 0.5 μM CbMan5B/Cel44A-TM2. At different time points, 200 μl of reaction mixture was sampled, quenched, diluted 50-fold in water, and subjected to HPAEC-PAD analysis. (B) The time course of hydrolysis of PASC by CbMan5B/Cel44A-TM2. PASC (5 mg/ml) was incubated with 1 μM CbMan5B/Cel44A-TM2, and at different time points, 200 μl of reaction mixture was sampled and the enzyme heat inactivated. Samples were then diluted 80-fold in water and subjected to HPAEC-PAD. The standards were glucose (G1), cellobiose (G2), cellotriose (G3), cellotetraose (G4), cellopentaose (G5), and cellohexaose (G6). (C) Based on the HPAEC-PAD profiles of the time course of hydrolysis of cellopentaose and PASC, the main cleavage pattern of CbMan5B/Cel44A-TM2 was proposed to be −4 to +n.
The β-sandwich domain is required for proper folding of the GH44 module.
The GH44 catalytic modules are composed of a TIM-like domain with an accompanying β-sandwich domain. The co-occurrence of a TIM-like domain with a β-sandwich domain is also found in GH5, GH30, GH39, and GH51 proteins (8, 13, 19, 25, 39). The β-sandwich domain is regarded as “a composite domain” for these proteins in the Structural Classification of Proteins database (32). It is composed of nine strands from the C terminus and one strand from the N terminus. The only exception is a human GH30 β-glucosidase, which contains two additional strands at the N-terminal region (13). The β-sandwich in these glycoside hydrolases shows structural similarity to domain C of several α-amylases and also to cellulose binding modules (15, 22, 36). In fact, the β-sandwich is considered a carbohydrate-binding module that has lost its binding function, a hypothesis supported by the observation that the β-sandwich domain from GH51 failed to bind to xylan (19). The results from the present report strengthen this hypothesis, since we also observed that TM4 (composed of only the GH44 module) did not bind to any of the polysaccharides tested (see Fig. S3 in the supplemental material). When we made truncated proteins lacking different β-sheets of the β-sandwich domain, i.e., CbMan5B/Cel44A-TM4Δ1β, CbMan5B/Cel44A-TM4Δ2β, CbMan5B/Cel44A-TM4Δ3β, and CbMan5B/Cel44A-TM4Δ9β, each deletion resulted in an insoluble protein, as depicted by the SDS-PAGE analysis in Fig. S4 in the supplemental material. This result suggests that the β-sandwich domain helps to fold the TIM-like domain or to stabilize the TIM-like domain structure, as proposed earlier by Kamitori et al. (22).
Synergy between CbMan5B/Cel44A-TM2 and three other endoglucanases from C. bescii.
A commonly accepted hypothesis for cellulose hydrolysis to glucose is that an endoglucanase cleaves bonds within chains located in amorphous and disordered regions of cellulose. Subsequently, a cellobiohydrolase acts on the ends generated by the endoglucanases to release cellobiose. A β-glucosidase then cleaves the cellobiose into two glucose units (28). Our analysis of the genome of C. bescii suggested that it encodes several potential endoglucanases in a cluster. Thus, to understand the potential contributions of these enzymes to cellulose hydrolysis by C. bescii, we investigated the capacity of TM2 to act synergistically with the recombinant forms of the cellulose-degrading enzymes in substrate hydrolysis. The architectures of the recombinant enzymes tested with TM2 are shown in Fig. 6A. For the analysis, PASC was used as the substrate. As shown in Fig. 6B, the same degree of synergy of 1.2-fold was observed when TM2 was used in combination with CbCel9B/Man5A-TM1 (35), CbMan5C/Cel5A-TM2 (GenBank accession no. ACM61039.1), and CbCel5B-TM1 (GenBank accession no. ACM59753.1). Note that the wild-type CbCel5B has surface layer homology (SLH) sequences at the C-terminal end, suggesting that it anchors to the cell after secretion. However, to make the recombinant CbCel5B-TM1, the SLH sequences were deleted during PCR amplification. Synergistic release of end products was not observed in binary combinations that lacked the TM2 protein from the present study. However, when TM2 was combined with these three endoglucanases, most of its cellotetraose and cellotriose end products were converted to cellobiose or glucose (Fig. 6B), a likely explanation for the observed synergy.
The mechanism of synergy was investigated further by analyzing the hydrolysis patterns in a time course experiment. Cellopentaose and PASC were used to investigate the end product release patterns of the three polypeptides (Fig. 7; also, see Table S2 in the supplemental material). Within 2 min, cellopentaose was hydrolyzed by CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1 predominantly into cellobiose, as well as some glucose and cellotriose. As the incubation time increased, CbCel9B/Man5A-TM1 converted the cellotriose end product into glucose and cellobiose rapidly (Fig. 7A-i; also, see Table S2), while CbMan5C/Cel5A-TM2 and CbCel5B-TM1 appeared to convert cellotriose into glucose and cellobiose more slowly (Fig. 7A-ii and A-iii; also, see Table S2). A similar result was observed when PASC was used as the substrate (Fig. 7B; also, see Table S2). Thus, these enzymes had similar hydrolysis patterns, producing mainly cellobiose, with a small amount of glucose and cellotriose. Since cellobiose is the normal product of cellobiohydrolases (18, 29), we considered whether the three endoglucanases in C. bescii were also cellobiohydrolases. However, large amounts of insoluble reducing ends were produced when Avicel was hydrolyzed with the individual enzymes (see Fig. S5 in the supplemental material), distinguishing the three enzymes from cellobiohydrolases (21). Note also that CbMan5B/Cel44A-TM2 released more insoluble reducing ends than the other three enzymes. Thus, in the experiments where CbMan5B/Cel44A-TM2 was coupled with the other endoglucanases, some of the soluble reducing ends released by this enzyme served as substrates for the cellulase activities in CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1.
Fig 7.

Time course hydrolysis of cellopentaose and PASC by CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1. (A-i, A-ii, and A-iii) Time course of hydrolysis of cellopentaose by CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1. Cellopentaose (5 mg/ml) was incubated with 0.5 μM each enzyme. Two hundred microliters of samples was removed, heat inactivated, diluted 50-fold, and analyzed by HPAEC-PAD. (B-i, B-ii, and B-iii) Time course of hydrolysis of PASC by CbCel9B/Man5A-TM1, CbMan5C/Cel5A-TM2, and CbCel5B-TM1. PASC (5 mg/ml) was incubated with each enzyme at 1 μM. Two hundred microliters of samples was removed, heat inactivated, diluted 80-fold, and analyzed by HPAEC-PAD. The standards were G1 to G6.
Although the GH44 catalytic module of CbMan5B/Cel44A may share phylogenetic and structural similarities with homologs previously characterized, including those from C. acetobutylicum and C. thermocellum, different characteristics were observed in the present homolog. These characteristics included a higher optimal temperature, higher thermostability, higher specific activity on Avicel, and a previously undocumented cleavage of β-1,4 mannosidic linkage, which are all important for the biofuel industry. Furthermore, CbMan5B/Cel44A exhibits 89 to 95% identity across the entire polypeptide with homologs in other members of the genus Caldicellulosiruptor (such as C. kronotskyensis; GenBank accession no. ADQ45728), and hence the results obtained in this study may be applicable to these two thermophilic proteins. Note that the modular organization of the Caldicellulosiruptor enzymes is different from that of the C. thermocellum enzyme. We also recently expressed from C. bescii an enzyme capable of degrading cellobiose into glucose (I. K. O. Cann, A. Miyagi, A. Asangba, and R. I. Mackie, unpublished data). The protein lacks a signal peptide, suggesting that it is intracellularly located, and it is highly conserved in other Caldicellulosiruptor spp. (C. kronotskyensis, GenBank accession no. ADQ47014; C. owensensis, GenBank accession no. ADQ03897; C. saccharolyticus, GenBank accession no. CAA31087; and C. hydrothermalis, GenBank accession no. ADQ07915). In C. saccharolyticus, the enzyme was designated a β-glucosidase (27). The amino acid sequence identities among these β-glucosidase homologs range from 92% to 95%. Therefore, based on our data, we hypothesize that in C. bescii, and perhaps other relatives, a secreted mixture of endoglucanases release cello-oligosaccharides (mostly cellobiose) and glucose from cellulose, as shown in Fig. S6A in the supplemental material, and these end products are transported into the cell, where a β-glucosidase (CbCdx1A; GenBank accession no. ACM59590) cleaves the cellobiose to generate more glucose (see Fig. S6A) for fermentation. A model of this hypothesis is provided in Fig. S6B in the supplemental material. From a biofuel production application perspective, this group of thermostable enzymes from C. bescii can serve as an important resource for assembling an enzyme cocktail that releases fermentable sugars (either glucose or a mixture of glucose and cellobiose) from cellulose at high temperatures for subsequent fermentation.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the Energy Biosciences Institute.
Footnotes
Published ahead of print 27 July 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Ahsan M, et al. 1997. Purification and characterization of the family J. catalytic domain derived from the Clostridium thermocellum endoglucanase CelJ. Biosci. Biotechnol. Biochem. 61: 427– 431 [DOI] [PubMed] [Google Scholar]
- 2. Ahsan MM, Kimura T, Karita S, Sakka K, Ohmiya K. 1996. Cloning, DNA sequencing, and expression of the gene encoding Clostridium thermocellum cellulase CelJ, the largest catalytic component of the cellulosome. J. Bacteriol. 178: 5732– 5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aziz RK, et al. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blumer-Schuette SE, Lewis DL, Kelly RM. 2010. Phylogenetic, microbiological, and glycoside hydrolase diversities within the extremely thermophilic, plant biomass-degrading genus Caldicellulosiruptor. Appl. Environ. Microbiol. 76: 8084– 8092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantarel BL, et al. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37: D233– D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho KM, et al. 2006. A cel44C-man26A gene of endophytic Paenibacillus polymyxa GS01 has multi-glycosyl hydrolases in two catalytic domains. Appl. Microbiol. Biotechnol. 73: 618– 630 [DOI] [PubMed] [Google Scholar]
- 7. Cho KM, et al. 2008. Changes in the activity of the multifunctional β-glycosyl hydrolase (Cel44C-Man26A) from Paenibacillus polymyxa by removal of the C-terminal region to minimum size. Biotechnol. Lett. 30: 1061– 1068 [DOI] [PubMed] [Google Scholar]
- 8. Czjzek M, et al. 2005. Enzyme-substrate complex structures of a GH39 β-xylosidase from Geobacillus stearothermophilus. J. Mol. Biol. 353: 838– 846 [DOI] [PubMed] [Google Scholar]
- 9. Dam P, et al. 2011. Insights into plant biomass conversion from the genome of the anaerobic thermophilic bacterium Caldicellulosiruptor bescii DSM 6725. Nucleic Acids Res. 39: 3240– 3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies GJ, Wilson KS, Henrissat B. 1997. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321: 557– 559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dodd D, Kiyonari S, Mackie RI, Cann IK. 2010. Functional diversity of four glycoside hydrolase family 3 enzymes from the rumen bacterium Prevotella bryantii B14. J. Bacteriol. 192: 2335– 2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doner LW, Irwin PL. 1992. Assay of reducing end-groups in oligosaccharide homologues with 2,2′-bicinchoninate. Anal. Biochem. 202: 50– 53 [DOI] [PubMed] [Google Scholar]
- 13. Dvir H, et al. 2003. X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 4: 704– 709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frangos T, Bullen D, Bergquist P, Daniel R. 1999. Hemicellulolytic and cellulolytic functions of the domains of a β-mannanase cloned from Caldicellulosiruptor saccharolyticus. Int. J. Biochem. Cell Biol. 31: 853– 859 [Google Scholar]
- 15. Fujimoto Z, et al. 1998. Crystal structure of a catalytic-site mutant α-amylase from Bacillus subtilis complexed with maltopentaose. J. Mol. Biol. 277: 393– 407 [DOI] [PubMed] [Google Scholar]
- 16. Gibbs MD, Saul DJ, Luthi E, Bergquist PL. 1992. The β-mannanase from “Caldocellum saccharolyticum” is part of a multidomain enzyme. Appl. Environ. Microbiol. 58: 3864– 3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansen CK, Diderichsen B, Jorgensen PL. 1992. CelA from Bacillus lautus PL236 encodes a novel cellulose-binding endo-β-1,4-glucanase. J. Bacteriol. 174: 3522– 3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harjunpaa V, et al. 1996. Cello-oligosaccharide hydrolysis by cellobiohydrolase II from Trichoderma reesei. Association and rate constants derived from an analysis of progress curves Eur. J. Biochem. 240: 584– 591 [DOI] [PubMed] [Google Scholar]
- 19. Hovel K, et al. 2003. Crystal structure and snapshots along the reaction pathway of a family 51 α-l-arabinofuranosidase. EMBO J. 22: 4922– 4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iakiviak M, Mackie RI, Cann IK. 2011. Functional analyses of multiple lichenin-degrading enzymes from the rumen bacterium Ruminococcus albus 8. Appl. Environ. Microbiol. 77: 7541– 7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irwin DC, Spezio M, Walker LP, Wilson DB. 1993. Activity studies of eight purified cellulases: Specificity, synergism, and binding domain effects. Biotechnol. Bioeng. 42: 1002– 1013 [DOI] [PubMed] [Google Scholar]
- 22. Kamitori S, et al. 1999. Crystal structure of Thermoactinomyces vulgaris R-47α-amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 A resolution. J. Mol. Biol. 287: 907– 921 [DOI] [PubMed] [Google Scholar]
- 23. Kitago Y, et al. 2007. Crystal structure of Cel44A, a glycoside hydrolase family 44 endoglucanase from Clostridium thermocellum. J. Biol. Chem. 282: 35703– 35711 [DOI] [PubMed] [Google Scholar]
- 24. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680– 685 [DOI] [PubMed] [Google Scholar]
- 25. Larson SB, Day J, Barba de la Rosa AP, Keen NT, McPherson A. 2003. First crystallographic structure of a xylanase from glycoside hydrolase family 5: implications for catalysis. Biochemistry 42: 8411– 8422 [DOI] [PubMed] [Google Scholar]
- 26. Lever M. 1972. A new reaction for colorimetric determination of carbohydrates. Anal. Biochem. 47: 273– 279 [DOI] [PubMed] [Google Scholar]
- 27. Love DR, Fisher R, Bergquist PL. 1988. Sequence structure and expression of a cloned β-glucosidase gene from an extreme thermophile. Mol. Gen. Genet. 213: 84– 92 [DOI] [PubMed] [Google Scholar]
- 28. Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66: 506– 577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Medve J, Karlsson J, Lee D, Tjerneld F. 1998. Hydrolysis of microcrystalline cellulose by cellobiohydrolase I and endoglucanase II from Trichoderma reesei: adsorption, sugar production pattern, and synergism of the enzymes. Biotechnol. Bioeng. 59: 621– 634 [PubMed] [Google Scholar]
- 30. Merino S, Cherry J. 2007. Progress and challenges in enzyme development for biomass utilization. Adv. Biochem. Eng. Biotechnol. 108: 95– 120 [DOI] [PubMed] [Google Scholar]
- 31. Moon YH, Iakiviak M, Bauer S, Mackie RI, Cann IK. 2011. Biochemical analyses of multiple endoxylanases from the rumen bacterium Ruminococcus albus 8 and their synergistic activities with accessory hemicellulose-degrading enzymes. Appl. Environ. Microbiol. 77: 5157– 5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murzin AG, Brenner SE, Hubbard T, Chothia C. 1995. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247: 536– 540 [DOI] [PubMed] [Google Scholar]
- 33. Ng TK, Weimer PJ, Zeikus JG. 1977. Cellulolytic and physiological properties of Clostridium thermocellum. Arch. Microbiol. 114: 1– 7 [DOI] [PubMed] [Google Scholar]
- 34. Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. 2010. CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20: 1574– 1584 [DOI] [PubMed] [Google Scholar]
- 35. Su X, Mackie RI, Cann IK. 2012. Biochemical and mutational analyses of a multi-domain cellulase/mannanase from Caldicellulosiruptor bescii. Appl. Environ. Microbiol. 78: 2230– 2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tormo J, et al. 1996. Crystal structure of a bacterial family-III cellulose-binding domain: a general mechanism for attachment to cellulose. EMBO J. 15: 5739– 5751 [PMC free article] [PubMed] [Google Scholar]
- 37. Warner CD, Go RM, Garcia-Salinas C, Ford C, Reilly PJ. 2011. Kinetic characterization of a glycoside hydrolase family 44 xyloglucanase/endoglucanase from Ruminococcus flavefaciens FD-1. Enzyme Microb. Technol. 48: 27– 32 [DOI] [PubMed] [Google Scholar]
- 38. Warner CD, et al. 2010. Tertiary structure and characterization of a glycoside hydrolase family 44 endoglucanase from Clostridium acetobutylicum. Appl. Environ. Microbiol. 76: 338– 346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang JK, et al. 2004. Crystal structure of β-d-xylosidase from Thermoanaerobacterium saccharolyticum, a family 39 glycoside hydrolase. J. Mol. Biol. 335: 155– 165 [DOI] [PubMed] [Google Scholar]
- 40. Yang SJ, et al. 2009. Efficient degradation of lignocellulosic plant biomass, without pretreatment, by the thermophilic anaerobe”Anaerocellum thermophilum“ DSM 6725. Appl. Environ. Microbiol. 75: 4762– 4769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang SJ, et al. 2010. Classification of ‘Anaerocellum thermophilum ’ strain DSM 6725 as Caldicellulosiruptor bescii sp. nov. Int. J. Syst. Evol. Microbiol. 60: 2011– 2015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.