

Abstract
Metabolic engineering of microorganisms has become a versatile tool to facilitate production of bulk chemicals, fuels, etc. Accordingly, CO2 has been exploited via cyanobacterial metabolism as a sustainable carbon source of biofuel and bioplastic precursors. Here we extended these observations by showing that integration of an ldh gene from Bacillus subtilis (encoding an l-lactate dehydrogenase) into the genome of Synechocystis sp. strain PCC6803 leads to l-lactic acid production, a phenotype which is shown to be stable for prolonged batch culturing. Coexpression of a heterologous soluble transhydrogenase leads to an even higher lactate production rate and yield (lactic acid accumulating up to a several-millimolar concentration in the extracellular medium) than those for the single ldh mutant. The expression of a transhydrogenase alone, however, appears to be harmful to the cells, and a mutant carrying such a gene is rapidly outcompeted by a revertant(s) with a wild-type growth phenotype. Furthermore, our results indicate that the introduction of a lactate dehydrogenase rescues this phenotype by preventing the reversion.
INTRODUCTION
Concerns about shrinking supplies of fossil fuel and about climate change have been a strong incentive during the past decade for the development of sustainable alternatives to provide fuel and chemical feedstock. Because of these concerns, several different ways to produce solar biofuel have been proposed. Initially the proposed procedures were based on the conversion of agricultural crops into short-chain alcohols. This unfortunately results in a direct competition for resources between the energy sector and food supply. Therefore, second-generation types of processes were proposed, in which one aims at the conversion of the triglyceride fraction from, e.g., green algae into biodiesel, which relaxes the competition between energy and the food supply. Nevertheless, the conversion of CO2 into biofuel, driven by solar energy, is still rather indirect even in this second-generation approach: CO2 is first converted into the complex building blocks of biomass, from which subsequently the triglyceride fraction must be extracted and converted into biodiesel.
For these reasons, a third-generation type of process has been proposed, in which CO2 is converted into biofuel directly, accomplished by the introduction of genes encoding a metabolic—e.g., fermentative—pathway into a cyanobacterium, resulting in the emergence of a “photofermentative” chimera. For several biofuel and chemical feedstock products, including ethanol, ethylene, isoprene, d-lactic acid, glucose, sucrose, isobutyraldehyde, and 1-butanol, the proof of principle of this approach has been provided (see references 3, 8, 10, 24, 25, 28, and 39 and references therein). Ideally, this process would be carried out in such a way that the majority of the CO2 that is fixed in the cyanobacterial Calvin-Benson cycle is converted into the selected product. This implies that the central metabolism in the selected chimera should be largely redirected toward the biofuel product, and ideally biomass would be formed only to compensate for the death rate of the organism in the producing cell suspension. The terms “photofermentative” and “photanol” (16) have been coined for this idea to contribute to the efficient and sustainable harvesting of solar biofuel.
As a model system for this approach, we concentrated on a very basic and well-defined fermentation end product: lactic acid, which is traditionally produced from sucrose by lactic acid bacteria and is used as a chemical feedstock for the production of, e.g., bioplastics, although other sugars, including sugars derived from lignocellulosic materials, are also used for this purpose (1). However, the NAD(P)H-dependent fermentative conversion of pyruvate to lactic acid is found in many additional organisms, including the enterobacteria and the bacilli (13). Most fermentative bacteria use NADH, rather than NADPH, as the redox cofactor for pyruvate reduction. Cyanobacteria, however, make reducing equivalents available, with the electrons derived from water splitting, primarily in the form of NADPH rather than NADH. Interestingly, the genome of Synechocystis sp. strain PCC6803 harbors a gene that shows similarity to genes encoding energy-linked transhydrogenases (26). Heterologous expression of a non-energy-linked transhydrogenase is therefore expected to increase the cellular NADH/NAD+ ratio, so that a heterologously expressed NADH-dependent lactate dehydrogenase (LDH) could benefit from this (compare with reference 28).
In this study, we show that the carbon flux in Synechocystis sp. PCC6803 can be partially redirected to a preferred fermentation product, i.e., lactic acid. The choice of the particular LDH selected strongly affects the partitioning of carbon over the biomass and (fermentation) product. Coexpression of a soluble transhydrogenase with a lactate dehydrogenase gives a significant further increase in lactic acid productivity. The introduction of the lactate dehydrogenase rescues the genetic instability of a mutant harboring a soluble transhydrogenase only.
Accordingly, we report significant improvement of the state of the art in the photofermentative approach for the production of lactic acid, and we discuss how to further enhance its production rate. This work may therefore contribute to the engineering of a cyanobacterial cell factory that can convert the majority of the CO2 that it fixes into a desired product.
MATERIALS AND METHODS
Strains and growth conditions.
All cloning procedures were carried out in Escherichia coli XL1 (blue) (Stratagene), grown in liquid LB medium at 37°C in a shaking incubator at 200 rpm or on solidified LB plates containing 1.5% (wt/vol) agar. When appropriate, media were supplemented with antibiotics needed for propagation of a specific plasmid(s) or marker(s). Concentrations of antibiotics used, alone or in combination, were 50 μg/ml kanamycin, 35 μg/ml chloramphenicol, and 100 μg/ml ampicillin.
Synechocystis sp. strain PCC6803 (a glucose-tolerant derivative, obtained from D. Bhaya, University of Stanford, Stanford, CA) was routinely cultivated in BG-11 medium (31) (Cyanobacteria BG-11 freshwater solution; Sigma) at 30°C in a shaking incubator at 120 rpm (Innova 43; New Brunswick Scientific) under constant moderate white-light illumination (30 μE/m2/s, measured with a LI-250 light meter; Li-Cor) provided by 15-W cool fluorescent white lights (F15T8-PL/AQ; General Electric). Where appropriate, cultures were supplemented with 50 mM NaHCO3 (pH 8.0) (Sigma). Kanamycin-resistant mutant strains were grown in medium containing 20 μg/ml kanamycin. For plates, BG-11 medium was supplemented with 1.5% (wt/vol) agar, 5 mM glucose, 0.3% (wt/vol) sodium thiosulfate (30), and 20 μg/ml kanamycin for resistant strains. Concentrated stocks of cells were routinely stored at −80°C in BG-11 medium supplemented with 5% dimethyl sulfoxide. Cells were restreaked on plates and grown to stationary phase in a preculture before they were rediluted to an optical density at 730 nm (OD730) (Lightwave II spectrophotometer; Biochrom) of 0.1 at the start of the experiment. Growth was monitored by recording the OD730.
Gene cassette construction.
Plasmids used for cloning and for transformation of Synechocystis were constructed using standard cloning techniques (33). The ZR Plasmid Miniprep-Classic kit (Epigenetics) was used for plasmid preparations according to the manufacturer's instructions. Genes of interest were amplified by PCR using Pwo proofreading DNA polymerase (Roche) from isolated genomic DNA of the respective organisms with specific primers (see Table S1 in the supplemental material). The DNeasy blood and tissue kit (Qiagen) was used for genomic DNA preparations according to the manufacturer's instructions. The primers were selected to introduce BioBrick-compatible restriction sites that were used for standard cloning procedures (19) and the “three-antibiotic based assembly strategy” (36). An l-lactate dehydrogenase (l-LDH) gene (ldh) was amplified from genomic DNA of Bacillus subtilis 168. A soluble transhydrogenase (sth) gene was amplified from Pseudomonas aeruginosa PAO1, and a d-lactate dehydrogenase (ldhA) gene was amplified from genomic DNA of the DH5α strain of E. coli K-12. Primers for genes of interest were designed to introduce a (consensus) ribosomal binding site (RBS). Oligonucleotides were purchased from Biolegio (The Netherlands). Restriction enzymes, T4 DNA ligase, and FastAP thermosensitive alkaline phosphatase were obtained from Fermentas. DNA fragments were purified using the MSB Spin PCRapace kit (Invitek) according to the manufacturer's instructions. The sequence of the promoter Ptrc was taken from reference 5 and chemically synthesized (GeneArt, Germany) such that it was flanked by BioBrick-compatible restriction sites. Ptrc has been successfully tested in Synechocystis (17). The transcriptional terminator used was a BioBrick (BBa_B0014), which, in combination with selected BioBrick plasmids, was obtained from the Registry of Standard Biological Parts (http://partsregistry.org). The plasmids used and constructed in this study are listed in Table 1.
Table 1.
Strains, plasmids, and BioBrick parts used in this study
| Strain, plasmid, or BioBrick part | Description | Source or reference |
|---|---|---|
| pBluescript II SK(+) | Backbone for integration plasmid | Stratagene |
| E. coli XL-1 (blue) | Cloning host strain | Stratagene |
| pSB1A3 | Standard BioBrick vector | partsregistry.org |
| pSB1AC3 | Standard BioBrick vector | partsregistry.org |
| Double terminator | Transcriptional terminator | BBa_B0014 (partsregistry.org) |
| Ptrc | trc promoter on pSEQ_Trc, chemically synthesized | This study; sequence from Brosius et al., 1985 (5) |
| Synechocystis sp. PCC6803 | Wild type, glucose tolerant, naturally transformable | D. Bhaya |
| pPSBA2KS | Source of kanamycin cassette | Lagarde et al., 2000 (23) |
| pHKH001 | Integration vector disrupting slr0168 in Synechocystis genome with kanamycin cassette as selection marker | This study |
| pHKH005 | Integration vector containing B. subtilis ldh cassette | This study |
| pHKH003 | Integration vector containing E. coli ldh cassette | This study |
| pHKH012 | Integration vector containing P. aeruginosa sth cassette | This study |
| pHKH015 | Integration vector containing B. subtilis ldh and P. aeruginosa sth cassettes | This study |
| SAA013 | Synechocystis Ptrc∷ldh∷Kmr, d-LDH of E. coli | This study |
| SAA015 | Synechocystis Ptrc∷ldhA∷Kmr, l-LDH of B. subtilis | This study |
| SAA016 | Synechocystis Ptrc∷sth∷Kmr,STH of P. aeruginosa | This study |
| SAA017 | Synechocystis Ptrc∷ldh∷Ptrc∷sth∷Kmr, l-LDH of B. subtilis, STH of P. aeruginosa | This study |
Integration plasmid.
Flanking homologous regions in the plasmid used for insertion of genes of interest into the cyanobacterial genome, via double homologous recombination, were amplified from isolated genomic DNA of Synechocystis. The two selected homologous regions (Hom1 and Hom2) target slr0168, a neutral site (21, 27) in the Synechocystis genome. pBluescript II SK(+) (Stratagene) served as the backbone for the integration plasmid. Hom1 stretches from chromosomal position 2300735 to 2301291 (18) and was cloned into the SacI and SacII sites of pBluescript II SK(+). Hom2 stretches from chromosomal position 2301292 to 2301835 and was cloned into the XhoI and KpnI sites of pBluescript II SK(+). The kanamycin resistance cassette (aphX), originating with the E. coli transposon Tn903, was amplified from pPSBA2KS (23) and cloned into the SalI site of pBluescript II SK(+). To ease further cloning, the remaining multiple-cloning site (MCS) of the integration plasmid was designed to be compatible with the BioBrick restriction sites.
Transformation of Synechocystis.
Natural transformation of Synechocystis was performed essentially as described in reference 42. Briefly, 5 to 10 μg of plasmid DNA was mixed with 100 μl of a concentrated cell suspension (OD730 = 20) from an exponentially growing culture and incubated for 5 h in the light, with shaking at 120 rpm. Then, the transformation mixture was plated on a 1.5% (wt/vol) agar plate containing BG-11 medium, supplemented with 5 mM glucose, 0.3% (wt/vol) sodium thiosulfate, and 3 μg/ml kanamycin. After isolation of individual colonies, segregation of the insert into the multiple copies of the cyanobacterial genome (15, 22) was achieved by restreaking the mutants stepwise on plates containing increasing concentrations of kanamycin until complete segregation routinely was accomplished at 20 μg/ml kanamycin. Recombinant Taq DNA polymerase (Fermentas) was used for nonpreparative PCR, such as colony PCR, to monitor the segregation process and to verify the correct incorporation of the DNA fragments into the Synechocystis genome.
Sequencing of DNA fragments was performed at Macrogen Europe (The Netherlands) with dedicated primers (see Table S1 in the supplemental material). The plasmids and strains that were constructed for this study are listed in Table 1.
d- and l-lactic acid assay.
Batch culture samples were harvested at selected time points. For measurement of the lactic acid concentration, a 500-μl aliquot was taken. Cells were removed by centrifugation for 2 min at 14,000 rpm at room temperature. Of the resulting supernatant, 100 μl was used in the d-/l-lactic acid (rapid) assay (Megazyme) to determine the l-lactate concentration according to the manufacturer's instructions. As a control, we also assayed for d-lactate in the cell-free supernatant of the wild type and the mutant strains. All indicated volumes of the assay kit were scaled down exactly 10 times for use in a 96-well plate reader at 30°C (Multiscan FC microplate photometer; Thermo Scientific).
Preparation of crude cell extract.
Synechocystis cells were harvested in the mid-exponential growth phase by centrifugation at 4,000 rpm for 10 min at 4°C. The pellets obtained were resuspended in prechilled buffer of the subsequent in vitro assay. Cells were disrupted by bead beating, and cell debris was removed by centrifugation at 12,500 rpm for 30 min at 4°C. Glass beads of 0.1 mm in diameter and a Precellys24 homogenizer (Berton) were used. The protein content of the crude cell-free extract (CFE) was determined with the colorimetric bicinchoninic acid (BCA) protein assay kit (Pierce). Absorption at 562 nm was measured and compared to a standard curve prepared with bovine serum albumin.
In vitro enzymatic activity assays.
Enzyme activity assays were performed in transparent 96-well plates. Assay conditions for the l-LDH originating from B. subtilis were adopted from reference 32. Briefly, the reaction mixture (total volume, 200 μl) containing 50 mM sodium phosphate, pH 6.5, 300 μM NADH, 2.5 mM MgCl2, and 500 μg or 1,000 μg CFE of the respective strains was preincubated at 30°C for 5 min. For preparation of CFE, cultures from the mid-exponential growth phase were used. To start the reaction, sodium pyruvate was added to a final concentration of 30 mM and the oxidation of NADH was monitored at 340 nm in a 96-well plate reader at 30°C (Multiscan FC microplate photometer; Thermo Scientific). Assay conditions for the LDH originating from E. coli were adopted from reference (41). Briefly, a reaction scheme similar to that described for the B. subtilis LDH was followed, the only exception being the reaction buffer, which in this case consisted of 100 mM potassium phosphate, pH 7.5.
Conditions for the assay of the soluble transhydrogenase were adopted from reference 12. Briefly, the reaction mixture (total volume, 200 μl), containing 100 mM Tris-HCl, pH 7.5, 100 μM thio-NAD+, and 500 μg or 1,000 μg CFE, was preincubated for 5 min at 30°C. CFE of cultures from mid-exponential growth phase was used. To start the reaction, NADPH was added to a final concentration of 100 μM, and the formation of thio-NADH was monitored at 405 nm in a 96-well plate reader at 30°C (Multiscan FC microplate photometer; Thermo Scientific).
RESULTS
Construction of ldh-carrying Synechocystis strains.
Based in part on the earlier work carried out by our research group in this area (6, 7, 11) and on the catalytic properties of the respective enzymes, we selected a lactate dehydrogenase (ldh) gene from Escherichia coli (ldhA, encoding a d-LDH, EC 1.1.1.28 [41]), from Bacillus subtilis (ldh, encoding an l-LDH, EC 1.1.1.27 [32]), and from Lactococcus lactis (ldh, encoding an l-LDH, EC 1.1.1.27 [13, 14]). The three genes were cloned using the “BioBrick” cloning strategy (19) and subsequently placed in (separate) integration vectors to achieve stable chromosomal incorporation of these genes within the neutral docking site slr0168 in Synechocystis sp. PCC6803 (henceforth referred to as Synechocystis) (21). The cloning strategy included the addition of a Ptrc promoter, as well as a transcriptional terminator, to each gene encoding the heterologous enzyme. A kanamycin resistance marker, carrying its endogenous regulatory sequences, was cloned in each of these plasmids (Fig. 1). Tests carried out during the cloning phase of this project revealed that the ldh gene from L. lactis consistently generated spontaneous mutations upon cloning in E. coli, and for this reason functional expression of this enzyme has not been achieved in either in E. coli or in Synechocystis.
Fig 1.

Heterologous gene insertion in the cyanobacterial genome at a neutral docking site. (A) Schematic presentation of the plasmid construction scheme for subsequent integration of two heterologous genes (B. subtilis ldh and P. aeruginosa sth) into the neutral docking site, slr0168, of Synechocystis sp. PCC6803, resulting in a disruption of the open reading frame (ORF) of slr0168. (B) Verification by PCR of the insertion of the genes into the Synechocystis genome and proof of complete segregation of the ensuing two types of genomes in single colonies of the organism with primers flanking the insertion site. Panels C (ldh) and D (sth) show the verification of the insertions with specific primers for the two genes of interest. PCR on plasmids used for transformation served as positive controls. NC, negative control; M, marker.
The remaining two ldh genes were transferred to a glucose tolerant axenic wild-type Synechocystis strain, through natural transformation, using the plasmids pHKH003 and pHKH005, respectively (Table 1). Repeated plating on agar plates with increasing antibiotic concentrations allowed isolation of fully segregated transformants in which the exogenous dehydrogenase gene, together with the kanamycin resistance marker, was stably incorporated in the targeted docking site (Fig. 1). On average, restreaking on plates three to four times with increasing kanamycin concentrations was necessary to achieve full chromosome segregation. The segregation process was monitored by colony PCR. The two strains that were isolated were named SAA013 and SAA015, for the cyanobacteria carrying the lactate dehydrogenase of E. coli ldh and B. subtilis ldh, respectively (Table 1).
Lactic acid production in the lactate dehydrogenase-carrying Synechocystis strains.
SAA013 and SAA015, together with the parent wild-type Synechocystis strain, were tested with respect to their capacity to synthesize lactic acid from CO2 and H2O. To this end, batch cultures of these strains were inoculated in carbonate-containing BG-11 medium with continuous white-light illumination. Wild-type cultures did not show d- or l-lactate formation. Results obtained with the wild type and with SAA013 were indistinguishable; of these two, only the latter will therefore be discussed. Surprisingly, only SAA015 produced significant amounts of lactic acid (which is 97 to 100% l-lactic acid, as determined by the selective enzymatic assay [Fig. 2A]). During prolonged incubation of SAA015 (10 days), l-lactic acid accumulated to a concentration of 0.7 mM, with a maximal rate (during late-exponential growth; Fig. 2A) of 0.0058 mmol lactate/g dry weight (DW)/h, which corresponds to 0.0174 mmol carbon/g DW/h. The amount of lactic acid in culture supernatants of SAA013 did not exceed the detection limit of the enzymatic assay for lactic acid, which was about 25 μM, even after prolonged incubation.
Fig 2.

Growth and extracellular lactate formation in engineered strains of Synechocystis sp. PCC6803, carrying a heterologous lactate dehydrogenase gene (ldh), cultured in continuous light (A) or in a light/dark cycle (B). Cells were grown in BG-11 medium supplemented with 50 mM NaHCO3. Filled symbols represent the OD730s; open symbols represent the lactate concentrations. SAA013, the strain with integrated E. coli ldh, is represented by diamonds (assaying for d-lactate); SAA015, the strain with integrated B. subtilis ldh, is represented by squares (assaying for l-lactate). Values are the averages for biological replicates; error bars indicate the standard deviations (n = 3); if error bars are not visible, they are smaller than the data point symbol. Cultures were grown under continuous white-light illumination (30 μE/m2/s; A) or under a light/dark cycle of 16 h of light (30 μE/m2/s) and 8 h of darkness (B). Gray-shaded areas represent the dark periods. SAA013 did not show l-lactate production, and SAA015 did not show d-lactate (data not shown).
Lactate formation was routinely measured in cultures incubated with white light at 30 μE/m2/s. Although the volumetric rate of lactate production varies slightly with light intensity, normalized to the biomass content, it is to a great extent independent of this parameter in the range between 10 and 100 μE/m2/s (data not shown). For large-scale application of a cyanobacterial cell factory aimed at the production of lactic acid from CO2 (see also references 2 and 4), it is of interest to know the temporal pattern of lactic acid production under a circadian regime of illumination. A batch culture of strain SAA015 was therefore subjected to a light-dark cycle of 16 h of illumination, followed by 8 h of darkness, for a period of 10 days. In such a regime, the increase of the OD730 is limited to the light periods (Fig. 2B). The OD730 decreases slightly in the dark, which may be due to the catabolism of glycogen that accumulated during the preceding period in the light. Furthermore, lactic acid production was also limited to the light period. Significantly, a decrease of the lactate concentration in the dark, conceivable due to, e.g., oxidative catabolism, was not observed, and the net rate of lactic acid production under these conditions over a period of several days was 0.0056 mmol lactate/g DW/h (as calculated during late-exponential growth [Fig. 2B]), which is almost as high as the rate in continuous light (Fig. 2A).
The lactate dehydrogenases show overcapacity in their activities relative to production rates.
To identify what caused the differences between strains SAA013 and SAA015, enzymatic assays of their crude cell extracts (CFE) were performed. CFEs were assayed under the conditions described previously (32, 41) in the presence of 30 mM pyruvate and 0.3 mM NADH, which approximate Vmax conditions for both enzymes (32, 41). LDH activities in CFE of SAA013 and SAA015 were 0.11 and 0.18 mmol/g DW/h, respectively (Table 2), which clearly shows that although lactate was not detected in the culture supernatant of SAA013, its CFE showed significant LDH activity (see Discussion for further details). Possible reasons why the actual rate of lactic acid production in SAA015 is about 30-fold lower than the maximal capacity of the cell extract of that strain to convert pyruvate into lactic acid are as follows: (i) suboptimal substrate concentrations of the reaction, (ii) the presence/absence of activity-modulating metabolites (like fructose 1,6-bis-phosphate for the aforementioned LDH from L. lactis) (14), (iii) suboptimal physiological conditions in the heterologous cytoplasm, or (iv) any combination of these three factors. However, because LDH from B. subtilis is relatively insensitive toward activity-modulating metabolites, we assume that the first of these factors is the most important (32).
Table 2.
Enzymatic activities of LDH and STH in cell extracts of wild-type Synechocystis and three engineered strainsa
| Strain | Enzyme | Activity (Vmax) (mmol/g DW/h) | Relative activity (%)b |
|---|---|---|---|
| Wild type | LDH | 0.0078 ± 0.001 | 0.21 ± 0.03 |
| Wild type | STH | ND | NA |
| SAA013 | LDH | 0.11 ± 0.012 | 2.9 ± 0.341 |
| SAA013 | STH | ND | NA |
| SAA015 | LDH | 0.18 ± 0.024 | 4.7 ± 0.707 |
| SAA015 | STH | ND | NA |
| SAA017 | LDH | 0.24 ± 0.038 | 6.5 ± 1.090 |
| SAA017 | STH | 0.23 ± 0.003 | 6.2 ± 0.079 |
The wild type does not display significant LDH activity. The wild type and both ldh-containing single mutants investigated (SAA013 and SAA15) lack detectable STH activity. ND, not detected; NA, not applicable.
See the supplemental material for calculations.
Construction of sth-carrying Synechocystis strains.
The relatively high affinity of the LDH from B. subtilis for nicotinamide nucleotides suggests that optimization of cofactor availability, in the form of an increase in the intracellular concentration of NADH, may further stimulate lactic acid production in transgenic Synechocystis strains. The LDH from B. subtilis uses the reducing equivalents from NADH for the conversion of pyruvate into lactic acid. However, the molecular machinery embedded in the thylakoid membrane of cyanobacteria generates reducing equivalents primarily in the form of NADPH. We therefore decided to test the additional integration of a transhydrogenase in the engineered Synechocystis strain, carrying the heterologous ldh. To this end, genes encoding a soluble (energy-independent) transhydrogenase were chosen from Pseudomonas aeruginosa (P. aeruginosa sth) (Table 1) and Azotobacter vinelandii. The A. vinelandii enzyme could not be stably expressed in E. coli, and its transfer to Synechocystis was therefore not further pursued.
Initially, for the sole insertion of the P. aeruginosa sth gene, an integration plasmid (pHKH012) with the sth gene under the control of Ptrc was constructed and subsequently transferred to the wild-type Synechocystis strain via natural transformation. However, antibiotic-resistant transformants from such experiments did require high antibiotic titers for complete genome segregation and, upon complete segregation, grew very slowly (Fig. 3). Significantly, from a lawn of such cells (SAA016), colonies with a wild-type-like growth phenotype that rapidly overgrew their parents emerged (Fig. 3). It was therefore a delicate issue to characterize SAA016 physiologically. The genetic basis of the phenotypic reversion of the transgenic strains, carrying solely the sth gene, was investigated in a limited number of strains. In these strains, we found a duplication of ∼160 bp in the sth gene, which generated premature stop codons (data not shown). However, before a conclusion can be drawn about the generality of this observation, more of these mutants should be analyzed. The overall genetic stability of Synechocystis is limited primarily by the frequency of occurrence of point mutations (20). Whether or not the impairment of the expression of the functional transhydrogenase in SAA016 is also dependent on this mechanism or in contrast depends on other molecular mechanisms of spontaneous mutagenesis will require more extensive screening of revertants of the type identified in this study.
Fig 3.
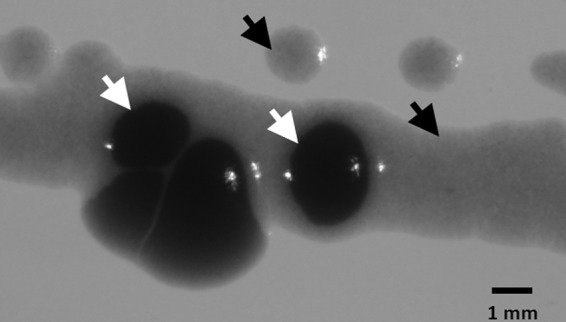
BG-11-containing agar plate showing growth of the strain with integrated P. aeruginosa sth (SAA016) interspersed with cells that reverted to a wild-type growth phenotype. SAA016 was grown for 2 weeks on a BG-11-containing agar plate, supplemented with kanamycin. SAA016 formed colonies or a lawn of slowly growing cells (black arrows), which are interspersed with cells that reverted to a faster-growth phenotype, i.e., resembling the wild type (white arrows).
In contrast to the strain with integrated P. aeruginosa sth only (SAA016), construction of a Synechocystis strain with both the B. subtilis ldh gene and the P. aeruginosa sth gene (both under the control of Ptrc and integrated at the slr0168 docking site [SAA017]) was achieved straightforwardly with the integration plasmid pHKH015. Stable transformants of this strain (see Fig. S1 in the supplemental material) showed considerable activities of the two heterologous enzymes in their CFE: 0.23 and 0.24 mmol/g DW/h for soluble transhydrogenase (STH) and LDH, respectively (Table 2; see also Fig. S2).
Lactic acid production in the transhydrogenase-carrying Synechocystis strain.
The comparison of strain SAA017 with strain SAA015 shows that introduction of the soluble transhydrogenase leads to an approximately 5-fold increase in lactic acid production, such that the culture reaches a final concentration of 3.2 mM lactic acid after a 2-week period (Fig. 4). A slight retardation in growth rate is observed, possibly due in part to the increased partitioning of carbon toward lactic acid. Wild-type Synechocystis under these specific conditions shows a growth rate of 0.068 h−1. Inhibitory effects of lactic acid on the growth rate of wild-type Synechocystis are observed only at concentrations of 100 mM and above (a 50% inhibitory concentration [IC50] of 235 mM was determined [see Fig. S3 in the supplemental material]). The actual partitioning of carbon between cellular biomass and the lactic acid product in the late-exponential growth phase (between days 5 and 8) (Fig. 4) can be derived from the maximal growth rate of strain SAA017, i.e., 0.047 h−1 (which equals 0.047 g biomass/g DW/h or 1.86 mmol carbon/g DW/h) and a production rate of 0.0178 mmol lactate/g DW/h (which corresponds to 0.0534 mmol carbon/g DW/h). Thus, the partitioning equals about 3% of the carbon fixed in the Calvin-Benson cycle being diverted to lactic acid, taking into account that about 4 mol carbon is needed to form 1 mol of biomass (see the supplemental material for calculations). These results show that even under these conditions of increased lactic acid production, the majority of the fixed carbon still partitions into biomass. On the assumption that the expression of the soluble transhydrogenase will saturate the lactate dehydrogenase with NADH, these values nonetheless do allow one to make an estimate of the cytoplasmic pyruvate levels under these conditions (see Discussion for further details), which has been hypothesized above to be the major limiting factor in the heterologous reaction employed.
Fig 4.

Growth and extracellular l-lactate formation in wild-type Synechocystis and in two of the engineered derivatives. Cells were grown in plain BG-11 medium under white-light illumination (30 μE/m2/s). Filled symbols represent the OD730; open symbols represent the lactate concentration. The wild type is represented by circles; SAA015, the strain with integrated B. subtilis ldh, is represented by squares, and SAA017, the strain with integrated B. subtilis ldh and P. aeruginosa sth, is represented by triangles. Values are the averages for biological replicates; error bars indicate the standard deviations (n = 3); if an error bar is not visible, it is smaller than the respective data point symbol.
DISCUSSION
With respect to the previous study of lactic acid production from CO2 and water, using a cyanobacterium as the catalyst and solar energy as the free energy source, higher final product levels are reported in this communication, in spite of the fact that we did not integrate a lactic acid transporter into the engineered cyanobacterium. Whereas Niederholtmeyer et al. (28) report levels of about 600 μM lactic acid maximally, the final concentrations reported here are more than 5-fold higher. The amount of extracellular lactic acid that is formed in SAA015 is comparable to the maximal amounts obtained by Niederholtmeyer et al. We consider this coincidental: there are many factors that may provide an explanation, like the intracellular NADH or pyruvate concentration (which may be different in Synechococcus and Synechocystis), enzyme expression levels, etc.
Several considerations have provided arguments to give the integration of a lactic acid transporter a lower priority. (i) Weak acids generally (and even amino acids [9]) have high membrane permeability, as is well known from the field of chemiosmotic bioenergetics. Furthermore, as we work at relatively alkaline pHs (i.e., between 8 and 10), the lactate gradient presumably will be such that the lactate concentration is high in the extracellular medium. Experiments aimed to measure intracellular lactate levels did not reveal a large cytoplasmic accumulation either (S. A. Angermayr, unpublished). (ii) From our studies of the heterologous expression of an acetoin reductase in Synechocystis, we know that millimolar concentrations of both acetoin and butanediol equilibrate within minutes across the cyanobacterial cell membrane (O. Borirak, unpublished). (iii) The dynamics of lactic acid production in a circadian illumination regime (Fig. 2B) are also consistent with the notion that lactic acid permeability is relatively high. These dynamics also suggest that the fermentative production of lactic acid in the dark period (compare with reference 37) does not significantly contribute to the overall lactic acid production (a contribution which might amount to a several-millimolar concentration, based on of the glycogen content of the organism).
Two previous studies did introduce a transporter for the photofermentation product into the cyanobacterium (10, 28), which, particularly for sucrose production, resulted in significant improvement. However, it should be kept in mind that bacterial transporters generally function in substrate uptake into the cytoplasm. We therefore think that considering the details of energy coupling in these transporters (38) may help in further optimization of a cyanobacterial cell factory, although this may be complicated for lactic acid, considering the uncertainty on the proton/lactate symport stoichiometry of the latter.
Quantitative comparison of the results obtained in this study with those for the two lactate dehydrogenases that were successfully introduced into Synechocystis (i.e., from E. coli and B. subtilis) leads to some interesting conclusions. The difference observed between the two dehydrogenases, most notably the lack of lactate production of strain SAA013 (see also Fig. 2), is presumably due to their different affinities (as expressed in the Km value) for pyruvate, which are ∼5 mM (exact value ranges from 4.4 to 7.2 mM, dependent on the cytoplasmic pH [41]) and 0.8 mM, respectively. Additionally, the slight affinity of the B. subtilis LDH enzyme for NADPH may also have contributed, but the relevant Km values (13 and 288 μM for NADH and NADPH, respectively [32]) suggest only a small effect. The E. coli enzyme is not known to be able to use NADPH as a source of reducing equivalents.
Assuming that the introduction of the soluble transhydrogenase saturated the lactate dehydrogenase with NADH, the comparison between the in vitro activity of the enzyme in SAA017 (0.24 mmol/g DW/h) (Table 2) and the in vivo rate of lactic acid production (0.0178 mmol/g DW/h) allows one to estimate that the intracellular pyruvate concentration must be approximately 50 μM. This agrees reasonably well with results from metabolomics studies on the wild-type organism (40). This pyruvate concentration, together with the Km of the E. coli lactate dehydrogenase for pyruvate, also rationalizes why, upon heterologous expression of the latter enzyme, no significant amount of lactic acid was produced. These considerations also suggest that modifications in central carbon metabolic pathways (i.e., leading to increased pyruvate levels) in the cell might lead to further optimization, approaching a lactate-producing cell factory.
To put the maximal turnover capacity of the LDHs in the cell extract into a quantitative perspective, it is relevant to realize that for Synechocystis to grow at μmax, i.e., with an 8-h doubling time, a minimal flux of carbon through the Calvin-Benson cycle of 3.70 mmol carbon/g DW/h is necessary (based on an estimated cellular composition of the biomass of C4H7O2N [29, 35]). Hence, the measured in vitro enzyme activity of the lactate dehydrogenases demonstrates that in order to achieve the goal of a true “cell factory” for lactic acid (a cell that is able to channel the large majority of the fixed carbon into lactic acid), at least 10-fold-higher LDH activity is required than was achieved in this study. This goal can be pursued via the use of, e.g., stronger promoters, multiple promoters, multiple structural genes, or enzymes with improved kcat. Regarding the latter aspect particularly, the lactate dehydrogenase from Homarus americanus stands out among numerous other lactate dehydrogenases characterized to date (34) and thus deserves further testing.
At high rates of production of lactic acid, it will also be of interest to test whether introduction of such an introduced sink for the fixed carbon in the cells will accelerate the rate at which cells fix CO2, as observed for the transgenic strains that produces sucrose (10) and ethanol (S. A. Angermayr et al., unpublished). This observations open exciting prospects for fundamental research (i.e., to resolve the molecular mechanism of the underlying dynamic feedback mechanisms) and for applied research (i.e., to optimize volumetric productivity in photobioreactors).
Impairment of growth by the introduction of a soluble transhydrogenase (Fig. 3) may be due to either decreasing NADPH or NAD+ concentrations. The rescued growth defect, the absence of reversions, and the stable high rate of lactic acid production in SAA017 may suggest that NAD+ shortage (Fig. 5) may have significantly contributed to the defects in SAA016. More-detailed conclusions on this point require information about the in vivo levels of the reduced and oxidized forms of the nicotinamide nucleotides in Synechocystis. Our aforementioned experiments with heterologously expressed acetoin reductases may prove important for this, because they may allow application of a “metabolic-indicator method” to provide this information (O. Borirak, P. E. Savakis, S. A. Angermayr, and K. J. Hellingwerf, unpublished).
Fig 5.

Working model of the proposed mechanism for the suppression of the genetic reversion occurring in SAA016, as opposed to SAA017. A hypothesized schematic overview of the nicotinamide-adenine dinucleotide metabolism in Synechocystis is given. (A) The wild-type organism, equilibrating its redox balance. (B) SAA016, the strain with integrated P. aeruginosa sth, showing growth reduction caused by STH activity. (C) SAA017, the strain with the additionally integrated B. subtilis ldh gene in which the genetic reversions are prevented, presumably due to the relaxation of the redox imbalance by the added NADH-consuming LDH. CO2 fixation occurs in the Calvin-Benson cycle; LDPS, light-dependent photosynthetic electron transport; E, native energy-coupled transhydrogenase activity; STH, soluble transhydrogenase activity; LDH, lactate dehydrogenase activity.
Calculations show that the decrease of the growth rate of SAA017, compared to that of the wild type, cannot be fully explained by the repartitioning of carbon (the growth rate is decreasing more than the 3% of the fixed carbon that accumulates in the fermentation product in SAA017; see Results for further details). This could be due to the following: (i) a shortage of NADPH for the Calvin-Benson cycle, (ii) a shortage of NAD+ for prominent metabolic pathways, such as the tricarboxylic acid (TCA) cycle, (iii) a remaining imbalance of redox pools of the cell, or (iv) any combination of these three factors (compare results shown in Fig. 5).
The ideal cell factory for lactic acid should have an LDH activity that is as high as the maximal capacity of the upstream metabolism (i.e., the Calvin-Benson cycle and part of glycolysis). The results of this study have clearly revealed that we have not (yet) reached this state. Recognizing this, and considering the results in reference 10, this raises high expectations on the possibility of engineering a true cell factory for lactic acid. A better-performing lactate dehydrogenase (34) may be important in this engineering. Such experiments will provide ample opportunity to find out how rate control over the flux of carbon is distributed in an organism that carries out oxygenic photosynthesis, both in wild-type organisms and in engineered cell factories. This approach will require synergistic application of systems and synthetic biology and will be very rewarding, both in basic scientific understanding and in societal application.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to our colleagues at the Molecular Microbial Physiology group for useful discussions and critical reading of the manuscript. We thank Hans P. C. Matthijs for advice on cyanobacterial growth. We thank Karl D. Brune for help with cloning and the introduction of the BioBrick system to our laboratory. We thank Anna Osnato for help with the lactate toxicity assays. We thank W. Vermaas (Arizona State University) for pPSBA2KS, the source of the kanamycin cassette. We are grateful to D. Bhaya (Stanford University, California) for the glucose-tolerant wild-type strain Synechocystis sp. PCC6803.
Footnotes
Published ahead of print 3 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Abdel-Rahman MA, Tashiro Y, Sonomoto K. 2010. Lactic acid production from lignocellulose-derived sugars using lactic acid bacteria: overview and limits. J. Biotechnol. 156: 286– 301 [DOI] [PubMed] [Google Scholar]
- 2. Angermayr SA, Hellingwerf KJ, Lindblad P, de Mattos MJT. 2009. Energy biotechnology with cyanobacteria. Curr. Opin. Biotechnol. 20: 257– 263 [DOI] [PubMed] [Google Scholar]
- 3. Atsumi S, Higashide W, Liao JC. 2009. Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat. Biotechnol. 27: 1177– 1180 [DOI] [PubMed] [Google Scholar]
- 4. Bentley FK, Melis A. 2012. Diffusion-based process for carbon dioxide uptake and isoprene emission in gaseous/aqueous two-phase photobioreactors by photosynthetic microorganisms. Biotechnol. Bioeng. 109: 100– 109 [DOI] [PubMed] [Google Scholar]
- 5. Brosius J, Erfle M, Storella J. 1985. Spacing of the −10 and −35 regions in the tac promoter. Effect on its in vivo activity. J. Biol. Chem. 260: 3539– 3541 [PubMed] [Google Scholar]
- 6. de Boer JP, et al. 1993. pH and glucose profiles in aggregates of Bacillus laevolacticus. Appl. Environ. Microbiol. 59: 2474– 2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Graef MR, Alexeeva S, Snoep JL, Teixeira de Mattos MJ. 1999. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 181: 2351– 2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng M-D, Coleman JR. 1999. Ethanol synthesis by genetic engineering in cyanobacteria. Appl. Environ. Microbiol. 65: 523– 528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Driessen AJ, Kodde J, de Jong S, Konings WN. 1987. Neutral amino acid transport by membrane vesicles of Streptococcus cremoris is subject to regulation by internal pH. J. Bacteriol. 169: 2748– 2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ducat DC, Avelar-Rivas JA, Way JC, Silver PA. 2012. Rerouting carbon flux to enhance photosynthetic productivity. Appl. Environ. Microbiol. 78: 2660– 2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fiedler T, et al. 2011. Characterization of three lactic acid bacteria and their isogenic ldh deletion mutants shows optimization for YATP (cell mass produced per mole of ATP) at their physiological pHs. Appl. Environ. Microbiol. 77: 612– 617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. French CE, Boonstra B, Bufton KA, Bruce NC. 1997. Cloning, sequence, and properties of the soluble pyridine nucleotide transhydrogenase of Pseudomonas fluorescens. J. Bacteriol. 179: 2761– 2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garvie EI. 1980. Bacterial lactate dehydrogenases. Microbiol. Rev. 44: 106– 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaspar P, et al. 2007. The lactate dehydrogenases encoded by the ldh and ldhB genes in Lactococcus lactis exhibit distinct regulation and catalytic properties—comparative modeling to probe the molecular basis. FEBS J. 274: 5924– 5936 [DOI] [PubMed] [Google Scholar]
- 15. Griese M, Lange C, Soppa J. 2011. Ploidy in cyanobacteria. FEMS Microbiol. Lett. 323: 124– 131 [DOI] [PubMed] [Google Scholar]
- 16. Hellingwerf KJ, Teixeira de Mattos MJ. 2009. Alternative routes to biofuels: light-driven biofuel formation from CO2 and water based on the “photanol” approach. J. Biotechnol. 142: 87– 90 [DOI] [PubMed] [Google Scholar]
- 17. Huang H-H, Camsund D, Lindblad P, Heidorn T. 2010. Design and characterization of molecular tools for a Synthetic Biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res. 38: 2577– 2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaneko T, Tabata S. 1997. Complete genome structure of the unicellular cyanobacterium Synechocystis sp. PCC6803. Plant Cell Physiol. 38: 1171– 1176 [DOI] [PubMed] [Google Scholar]
- 19. Knight T. 2003. Idempotent vector design for standard assembly of biobricks. Massachusetts Institute of Technology, Cambridge, MA [Google Scholar]
- 20. Kufryk GI, Sachet M, Schmetterer G, Vermaas WFJ. 2002. Transformation of the cyanobacterium Synechocystis sp. PCC 6803 as a tool for genetic mapping: optimization of efficiency. FEMS Microbiol. Lett. 206: 215– 219 [DOI] [PubMed] [Google Scholar]
- 21. Kunert A, Hagemann M, Erdmann N. 2000. Construction of promoter probe vectors for Synechocystis sp. PCC 6803 using the light-emitting reporter systems Gfp and LuxAB. J. Microbiol. Methods 41: 185– 194 [DOI] [PubMed] [Google Scholar]
- 22. Labarre J, Chauvat F, Thuriaux P. 1989. Insertional mutagenesis by random cloning of antibiotic resistance genes into the genome of the cyanobacterium Synechocystis strain PCC 6803. J. Bacteriol. 171: 3449– 3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lagarde D, Beuf L, Vermaas W. 2000. Increased production of zeaxanthin and other pigments by application of genetic engineering techniques to Synechocystis sp. strain PCC 6803. Appl. Environ. Microbiol. 66: 64– 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan EI, Liao JC. 2012. ATP drives direct photosynthetic production of 1-butanol in cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 109: 6018– 6023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindberg P, Park S, Melis A. 2010. Engineering a platform for photosynthetic isoprene production in cyanobacteria, using Synechocystis as the model organism. Metab. Eng. 12: 70– 79 [DOI] [PubMed] [Google Scholar]
- 26. Mulkidjanian AY, et al. 2006. The cyanobacterial genome core and the origin of photosynthesis. Proc. Natl. Acad. Sci. U. S. A. 103: 13126– 13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagarajan A, Winter R, Eaton-Rye J, Burnap R. 2011. A synthetic DNA and fusion PCR approach to the ectopic expression of high levels of the D1 protein of photosystem II in Synechocystis sp. PCC 6803. J. Photochem. Photobiol. B 104: 212– 219 [DOI] [PubMed] [Google Scholar]
- 28. Niederholtmeyer H, Wolfstädter BT, Savage DF, Silver PA, Way JC. 2010. Engineering cyanobacteria to synthesize and export hydrophilic products. Appl. Environ. Microbiol. 76: 3462– 3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nogales J, Gudmundsson S, Knight EM, Palsson BO, Thiele I. 30 January 2012. Detailing the optimality of photosynthesis in cyanobacteria through systems biology analysis. Proc. Natl. Acad. Sci. U. S. A. doi:10.1073/pnas.1117907109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pakrasi HB, Williams JG, Arntzen CJ. 1988. Targeted mutagenesis of the psbE and psbF genes blocks photosynthetic electron transport: evidence for a functional role of cytochrome b559 in photosystem II. EMBO J. 7: 325– 332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. 1979. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111: 1– 61 [Google Scholar]
- 32. Romero S, Merino E, Bolívar F, Gosset G, Martinez A. 2007. Metabolic engineering of Bacillus subtilis for ethanol production: lactate dehydrogenase plays a key role in fermentative metabolism. Appl. Environ. Microbiol. 73: 5190– 5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 34. Scheer M, et al. 2011. BRENDA, the enzyme information system in 2011. Nucleic Acids Res. 39: D670– D676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shastri AA, Morgan JA. 2005. Flux balance analysis of photoautotrophic metabolism. Biotechnol. Prog. 21: 1617– 1626 [DOI] [PubMed] [Google Scholar]
- 36. Shetty RP, Endy D, Knight TF. 2008. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stal LJ, Moezelaar R. 1997. Fermentation in cyanobacteria. FEMS Microbiol. Rev. 21: 179– 211 [Google Scholar]
- 38. Sun J, Wu J, Carrasco N, Kaback HR. 1996. Identification of the epitope for monoclonal antibody 4B1 which uncouples lactose and proton translocation in the lactose permease of Escherichia coli. Biochemistry 35: 990– 998 [DOI] [PubMed] [Google Scholar]
- 39. Takahama K, Matsuoka M, Nagahama K, Ogawa T. 2003. Construction and analysis of a recombinant cyanobacterium expressing a chromosomally inserted gene for an ethylene-forming enzyme at the psbAI locus. J. Biosci. Bioeng. 95: 302– 305 [DOI] [PubMed] [Google Scholar]
- 40. Takahashi H, Uchimiya H, Hihara Y. 2008. Difference in metabolite levels between photoautotrophic and photomixotrophic cultures of Synechocystis sp. PCC 6803 examined by capillary electrophoresis electrospray ionization mass spectrometry. J. Exp. Bot. 59: 3009– 3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tarmy EM, Kaplan NO. 1968. Kinetics of Escherichia coli B d-lactate dehydrogenase and evidence for pyruvate-controlled change in conformation. J. Biol. Chem.. 243: 2587– 2596 [PubMed] [Google Scholar]
- 42. Vermaas W. 1996. Molecular genetics of the cyanobacterium Synechocystis sp. PCC 6803: principles and possible biotechnology applications. J. Appl. Phycol. 8: 263– 273 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.