

Abstract
Stress-induced hypertrophic growth of the heart predisposes the heart to arrhythmia, contractile dysfunction, and clinical heart failure. FHL2 (four-and-a-half LIM domain protein 2) is expressed predominantly in the heart, and inactivation of the gene coding for FHL2 leads to exaggerated responsiveness to adrenergic stress. Activation of calcineurin occurs downstream of β-adrenergic signaling and is required for isoproterenol-induced myocardial hypertrophy. Based on these facts, we hypothesized that FHL2 suppresses stress-induced activation of calcineurin. FHL2 is upregulated in mouse hearts exposed to isoproterenol, a β-adrenergic agonist, and isoproterenol-induced increases in the NFAT target genes RCAN1.4 and BNP were amplified significantly in FHL2 knockout (FHL2−/−) mice compared with levels in wild-type (WT) mice. To determine whether the effect of FHL2 on NFAT target gene transcript levels occurred at the level of transcription, HEK 293 cells and neonatal rat ventricular myocytes (NRVMs) were transfected with a luciferase reporter construct harboring the NFAT-dependent promoters of either RCAN1 or interleukin 2 (IL-2). Consistent with the in vivo data, small interfering RNA (siRNA) knockdown of FHL2 led to increased activation of these promoters by constitutively active calcineurin or the calcium ionophore ionomycin. Importantly, activation of the RCAN1 promoter by ionomycin, in control and FHL2 knockdown cells, was abolished by the calcineurin inhibitor cyclosporine, confirming the calcineurin dependence of the response. Overexpression of FHL2 inhibited activation of both NFAT reporter constructs. Furthermore, NRVMs overexpressing FHL2 exhibited reduced hypertrophic growth in response to constitutively active calcineurin, as measured by cell cross-sectional area and fetal gene expression. Finally, immunostaining in isolated adult cardiomyocytes revealed colocalization of FHL2 and calcineurin predominantly at the sarcomere and activation of calcineurin by endothelin-1-facilitated interaction between FHL2 and calcineurin. FHL2 is an endogenous, agonist-dependent suppressor of calcineurin.
INTRODUCTION
Epidemiological evidence links left ventricular (LV) hypertrophy with adverse cardiovascular events, including heart failure and death (1, 13, 35, 36). Consistent with this, therapies that improve clinical outcomes are often associated with regression of ventricular hypertrophy (11, 19, 46). However, whereas significant strides have been made in elucidating the molecular circuitry governing pathological cardiac remodeling (23), few therapies in clinical use target cell growth mechanisms directly.
Hypertrophic growth of the heart in response to a variety of pathological stresses is an initially adaptive response that, left unchecked, often progresses to heart failure (25). In many instances, the intracellular protein phosphatase calcineurin is a major mediator of stress-induced cardiac hypertrophy. Upon activation, calcineurin dephosphorylates NFAT (nuclear factor of activated T cells), which, in turn, translocates into the nucleus and activates expression from target promoters. Transgenic mice overexpressing calcineurin (2, 37, 43) or NFAT (37) develop substantial ventricular hypertrophy, followed by rapid progression to ventricular dilation, systolic dysfunction, and heart failure. Inhibition of calcineurin genetically or pharmacologically is sufficient to block hypertrophic growth in response to pressure overload or neurohormonal stimulation as well as in transgenic models of hypertrophy (reviewed in reference 64).
FHL2 (four-and-a-half LIM domain family protein 2), a LIM-only protein, was first identified from a subtractive cDNA hybridization screen of normal myoblasts and rhabdomyosarcoma cells. Subsequent expression analyses of human and mouse tissues, however, demonstrated that FHL2 is expressed primarily in the heart (18). FHL2 is expressed early in cardiogenesis and remains at high levels throughout adulthood. Its function in the heart is unknown.
LIM domains have been implicated in protein-protein interactions, and over 50 FHL2 binding partners have been identified (reviewed in reference 27). FHL2 is involved in many processes, including cell cycle regulation (31, 41), apoptosis (55, 60), differentiation (21, 32, 40, 63), extracellular matrix assembly (48), bone formation (20), and wound healing (28, 65). Although the highest expression of FHL2 occurs in the heart, knockout mice are viable and display no overt cardiac phenotype under basal conditions (5, 30). However, when treated with the β-adrenergic agonist isoproterenol, FHL2 knockout mice develop an exaggerated hypertrophic phenotype (30).
These facts led us to hypothesize that FHL2 can act as a governor of calcineurin, suppressing its activation by growth stimuli. Here, we present studies designed to test this hypothesis and define underlying mechanisms.
MATERIALS AND METHODS
Chronic isoproterenol infusion.
FHL2−/− mice were engineered and characterized previously (30). Briefly, a mutant FHL2 allele in which protein coding sequences from exon 2 of the native gene, including the translation initiation codon ATG, the N-terminal half LIM motif, part of the LIM1 domain, and 0.5 kb of intronic sequence, were replaced with a lacZ reporter gene was engineered (30). These animals, along with male wild-type (WT) controls, were subjected to isoproterenol infusion. Briefly, 7-day Alzet minipumps containing 200 μl of isoproterenol (32 mg/kg of body weight/day) or saline (control) were inserted subcutaneously into 8-week-old wild-type and FHL2−/− mice. Hearts were harvested at day 7 and subjected to necropsy and molecular analyses.
Cell culture and transfections.
HEK 293 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were transfected at 60 to 70% confluence with Lipofectamine 2000 reagent (Invitrogen). Cells were harvested after 24 h and lysed, and a luciferase assay was performed according to the manufacturer's protocol (Promega). Normalization for transfection efficiency was accomplished by cotransfecting cells with pCMV-LacZ or pRL-CVM (Promega). Small interfering RNAs (siRNAs) were transfected at 30% confluence, the reporter plasmid was transfected at 24 h, and cells were harvested and analyzed at 48 h. The negative-control and FHL2 siRNA was obtained from Ambion.
siRNAs used for knockdown experiments were as follows: negative control number 1, catalog number 4390844; FHL2-1, sense (5′-CAACGACUGCUUUAACUGUtt-3′ [lowercase indicates overhanging nucleotides]) and antisense (5′-ACAGUUAAAGCAGUCGUUGtg-3′); FHL2-2, sense (5′-GCAAGGACUUGUCCUACAAtt-3′) and antisense (5′-UUGUAGGACAAGUCCUUGCtt-3′).
Infection of neonatal rat ventricular myocytes (NRVMs).
Cardiomyocytes were isolated from the ventricles of 1- to 2-day-old Sprague-Dawley rat pups and plated as described previously (59) at a density of 1,250 cells/mm2 in medium containing 10% fetal calf serum (FCS). Cells were infected with either AdFHL2 (Vector Biolabs) or AdCMVβgal in serum-free Opti-MEM (Gibco) medium 48 h after plating. Cells were coinfected with either constitutively active calcineurin (AdCMVCnA*iresGFP) or green fluorescent protein (GFP) control. Experiments were performed at a multiplicity of infection (MOI) of 30 (≥90% infection efficiency visualized by GFP expression), and cells were fixed for immunostaining or harvested for RNA isolation 48 h after infection.
Immunostaining.
Cultured cardiomyocytes were washed with phosphate-buffered saline (PBS) 3 times, fixed with 4% paraformaldehyde (PFA) for 10 min, and permeabilized in 0.05% Triton-X for 15 min. Cells were blocked in 3% normal goat serum and 1% bovine serum albumin prepared in PBS for 30 min. α-Actinin antibody (Sigma) was diluted 1/100 in blocking solution and incubated for 1 h at room temperature, followed by 1 h of exposure to Cy-5-conjugated secondary antibody. Photographs were acquired on a Leica DM2000 microscope. A cross-sectional area was measured using ImageJ software.
The subcellular localizations of FHL2 and calcineurin were analyzed in adult heart sections. Heart tissue was fixed by sequential perfusion with 30 ml of heparinized PBS and 30 ml of 4% paraformaldehyde (PFA), followed by overnight incubation in 4% PFA at 4°C with agitation before routine paraffin processing. Immunostaining was performed as described previously (61). The following antibodies were used for immunostaining: anticalcineurin (Stressgen and Sigma) and anti-FHL2 (MBL International).
Immunoprecipitation.
Following 48 h of culture in 1% FBS medium, NRVMs were treated with 100 nm endothelin-1 (ET-1) (1 h). Cells were washed 3 times with PBS and harvested in NP-40 lysis buffer (50 mM Tris-HCl,150 mM NaCl, 1% NP-40) supplemented with Complete protease inhibitor (Roche). Cell debris was pelleted down, and the supernatant was used for immunoprecipitation. Calcineurin antibody (Stressgen; 1/100) was harvested using TrueBlot anti-rabbit antibody beads (eBioscience). Western blot analysis was performed using calcineurin (1/1,000) and FHL2 (MBL; 1/1,000) antibodies and TrueBlot secondary antibodies (1/2,000).
RNA purification and real-time reverse transcription-PCR (RT-PCR).
RNA isolation from tissues and cells was performed using TRIzol according to the manufacturer's protocol (Invitrogen), and RNA concentrations were measured (NanoDrop 1000; Thermo Scientific). Two micrograms of RNA from each sample was used to make cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCR was performed using SYBR green mix (Applied Biosystems) on an ABI-PE Prism 7000 sequence detection system with triplicates for each sample. Primers used are listed in Table 1. The relative quantities of mRNA were determined and normalized to levels of cyclophilin A or 18S RNA.
Table 1.
RT-PCR primers
| Target | Forward primer | Reverse primer |
|---|---|---|
| FHL2 | TCACAGCACGGGATGAGTTTC | GTGCCACCCAGACCACTAATG |
| RCAN1.4 | CCCGTGAAAAAGCAGAATGC | TCCTTGTCATATGTTCTGAAGAGGG |
| RCAN1.1 | GACCCGCGCGTGTTC | TGTCATATGTTCTGAAGAGGGATTC |
| BNP | CACCGCTGGGAGGTCACT | GTGAGGCCTTGGTCCTTCAA |
| ANP | CTTCTTCCTCTTCCTGGCCT | TTCATCGGTCTGCTCGCTCA |
| β-MHC | CTGACTGAACAGCTGGGCTC | AACTCTGGAGGCTCTTCACT |
| Cyclophilin A | CAGACGCCACTGTCGCTTT | TGTCTTTGGAACTTTGTCTGCAA |
| 18S | AAACGGCTACCACATCCAAG | CCTCCAATGGATCCTCGTTA |
Statistical methods and data handling.
All values are presented as means and standard errors of the mean (SEM). Statistical significance was analyzed using Student's unpaired, 2-tailed t test (comparison of 2 groups).
RESULTS
Increased FHL2 expression in hearts exposed to isoproterenol.
Inactivation of the FHL2 gene leads to an exaggerated hypertrophic response to β-adrenergic stimulation, suggesting that FHL2 is involved in modifying this response (30). First, we analyzed whether the expression of FHL2 is regulated during isoproterenol-induced hypertrophy. Eight-week-old wild-type mice were treated with isoproterenol (32 mg/kg/day) or saline (diluent control) by minipump infusion. After the 7th day of treatment, left ventricular (LV) tissue was collected for both protein and RNA isolation. Western blot analysis using an antibody specific to FHL2 revealed a nearly 2-fold increase over the saline control in the abundance of FHL2 protein after isoproterenol treatment (Fig. 1A and B). Real-time RT-PCR analysis of FHL2 mRNA levels showed a similar increase, with the isoproterenol-treated group displaying consistently higher levels of FHL2 transcripts (Fig. 1C). These data, then, reveal that FHL2 is upregulated during isoproterenol-induced hypertrophy and suggest that this upregulation plays a role in modifying the hypertrophic response.
Fig 1.
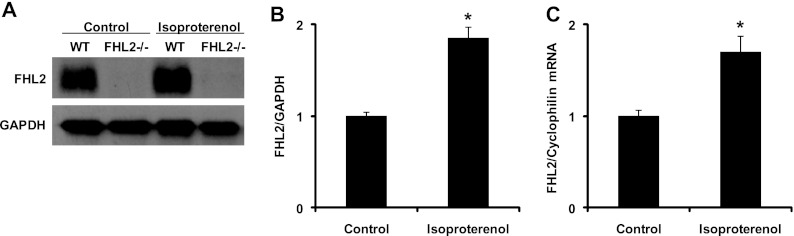
Increased expression of FHL2 in isoproterenol-treated hearts. (A) Western blots for FHL2 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (control) in samples prepared from control and isoproterenol-treated hearts. (B) Quantitative analysis of FHL2 protein levels by Western blotting. n = 3. *, P < 0.05. (C) FHL2 transcript levels in control and isoproterenol-treated hearts were measured by real-time RT-PCR and normalized to cyclophilin mRNA levels. n = 3. *, P < 0.05.
Increased expression of isoproterenol-activated NFAT target genes in FHL2−/− mice.
The calcineurin/NFAT pathway is a central regulator of hypertrophy, and inhibition of this signaling cascade blunts hypertrophic growth in cardiac myocytes. We next examined whether calcineurin signaling was altered in the setting of FHL2 gene silencing. Eight-week-old WT and FHL2−/− mice were treated with saline infusion (control) or isoproterenol for 7 days. The extent of the hypertrophic response was documented by ratios of heart weight to body weight (HW/BW). The expression of NFAT-responsive genes was analyzed by real-time RT-PCR from RNA isolated from heart tissue.
In saline-treated controls, HW/BW ratios were similar in WT and FHL2−/− mice, confirming that FHL2−/− mice have no overt hypertrophic phenotype under basal conditions. Isoproterenol infusion induced robust cardiac hypertrophy in both WT and FHL2−/− mice. In agreement with earlier studies (30), FHL2−/− mice manifested amplified hypertrophic growth compared to that of the WT (P < 0.05), with increases in HW/BW ratios of 39% and 27%, respectively (Fig. 2A). Body mass (BW) was unchanged in the 4 treatment groups, with the following values for each group: WT + vehicle (Veh), 25.0 ± 0.2 g, n = 3; FHL2−/− + Veh, 25.8 ± 1.9 g, n = 3; WT + isoproterenol (ISO), 25.6 ± 0.8 g, n = 3; and FHL2−/− KO + ISO, 25.2 ± 0.1 g, n = 3. All P values were not significant (NS).
Fig 2.

Increased isoproterenol-induced expression of NFAT target genes in FHL2 knockout mice. WT and FHL2−/− mice were treated with isoproterenol or vehicle for 7 days, and hearts were collected at the end of day 7. (A) Heart-weight-to-body-weight ratios of WT and FHL2−/− mice after 7 days of isoproterenol stimulation. n = 3 in each group. *, P value of <0.01 versus the WT; #, P value of <0.05 versus WT mice treated with isoproterenol. (B, C, and D) mRNA was isolated from hearts, and expression of RCAN1.4 (B), BNP (C), and RCAN1.1 (D) was measured using real-time RT-PCR. *, P value of <0.05 versus the WT; #, P value of <0.05 versus WT mice treated with isoproterenol.
To determine whether this exaggerated hypertrophic growth response derived from increased calcineurin/NFAT signaling, we measured transcript levels of two NFAT target genes, RCAN1.4 and BNP. Real-time RT-PCR revealed that in the absence of isoproterenol treatment, FHL2−/− mice manifested no change relative to the WT in the mRNA levels of RCAN1.4 or BNP. In contrast, isoproterenol treatment resulted in a significant increase (P < 0.05) in both RCAN1.4 and BNP expression in FHL2−/− mice compared to that of the WT (Fig. 2B and C). In contrast, expression of the ubiquitous RCAN isoform RCAN1.1 was not significantly altered in the isoproterenol treatment group compared to that in the control in either genotype (Fig. 2D). Together, these data are consistent with a model in which the increased cardiac hypertrophy in mice lacking FHL2 derives from the release of FHL2-dependent repression of calcineurin/NFAT signaling.
Knockdown of FHL2 protein enhances calcineurin-dependent NFAT promoter activation.
Our in vivo data suggested that loss of the FHL2 protein results in increased stress-triggered calcineurin/NFAT signaling. To test this directly, we employed a luciferase reporter construct driven by the RCAN1 promoter, which harbors an NFAT binding site and is activated by calcineurin. Promoter activity was measured as a ratio of luciferase activity to that of an internal control (beta-galactosidase [β-Gal]), with the control extract set to 1. NRVMs were infected with an adenoviral vector expressing constitutively active calcineurin (lacking the C-terminal autoinhibitory domain) to activate the NFAT pathway. FHL2 steady-state protein levels were suppressed using two sequence-independent siRNA constructs (Fig. 3A). Cells exposed to a control RNA interference (RNAi) construct manifested an ∼3-fold increase in calcineurin-stimulated RCAN1 reporter activity (Fig. 3B). In contrast, cells harboring either FHL2-targeting RNAi construct displayed an amplified response to calcineurin which was nearly doubled relative to that of the control RNAi (Fig. 3B). To confirm the inhibitory role of FHL2 on calcineurin/NFAT signaling, NRVMs were infected with constitutively active calcineurin with or without FHL2. Calcineurin induced robust activation of the RCAN1 promoter which was suppressed by FHL2 (Fig. 3C, black bars).
Fig 3.

FHL2 knockdown augments calcineurin activation. (A) Neonatal cardiomyocytes transfected with two sequence-independent FHL2 siRNA constructs manifest diminished levels of FHL2. (B) Luciferase expression driven by constitutively active calcineurin acting on the RCAN1 promoter is amplified in FHL2 siRNA-transfected cells compared with that in control siRNA-transfected cells. *, P value of <0.01 versus siControl plus GFP; #, P value of <0.05 versus siControl plus calcineurin; OD, optical density. (C) FHL2 inhibits calcineurin-driven transcription of the RCAN1 promoter. This inhibition was not modified by the addition of UO126 (5 μM). *, P value of <0.01 versus the control; #, P value of <0.05 versus calcineurin. (D) Western blot representing the phosphorylation status of ERK1/2 in cells treated with UO126. (E) Increased activation of the RCAN1 promoter by ionomycin in FHL2 siRNA-transfected cardiomyocytes was abolished by CsA. *, P value of <0.01 versus siControl; #, P value of <0.01 versus siControl plus ionomycin.
The calcineurin/NFAT and MEK/ERK signaling pathways, each a positive regulator of cardiac hypertrophy, are interdependent, such that inhibition of one pathway blunts the hypertrophic response triggered by the other (54). It has been demonstrated that FHL2 suppresses MEK/ERK signaling in cardiomyocytes (49), so we questioned whether the effect of FHL2 on calcineurin/NFAT-dependent events occurred through inhibition of the MEK/ERK pathway. To accomplish this, we determined whether UO126, a specific MEK1/2 inhibitor, altered the inhibition of calcineurin mediated by FHL2. As expected, UO126 reduced ERK1/2 phosphorylation (Fig. 3D) and RCAN1 promoter activity (Fig. 3C, white bars). However, the repressive effects of FHL2 on RCAN1 promoter activity were similar in the presence or absence of UO126 (Fig. 3C), suggesting that FHL2 suppresses calcineurin signaling independently of ERK1/2.
We next set out to determine whether knockdown of FHL2 was similarly capable of enhancing the effect of endogenous calcineurin on NFAT promoter activation. To do this, we activated endogenous calcineurin by treating the cells with the calcium ionophore ionomycin. FHL2 levels were suppressed by siRNA treatment for 48 h, followed by exposure of the cells to ionomycin for 4 h. Ionomycin activated the RCAN1 promoter ∼4-fold in control cells, whereas FHL2 knockdown resulted in a significant increase (P < 0.05) in the responsiveness of the RCAN1 promoter to ionomycin stimulation (Fig. 3E). Ionomycin-dependent activation of the RCAN1 promoter in FHL2-knockdown cells was abolished by cyclosporine (CsA) (Fig. 3E), confirming calcineurin dependence. Together, these data show that suppressing FHL2 increases cellular responsiveness to calcineurin/NFAT signaling.
FHL2 modulates calcineurin-dependent activation of NFAT target genes.
Our in vivo and in vitro data suggested that steady-state levels of FHL2 protein play a role in modulating calcineurin/NFAT signaling to blunt the response to hypertrophic stimuli. To determine whether these effects are direct, as opposed to secondary to the hypertrophic growth response, we studied HEK 293 cells in which FHL2 levels were modulated, first by siRNA knockdown and then by overexpression using a cytomegalovirus (CMV)-driven FHL2 expression construct. Promoter activity was measured as a ratio of luciferase activity to that of an internal control (Renilla luciferase). FHL2 levels were decreased by siRNA treatment (48 h) (Fig. 4A) or increased by transfection with the FHL2 construct (24 h), followed by treatment of the cells with ionomycin (4 h). Ionomycin treatment activated the RCAN1 promoter by 40% (±0.07%; n = 6) in control cells (Fig. 4B). Similar to our findings in NRVMs, decreasing FHL2 levels in HEK 293 cells resulted in a dramatic increase in the responsiveness of the RCAN1 promoter to stimulation by ionomycin (P < 0.05) (Fig. 4B). This activity was dose responsive in that titration of the FHL2 expression plasmid resulted in increasing levels of FHL2 protein and decreased levels of ionomycin-induced RCAN1 promoter activity (Fig. 4C). Importantly, overexpression of FHL2 decreased the ability of constitutively active calcineurin to activate expression from the RCAN1 promoter, confirming that the inhibitory effect of FHL2 on NFAT promoter activation occurs downstream of calcineurin activation (Fig. 4D). The overexpression results were confirmed and extended using an interleukin 2 (IL-2) promoter construct (Fig. 4E). In aggregate, these data show that FHL2 governs cellular responsiveness to calcineurin/NFAT signaling.
Fig 4.

FHL2 overexpression inhibits ionomycin-triggered calcineurin activation. (A) Decreased FHL2 protein levels in HEK 293 cells transfected with siRNA targeting FHL2. (B) Increased activation of the RCAN1 promoter by ionomycin in FHL2 siRNA-transfected HEK 293 cells. *, P value of <0.05 versus siControl; #, P value of <0.05 versus siControl plus ionomycin. (C) Increasing levels of FHL2 progressively inhibits activation of the RCAN1 promoter by ionomycin in HEK 293 cells. (D) FHL2 inhibits calcineurin-driven transcription of the RCAN1 promoter. *, P value of <0.01 versus the control; #, P value of <0.05 versus ionomycin; ##, P value of <0.05 versus calcineurin. (E) FHL2 inhibits calcineurin-driven transcription of the IL-2 promoter. *, P value of <0.01 versus the control; ##, P value of <0.01 versus calcineurin.
FHL2 inhibits the calcineurin-dependent hypertrophic growth response.
It has been demonstrated that constitutively active calcineurin is sufficient to induce a hypertrophic response in neonatal (43) and adult (2) ventricular myocytes. Data presented here demonstrate that overexpression of FHL2 blunts activation of NFAT-dependent promoters, suggesting that FHL2 can inhibit calcineurin/NFAT-dependent gene expression and subsequent hypertrophic growth. To test this further, we expressed constitutively active calcineurin along with either FHL2 or control protein (β-Gal). Cells were infected, and hypertrophic growth was assessed at 48 h as cell cross-sectional area (CSA) and fetal gene expression. As anticipated, FHL2 significantly inhibited hypertrophic growth induced by calcineurin as determined by CSA (Fig. 5A and B). In addition, expression of the hypertrophic genes ANF and BNP, elevated in calcineurin-infected cells, was blunted by FHL2 overexpression (Fig. 5C and D). Together, these results support a model in which FHL2 blunts myocyte hypertrophy through the suppression of the calcineurin/NFAT signaling cascade.
Fig 5.

FHL2 inhibits hypertrophic growth induced by calcineurin in neonatal rat ventricular myocytes. (A) NRVMs infected with β-Gal (control) or FHL2 were coinfected with GFP or calcineurin. Forty-eight hours after infection, cells were fixed and immunostained with α-actinin antibody. Scale bar, 20 μm. (B) α-Actinin-positive cell cross-sectional areas. n = ∼180 cells in each group. *, P value of <0.01 versus β-Gal plus GFP; #, P value of <0.01 versus β-Gal plus calcineurin. (C and D) Expression of hypertrophic marker genes was analyzed by real-time RT-PCR. **, P value of <0.05 versus β-Gal plus GFP; ##, P value of <0.05 versus β-Gal plus calcineurin.
FHL2 interacts with activated calcineurin.
Results presented so far are consistent with a model in which FHL2 acts downstream of calcineurin activation to inhibit NFAT target gene expression. To examine whether this inhibition occurs via direct interaction with calcineurin, we first tested for colocalization of the two proteins. In recent years, it has been revealed that Z disks harbor a wide array of signaling molecules, including calcineurin (15), and FHL2 localizes at, or in close proximity to, Z disks (34). To test whether FHL2 and calcineurin colocalize to similar regions of the sarcomere, adult heart tissues were coimmunostained with FHL2 and calcineurin antibodies, demonstrating significant colocalization of the two proteins (Fig. 6A and D).
Fig 6.

FHL2 colocalizes with calcineurin. (A) Costaining of WT heart tissues with calcineurin and FHL2 antibodies. Scale bar, 5 μm. (B) FHL2 immunoprecipitates with constitutively active calcineurin in HEK 293 cells. (C) Immunoprecipitation of endogenous calcineurin from NRVMs, immunoblotted with FHL2 antibody. (D) Staining of WT and FHL2−/− heart tissues with calcineurin antibody. Scale bar, 5 μm.
LIM domains have been implicated in protein-protein interactions (10). Indeed, the LIM protein MLP interacts with calcineurin (23). To determine whether FHL2 can also interact with calcineurin, we expressed in HEK 293 cells either full-length, constitutively active calcineurin (calcineurin 398, lacking the autoinhibitory domain and part of the calmodulin binding region) or additionally truncated calcineurin (calcineurin 342, lacking the autoinhibitory domain and regions that bind calmodulin and calcineurin B) along with GFP-FHL2. In these experiments, FHL2 coprecipitated specifically with the constitutively active calcineurin mutant (Fig. 6B).
These data suggest that FHL2 inhibits NFAT gene activation through a direct interaction with activated calcineurin. To evaluate this further, we immunoprecipitated endogenous proteins from neonatal rat cardiomyocytes, comparing cells in which calcineurin was activated by exposure to endothelin-1 (ET-1; 1 h) to those not activated. In untreated cells, the calcineurin antibody failed to pull down detectable amounts of FHL2 (Fig. 6C). In contrast, however, under conditions where calcineurin is activated, calcineurin and FHL2 manifested a significant interaction (Fig. 6C). As a positive control, we pulled down FHL2 with an antibody to sphingosine kinase 1 (SK1), a molecule previously shown to bind FHL2 in a manner inhibited by endothelin-1 (60). Here, we observed a reciprocal interaction in which binding of FHL2 was repressed by ET-1 (Fig. 6C). In aggregate, these data suggest strongly that FHL2 represses NFAT signaling through interaction with activated calcineurin protein.
DISCUSSION
In recent years, calcineurin has emerged as a major mechanism of pathological cardiac remodeling, active in numerous forms of disease. As such, this molecule has generated much interest as a target of therapy. In this study, we elucidated the role of FHL2, a molecule implicated in the control of multiple signaling cascades, in regulating calcineurin. We demonstrate that expression of FHL2 increases in the setting of β-adrenergic signaling-induced calcineurin activation. FHL2, in turn, represses calcineurin signaling and consequent NFAT activation, myocyte growth, and fetal gene expression. We show that FHL2 colocalizes with calcineurin in native cardiomyocytes and interacts with the enzyme in both experimental and native conditions. We go on to provide evidence that FHL2 preferentially targets calcineurin in the active state. Together, these findings shed new light on a novel mechanism of calcineurin regulation, thereby raising yet further the prospects of targeting this molecule for therapeutic gain.
FHL2 expression is selectively increased with β-adrenergic drive.
FHL2 protein and mRNA are most abundant in the heart, with low but detectable levels found in other tissues (reviewed in reference 27). Cardiac expression of FHL2 initiates during embryogenesis and remains high throughout adulthood (30). Indeed, FHL2 expression is an early marker of the cardiomyocyte lineage and is selectively expressed in both the atrial and ventricular myocardium in the adult heart.
FHL2 levels are unchanged in adult hearts subjected to pressure overload-induced hypertrophy (5, 6). FHL2 expression, however, is increased in hearts after cardiopulmonary bypass (62), and its protein abundance is decreased in failing human hearts (3). Here, we demonstrate that FHL2 expression is increased in isoproterenol-induced hypertrophy at both the protein and mRNA levels. Together, these data point to complex regulation of FHL2 gene expression. For example, pressure overload entails a global set of growth cues that together activate diverse hypertrophic signaling pathways. In contrast, isoproterenol-induced β-adrenergic activation is a more focused response which is largely reliant on calcineurin/NFAT signaling (4, 12, 50). Consistent with this, mechanical stress activates α-adrenergic receptors (Ang II and ET-1) and β-adrenergic receptors (epinephrine and norepinephrine). That said, mice null for either α- or β-adrenergic (47) receptors still develop hypertrophy in response to thoracic aortic constriction (TAC), supporting involvement of a global response rather than dependence on a single pathway.
In contrast to the isoproterenol response, FHL2 knockout mice develop hypertrophy comparable to that of the WT in response to pressure overload (B. Hojayev and J. A. Hill, unpublished data). One possible explanation for this differential growth response to TAC and isoproterenol in FHL2−/− mice relates to increased expression of FHL2 in isoproterenol-treated hearts, a finding not seen in TAC. The effect of FHL2 on transcriptional regulation of gene expression is well established in vitro (14, 44, 45), where nuclear localization of the protein is sometimes observed. However, there are no reports of regulation of transcription in cardiomyocytes by FHL2, and data reported here lend credence to this notion, as we detect FHL2 exclusively at the sarcomere in adult myocytes. Indeed, nuclear localization of FHL2 was not detected in cardiomyocytes even in the setting of hypertrophic stimulation induced by TAC (Hojayev and Hill, unpublished data).
The tumor suppressor p53 is a regulator of FHL2 gene transcription (18). FHL2 was first isolated from rhabdomyosarcoma, which expresses mutant p53 (another name for the protein is DRAL [downregulated in rhabdomyosarcoma LIM protein]) (18). Overexpression of wild-type p53 in this cell line restored FHL2 expression (55). Similarly, gamma irradiation increases expression of FHL2 in a p53-dependent manner (55). FHL2 expression is turned on early in embryogenesis, and it remains expressed through adulthood (30), coinciding with the expression of Nkx2.5, a homeodomain transcription factor (29) and the earliest known marker of vertebrate heart development. In fact, the putative FHL2 promoter contains multiple potential binding sites for Nkx2.5 (27), and expression of Nkx2.5 is upregulated in adrenergic agonist-induced cardiac hypertrophy (52). At present, the roles of p53 and Nkx2.5 in β-adrenergic signaling-induced activation of FHL2 in cardiomyocytes are unclear.
FHL2 represses calcineurin.
Calcineurin is a Ca2+-dependent protein phosphatase that dephosphorylates NFAT family transcription factors, leading to their nuclear translocation and activation of target genes. Calcineurin is activated in a majority of models of pathological cardiac hypertrophy. Constitutively active calcineurin, lacking the autoinhibitory domain, is sufficient to induce hypertrophic growth in vitro and in vivo (2, 43). As a result, calcineurin signaling is a promising target for therapeutic intervention in heart disease (17).
In studies reported here, we find that calcineurin/NFAT signaling is activated in FHL2−/− mice. RCAN1.4 and BNP transcripts were analyzed as readouts, as they are direct targets of NFAT (8, 43, 51), and both are activated in hypertrophy (22, 26). We report that increases in RCAN1.4 and BNP levels are amplified in isoproterenol-treated FHL2−/− mice. Knockdown of FHL2 resulted in augmented activation of the RCAN1.4 promoter by calcineurin or ionomycin in a cyclosporine-repressible manner. Conversely, overexpression of FHL2 inhibited activation of the RCAN1.4 promoter and blunted hypertrophic growth in cultured cardiomyocytes induced by constitutively active calcineurin. In aggregate, these data support a model in which FHL2 inhibits activation of NFAT target genes by repressing calcineurin.
FHL1 and FHL2 have similar structures and share 50% amino acid sequence identity, yet they play opposite roles in cardiac remodeling. Whereas FHL2 knockout mice display exaggerated hypertrophy in response to isoproterenol, FHL1-null mice exhibit blunted hypertrophic growth induced by either TAC or constitutively active Gq (56). It has been proposed that the blunted phenotype in FHL1 knockout mice is due to reduced activation of the ERK pathway, suggesting that FHL1 promotes activation of ERK. In another study, FHL1 activated NFAT-dependent gene expression in C2C12 cells and in FHL1 transgenic mice (9). However, consistent with the in vivo phenotype, FHL2 inhibits both ERK (49) and calcineurin-NFAT-dependent gene expression. Collectively, these data suggest that FHL1 and FHL2 have opposite effects on MEK/ERK and calcineurin-NFAT signaling, which underlies their opposing effects on hypertrophic phenotypes.
Localization to sarcomere.
It has been reported that FHL2 localizes to the Z disk and M band in cardiomyocytes via association with the N2B and is2 regions of titin (34). However, we find that FHL2 is localized predominantly at Z disks, with very low or undetectable levels detected at the M band. Cardiomyocyte Z disks are hubs of multiple signaling pathways, and many regulatory proteins, including proteins involved in hypertrophy, such as calcineurin, ERK, MLP, PKC, and PKA, localize there. In addition, the elusive cardiac stretch-sensing protein/complex is believed to localize to the Z disk, triggering hypertrophic pathways in pressure overload models. Consistent with the function of the LIM domain, FHL2 may act as a Z disk scaffolding protein. However, FHL2 is not required for sarcomeric localization of either ERK1/2 (Hojayev and Hill, unpublished data) or calcineurin (Fig. 6D) under normal conditions, as demonstrated by immunostaining of wild-type and FHL2 knockout hearts. Moreover, FHL2 does not appear to contribute to mechanosensing, as FHL2 knockout mice develop hypertrophy comparable to that of the WT in response to pressure overload (5) (Hojayev and Hill, unpublished data).
FHL2-calcineurin interaction.
We have shown that FHL2 and calcineurin colocalize and form a complex with each other. Other proteins that interact with calcineurin and repress its activity include RCAN1.4, cyclophilin (complexed with CsA) (38), FKBP12 (complexed with FK506) (38), AKAP79 (7), Cain (33), Bcl-2 (57), and calsarcin (16). Interaction between NFAT and calcineurin depends on 2 conserved sites within the NFAT sequence, viz., PxIxIT and LxVP (42). AKAP79 and Cain similarly inhibit calcineurin through their PxIxIT motif (58). RCAN1.4 has a similar motif in addition to the calcineurin inhibitor calcipressin 1 (CIC) motif (58). CsA and FK506 bind to cyclophilin and FKBP12, respectively, which, in turn, bind and inhibit calcineurin through their LxVP motifs (39). However, neither calsarcin nor Bcl-2 harbors these sequence motifs. Similarly, analysis of the FHL2 sequence reveals that FHL2 does not contain those motifs either.
It has been proposed that Bcl-2 inhibits calcineurin/NFAT by sequestering calcineurin within the endoplasmic reticulum (ER) or the mitochondrial membrane (57). This model raises the prospect that inhibition by FHL2 is accomplished by sequestration of calcineurin at the sarcomere, consistent with our observation of increased affinity between FHL2 and calcineurin following adrenergic stimulation. However, the exact mechanism of inhibition of calcineurin by FHL2 requires further study, including interaction mapping studies.
We report that FHL2 colocalizes with calcineurin in both immunocytochemical and immunoprecipitation assays. Of course, these data do not demonstrate a bimolecular, protein-protein interaction between FHL2 and calcineurin. That said, calcineurin has been shown to interact with MLP (muscle LIM protein) (24), a protein harboring 2 LIM domains. We have found using overexpression strategies that FHL2 preferentially binds a constitutively active calcineurin mutant. Similarly, in the setting of endogenous calcineurin and FHL2 in NRVMs, interaction between the proteins was almost undetectable under conditions where calcineurin was inactive. Stimulation with ET-1 and consequent activation of calcineurin, however, increased the amount of FHL2 immunoprecipitated with calcineurin, suggesting that FHL2 has higher affinity for the activated conformation of calcineurin protein. In parallel, immunoprecipitation of SK-1 from the same lysates revealed that FHL2 binds SK-1 under basal conditions and dissociates when treated with ET-1, as reported previously (60). Consistent with this mechanism of interaction, FHL2 has low affinity for unphosphorylated ERK2 (inactive) and high affinity for phosphorylated ERK2 (active) (49). Heregulin (HRG; epidermal growth factor [EGF]-like growth factor) stimulation triggers differentiation of the MCF-7 breast cancer cell line and increased the association between FHL2 and c-Fos (53).
Summary and perspective.
Here, we present findings that implicate β-adrenergic signaling-triggered FHL2 interaction with calcineurin as a novel mechanism regulating calcineurin activation. Together, these data shed new light on LIM domain protein-dependent governance of signal transduction in the heart, thereby opening the way for therapeutic interventions of potential clinical relevance.
ACKNOWLEDGMENTS
We thank members of the Hill lab for constructive comments and support.
We had full access to, and take full responsibility for, the integrity of these data. All authors have read and agree to the manuscript as written.
There are no conflicts of interest to disclose.
This work was supported by grants from the NIH (HL-075173 to J.A.H., HL-080144 to J.A.H., HL-090842 to J.A.H., HL-072016 to B.A.R., and HL-097768 to B.A.R.), AHA (0640084N to J.A.H. and 0655202Y to B.A.R.), ADA (7-08-MN-21-ADA to J.A.H.), and the AHA-Jon Holden DeHaan Foundation (0970518N to J.A.H.).
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Berenji K, Drazner MH, Rothermel BA, Hill JA. 2005. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am. J. Physiol. Heart Circ. Physiol. 289:H8–H16 [DOI] [PubMed] [Google Scholar]
- 2. Berry JM, et al. 2011. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ. Res. 109:407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bovill E, Westaby S, Crisp A, Jacobs S, Shaw T. 2009. Reduction of four-and-a-half LIM-protein 2 expression occurs in human left ventricular failure and leads to altered localization and reduced activity of metabolic enzymes. J. Thorac. Cardiovasc. Surg. 137:853–861 [DOI] [PubMed] [Google Scholar]
- 4. Bueno OF, et al. 2002. Impaired cardiac hypertrophic response in calcineurin Aβ-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 99:4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chu PH, Bardwell WM, Gu Y, Ross J, Jr, Chen J. 2000. FHL2 (SLIM3) is not essential for cardiac development and function. Mol. Cell. Biol. 20:7460–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. 2000. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech. Dev. 95:259–265 [DOI] [PubMed] [Google Scholar]
- 7. Coghlan VM, et al. 1995. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267:108–111 [DOI] [PubMed] [Google Scholar]
- 8. Cohen TV, Randall WR. 2004. NFATc1 activates the acetylcholinesterase promoter in rat muscle. J. Neurochem. 90:1059–1067 [DOI] [PubMed] [Google Scholar]
- 9. Cowling BS, et al. 2008. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J. Cell Biol. 183:1033–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dawid IB, Breen JJ, Toyama R. 1998. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 14:156–162 [DOI] [PubMed] [Google Scholar]
- 11. Devereux RB, et al. 2004. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 292:2350–2356 [DOI] [PubMed] [Google Scholar]
- 12. De Windt LJ, et al. 2001. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U. S. A. 98:3322–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drazner MH, et al. 2004. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: the Cardiovascular Health Study. J. Am. Coll. Cardiol. 43:2207–2215 [DOI] [PubMed] [Google Scholar]
- 14. Du X, et al. 2002. The LIM-only coactivator FHL2 modulates WT1 transcriptional activity during gonadal differentiation. Biochim. Biophys. Acta 1577:93–101 [DOI] [PubMed] [Google Scholar]
- 15. Frank D, Kuhn C, Katus HA, Frey N. 2006. The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. 84:446–468 [DOI] [PubMed] [Google Scholar]
- 16. Frey N, et al. 2004. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 10:1336–1343 [DOI] [PubMed] [Google Scholar]
- 17. Frey N, Katus HA, Olson EN, Hill JA. 2004. Hypertrophy of the heart: a new therapeutic target? Circulation 109:1580–1589 [DOI] [PubMed] [Google Scholar]
- 18. Genini M, et al. 1997. Subtractive cloning and characterization of DRAL, a novel LIM-domain protein down-regulated in rhabdomyosarcoma. DNA Cell Biol. 16:433–442 [DOI] [PubMed] [Google Scholar]
- 19. Groenning BA, et al. 2000. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J. Am. Coll. Cardiol. 36:2072–2080 [DOI] [PubMed] [Google Scholar]
- 20. Gunther T, et al. 2005. Fhl2 deficiency results in osteopenia due to decreased activity of osteoblasts. EMBO J. 24:3049–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han W, et al. 2009. FHL2 interacts with and acts as a functional repressor of Id2 in human neuroblastoma cells. Nucleic Acids Res. 37:3996–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasegawa K, et al. 1993. Ventricular expression of atrial and brain natriuretic peptides in dilated cardiomyopathy. An immunohistocytochemical study of the endomyocardial biopsy specimens using specific monoclonal antibodies. Am. J. Pathol. 142:107–116 [PMC free article] [PubMed] [Google Scholar]
- 23. Heineke J, Molkentin JD. 2006. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7:589–600 [DOI] [PubMed] [Google Scholar]
- 24. Heineke J, et al. 2005. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc. Natl. Acad. Sci. U. S. A. 102:1655–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hill JA, Olson EN. 2008. Cardiac plasticity. N. Engl. J. Med. 358:1370–1380 [DOI] [PubMed] [Google Scholar]
- 26. Hill JA, et al. 2002. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J. Biol. Chem. 277:10251–10255 [DOI] [PubMed] [Google Scholar]
- 27. Johannessen M, Moller S, Hansen T, Moens U, Van Ghelue M. 2006. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell. Mol. Life Sci. 63:268–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kirfel J, et al. 2008. Impaired intestinal wound healing in Fhl2-deficient mice is due to disturbed collagen metabolism. Exp. Cell Res. 314:3684–3691 [DOI] [PubMed] [Google Scholar]
- 29. Komuro I, Izumo S. 1993. Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc. Natl. Acad. Sci. U. S. A. 90:8145–8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kong Y, et al. 2001. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation 103:2731–2738 [DOI] [PubMed] [Google Scholar]
- 31. Labalette C, et al. 2008. The LIM-only protein FHL2 regulates cyclin D1 expression and cell proliferation. J. Biol. Chem. 283:15201–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lai CF, et al. 2006. Four and half lim protein 2 (FHL2) stimulates osteoblast differentiation. J. Bone Miner. Res. 21:17–28 [DOI] [PubMed] [Google Scholar]
- 33. Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. 1998. Cain, a novel physiologic protein inhibitor of calcineurin. J. Biol. Chem. 273:18325–18331 [DOI] [PubMed] [Google Scholar]
- 34. Lange S, et al. 2002. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J. Cell Sci. 115:4925–4936 [DOI] [PubMed] [Google Scholar]
- 35. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. 1990. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 322:1561–1566 [DOI] [PubMed] [Google Scholar]
- 36. Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK. 1996. The progression from hypertension to congestive heart failure. JAMA 275:1557–1562 [PubMed] [Google Scholar]
- 37. Lim HW, et al. 2000. Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. J. Mol. Cell. Cardiol. 32:697–709 [DOI] [PubMed] [Google Scholar]
- 38. Liu J, et al. 1991. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807–815 [DOI] [PubMed] [Google Scholar]
- 39. Liu JO, Nacev BA, Xu J, Bhat S. 2009. It takes two binding sites for calcineurin and NFAT to tango. Mol. Cell 33:676–678 [DOI] [PubMed] [Google Scholar]
- 40. Martin B, et al. 2002. The LIM-only protein FHL2 interacts with beta-catenin and promotes differentiation of mouse myoblasts. J. Cell Biol. 159:113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin BT, et al. 2007. FHL2 regulates cell cycle-dependent and doxorubicin-induced p21Cip1/Waf1 expression in breast cancer cells. Cell Cycle 6:1779–1788 [DOI] [PubMed] [Google Scholar]
- 42. Martinez-Martinez S, et al. 2006. Blockade of NFAT activation by the second calcineurin binding site. J. Biol. Chem. 281:6227–6235 [DOI] [PubMed] [Google Scholar]
- 43. Molkentin JD, et al. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muller JM, et al. 2000. FHL2, a novel tissue-specific coactivator of the androgen receptor. EMBO J. 19:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muller JM, et al. 2002. The transcriptional coactivator FHL2 transmits Rho signals from the cell membrane into the nucleus. EMBO J. 21:736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Okin PM, et al. 2004. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 292:2343–2349 [DOI] [PubMed] [Google Scholar]
- 47. Palazzesi S, et al. 2006. Pressure overload causes cardiac hypertrophy in β1-adrenergic and β2-adrenergic receptor double knockout mice. J. Hypertens. 24:563–571 [DOI] [PubMed] [Google Scholar]
- 48. Park J, et al. 2008. Deficiency in the LIM-only protein FHL2 impairs assembly of extracellular matrix proteins. FASEB J. 22:2508–2520 [DOI] [PubMed] [Google Scholar]
- 49. Purcell NH, et al. 2004. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Mol. Cell. Biol. 24:1081–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rothermel BA, et al. 2001. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U. S. A. 98:3328–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rothermel BA, Vega RB, Williams RS. 2003. The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc. Med. 13:15–21 [DOI] [PubMed] [Google Scholar]
- 52. Saadane N, Alpert L, Chalifour LE. 1999. Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br. J. Pharmacol. 127:1165–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saeki Y, et al. 2009. Ligand-specific sequential regulation of transcription factors for differentiation of MCF-7 cells. BMC Genomics 10:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanna B, Bueno OF, Dai YS, Wilkins BJ, Molkentin JD. 2005. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol. Cell. Biol. 25:865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scholl FA, McLoughlin P, Ehler E, de Giovanni C, Schafer BW. 2000. DRAL is a p53-responsive gene whose four and a half LIM domain protein product induces apoptosis. J. Cell Biol. 151:495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sheikh F, et al. 2008. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 118:3870–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shibasaki F, Kondo E, Akagi T, McKeon F. 1997. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature 386:728–731 [DOI] [PubMed] [Google Scholar]
- 58. Sieber M, Baumgrass R. 2009. Novel inhibitors of the calcineurin/NFATc hub—alternatives to CsA and FK506? Cell Commun. Signal 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Simpson P, McGrath A, Savion S. 1982. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ. Res. 51:787–801 [DOI] [PubMed] [Google Scholar]
- 60. Sun J, Yan G, Ren A, You B, Liao JK. 2006. FHL2/SLIM3 decreases cardiomyocyte survival by inhibitory interaction with sphingosine kinase-1. Circ. Res. 99:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tannous P, et al. 2008. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 105:9745–9750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wan S, et al. 2002. Expression of FHL2 and cytokine messenger RNAs in human myocardium after cardiopulmonary bypass. Int. J. Cardiol. 86:265–272 [DOI] [PubMed] [Google Scholar]
- 63. Wang J, et al. 2007. Suppression of FHL2 expression induces cell differentiation and inhibits gastric and colon carcinogenesis. Gastroenterology 132:1066–1076 [DOI] [PubMed] [Google Scholar]
- 64. Wilkins BJ, Molkentin JD. 2004. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 322:1178–1191 [DOI] [PubMed] [Google Scholar]
- 65. Wixler V, et al. 2007. Deficiency in the LIM-only protein Fhl2 impairs skin wound healing. J. Cell Biol. 177:163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]