

Abstract
When explanted into culture, normal human cells exhibit a finite number of cell divisions before entering a proliferative arrest termed replicative senescence. To identify genes essential for entry into replicative senescence, we performed an RNA interference (RNAi)-based loss-of-function screen and found that suppression of the Never in Mitosis Gene A (NIMA)-related protein kinase gene NEK4 disrupted timely entry into senescence. NEK4 suppression extended the number of population doublings required to reach replicative senescence in several human fibroblast strains and resulted in decreased transcription of the cyclin-dependent kinase inhibitor p21. NEK4-suppressed cells displayed impaired cell cycle arrest in response to double-stranded DNA damage, and mass spectrometric analysis of Nek4 immune complexes identified a complex containing DNA-dependent protein kinase catalytic subunit [DNA-PK(cs)], Ku70, and Ku80. NEK4 suppression causes defects in the recruitment of DNA-PK(cs) to DNA upon induction of double-stranded DNA damage, resulting in reduced p53 activation and H2AX phosphorylation. Together, these observations implicate Nek4 as a novel regulator of replicative senescence and the response to double-stranded DNA damage.
INTRODUCTION
When explanted into culture, human cells derived from normal tissues exhibit a finite number of cell divisions (29). After extended passage in culture, human cells begin to divide more slowly and eventually enter an irreversible proliferative arrest termed replicative senescence (58). Senescent cells exhibit a characteristic large, flattened morphology and, although metabolically active, have permanently exited the cell cycle. Since senescence prevents cells from exceeding a defined replicative limit, replicative senescence has been implicated in aging, as well as in tumor suppression. In support of this hypothesis, several studies have identified senescent cells in premalignant lesions (8, 14–16, 41), and genetic mutations commonly found in human cancers, such as loss of the TP53 and RB1 tumor suppressors or constitutive expression of telomerase, have also been shown to interfere with replicative senescence (26, 32).
Oncogenic stress, DNA damage, replication fork stalling, acute telomere uncapping, and other types of cellular stress can induce an acute senescence response with hallmarks similar to those seen in cells that have entered replicative senescence (37, 39, 50). Several lines of evidence implicate a common set of pathways in the regulation of acute and replicative senescence, although the extent of overlapping pathways is not clear. The expression of the simian virus 40 (SV40) oncoprotein large T antigen (LT) permits human cells to bypass replicative senescence (51). LT binds and inactivates p53 and RB, resulting in a loss of cell cycle checkpoints required to enforce the senescent state. However, in some cell types, senescence is dependent solely upon the tumor suppressor p53, and inactivation of p53 alone is sufficient to bypass replicative senescence or to extend the time to senescence (3, 7). Others have identified cell lines in which p53 inactivation results in an extension of the time to replicative senescence, but this second proliferative barrier is now dependent upon the retinoblastoma protein (RB) pathway (6, 56).
In addition, overexpression of the telomerase catalytic subunit human telomerase reverse transcriptase (hTERT) in presenescent human cells leads to telomere length stability and facilitates immortalization (4, 17, 19, 33, 48). Certain cell types that are solely dependent upon p53 inactivation to bypass replicative senescence are capable of being immortalized by hTERT overexpression, leading to the hypothesis that telomere erosion triggers replicative senescence enforced by p53 (3). However, complicating this analysis, some human cell types, such as keratinocytes, also appear to require inactivation of the RB pathway, together with loss of p53 function and constitutive hTERT expression, to achieve immortality (17). Other types of cellular stress, including accumulated DNA and organelle damage, oxidative stress, and the stresses of cell culture itself, have also been implicated in triggering RB pathway-dependent senescence (10). Therefore, although the p53 and RB pathways clearly play significant roles, the molecular events that ultimately induce and enforce replicative senescence remain unclear.
To identify genes required for entry into replicative senescence, we performed a loss-of-function genetic screen using a pooled version of the RNAi Consortium (TRC) short hairpin RNA (shRNA) library (43) and identified the Never in Mitosis Gene A (NIMA)-related protein kinase gene NEK4 as a regulator of replicative senescence.
MATERIALS AND METHODS
Cell culture and lentiviral and retroviral constructs.
Culture conditions and retroviral or lentiviral infections were as described during the time in which the construction of the original TRC shRNA library was being developed (27, 43, 46). shRNAs in the TRC library were created by ligating oligonucleotides constructed on solid surfaces and then sequencing the products. Therefore, the order in which shRNAs were produced did not correspond to particular classes of genes at that time. Human foreskin fibroblasts (HFF) were isolated from human neonatal foreskin by dispase-mediated separation of the dermis from the epidermis. BJ fibroblasts were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 20% Medium 199 (Gibco), 15% fetal bovine serum (Sigma), glutamine, and penicillin/streptomycin (Gibco). 293T cells were cultured in high-glucose DMEM (Gibco) containing 10% fetal bovine serum (Sigma), glutamine, and penicillin/streptomycin (Gibco). The shRNA sequences used in the previously described (43) lentiviral vector pLKO.1-puro are listed in Table S2 in the supplemental material. Infections were performed at the minimal titer required to achieve ∼100% infection efficiency to ensure that simultaneously infected cultures had undergone the same number of population doublings (PD), unless otherwise specified. Validation experiments were performed using independently derived and propagated BJ fibroblasts expressing the individual shRNAs as described below. The Nek4 cDNA was cloned from 293T cell total cDNA by PCR and ligated into pBabe retroviral vectors engineered to form Flag fusions, and the constructs were sequence verified.
Immunoblotting, immunoprecipitation (IP), and reverse transcription (RT)-PCR.
Immunoblotting was performed on whole-cell extracts isolated in a buffer containing 1.25% NP-40, 1.25% SDS, 12.5 mM NaPO4 (pH 7.2), 2 mM EDTA or in a buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 0.5% NP-40, and 1 mM EDTA and sonicated before separation by SDS-PAGE and incubation with antibodies against β-actin–horseradish peroxidase (HRP) (Santa Cruz), p53 (Ab-6; Calbiochem), H-Ras (F235; Santa Cruz), DNA-dependent protein kinase catalytic subunit [DNA-PK(cs)] (Ab-4; NeoMarkers), p21 (C-19; Santa Cruz), SV40 LT (sc-148; Santa Cruz), Ku70 (H-308; Santa Cruz), Ku80 (B-1; Santa Cruz), hemagglutinin (HA) (clone 12CA5; Roche), Flag (M2; Sigma) or cyclin-dependent kinase 2 (CDK2) (D-12; Santa Cruz), ataxia-telangiectasia mutated (ATM) (NB100-104; Novus), pSer1981 ATM (10H11.E12; Rockland), and pRb (G3-245; BD Biosciences). The Nek4 polyclonal antibody was generated by immunization of a rabbit with the peptide N-CSEPSLSRQRRQKQQEQ-C, corresponding to amino acids 528 to 543 of the Nek4 protein sequence, followed by affinity purification of harvested antiserum (YenZym Antibodies).
Lysates for immunoprecipitation were prepared in a buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 0.5% NP-40, and 1 mM EDTA, followed by sonication, and immunoprecipitation was performed using Flag M2-conjugated agarose beads (Sigma). Immune complexes were eluted with the Flag peptide as recommended by the manufacturer (Sigma). For immunoprecipitations of Nek4 complexes, Nek4 (2631C1a; Santa Cruz) antibody was conjugated to Dynabeads Protein G (Invitrogen). Immunoprecipitates were incubated for >4 hours at 4°C before being washed three times with lysis buffer. Immune complexes were eluted with SDS-PAGE sample loading buffer and boiled before separation by SDS-PAGE.
The following primer pairs were used for quantitative RT-PCR (qRT-PCR): Nek4 (5′-GGAGCTATGGAGAGGTGACG-3′; 5′-CACAGAAGCCCATGACAATG-3′), p53 (5′-CCGCAGTCAGATCCTAGCG-3′; 5′-AATCATCCATTGCTTGGGACG-3′), p21 (5′-GAGGCCGGGATGAGTTGGGAGGAC-3′; 5′-CAGCCGGCGTTTGGAGTGGTAGAA-3′), p16 (5′-ATGGAGCCTTCGGCTGACT-3′; 5′-GTAACTATTCGGTGCGTTGGG-3′), HDM2 (5′-GGATCCTTTGCAAGCGCCAC-3′; 5′-TCAAAGGACAGGGACCTGCG-3′), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (5′-GAAGGTGAAGGTCGGAGTCA-3′; 5′-GACAAGCTTCCCGTTCTCAG-3′). qRT-PCR was performed in triplicate using SYBR green master mix (Applied Biosystems; ABI 7300). Threshold cycle (CT) values were determined using ABI Prism software, which includes automatic background correction and threshold selection.
Analysis of telomere length and telomerase activity.
Telomere restriction fragment (TRF) Southern blotting was performed as described previously (28), and the mean TRF length was measured as described previously (27) using 0.5% agarose gels. The program Image J was used (http://rsb.info.nih.gov/ij/index.html) to quantify the signal intensity of the images shown. Telomerase activity was measured by a telomere repeat amplification protocol as described previously (32).
Proliferation, BrdU incorporation, and SA β-Gal assays.
Proliferation assays were performed in triplicate using a Coulter Particle Counter every 5 to 10 days for rapidly dividing cultures and every 10 to 20 days for presenescent cultures. Population doublings were defined as log2(cells counted/cells plated). P values were determined by Student's paired two-sided t test. The doubling time was defined as days/cumulative PD over 13 to 17 days. Bromodeoxyuridine (BrdU) incorporation was measured by indirect immunofluorescence of cells that had been cultured with 30 μg/ml BrdU for the indicated amount of time using a fluorescein isothiocyanate (FITC)-conjugated anti-BrdU antibody (BU1/75 [ICR1]; Abcam). At least 100 cells were counted in each triplicate experiment. Senescence-associated β-galactosidase (SA β-Gal) staining was performed as described previously (18). Briefly, cells were fixed with 0.2% glutaraldehyde for 5 min at room temperature. After washing with PBS, the cells were stained with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) overnight at 37°C. The proportion of blue cells was determined by counting at least 100 cells from each triplicate well at the indicated time point or 4 PD after selection.
Colony formation assays.
Cells were trypsinized and seeded at 500 cells per well of a six-well plate in triplicate. Twenty-four hours after seeding, the cells were treated with the indicated doses of drug or gamma irradiation. For drug treatment, cells were treated for 1 h before changing to fresh medium. After approximately 8 days, the cells were fixed with methanol and stained with Giemsa stain (Sigma). Colonies containing more than 50 cells were scored.
Cell fractionation, extraction, and flow cytometry.
Cell fractionation was performed as previously described (52). Cells were trypsinized, washed with PBS, and resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 10 mM NaF, 340 mM sucrose, 10% glycerol, 1 mM dithiothreitol [DTT], 0.1% Triton X-100, and protease inhibitors [Roche]). Following low-speed centrifugation at 1,300 × g, the supernatant was collected as the cytoplasmic fraction. The pellet was resuspended in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease inhibitors [Roche]). Following low-speed centrifugation at 1,700 × g, the supernatant was collected as the soluble nuclear fraction. The pellet was then resuspended in SDS-PAGE sample loading buffer and sonicated, representing the insoluble chromatin fraction.
For extraction of cells prior to flow cytometric analysis, the cell pellets were resuspended in 500 μl of extraction buffer containing 0.5% Triton X-100, 0.2 μg/ml EDTA, 1% bovine serum albumin (BSA) in PBS. After 10 min on ice, samples were fixed with 3 ml methanol at −20°C. The cells were stained with antibodies specific for DNA-PK(cs) (18-2; Abcam) or γH2AX (JBW301; Millipore), followed by a fluorescent secondary antibody for analysis via flow cytometry. Analysis was performed on a BD FACScan.
Immunofluorescence assay.
Immunofluorescence assays were performed as previously described (47). Briefly, cells were plated onto coverslips, fixed with either 4% formaldehyde or methanol, permeabilized with 1% Triton X-100, and blocked with 10% serum and 0.1% cold-water fish skin gelatin (Sigma) in PBS plus 1% BSA, 0.02% saponin, and 0.05% sodium azide. The primary antibody used in this study was γH2AX (JBW301; Millipore), followed by secondary mouse antibody-Alexa Fluor 488 (Invitrogen). Nuclei were counterstained with Hoechst 33258 dye.
Comet assay.
Comet assays were performed using the CometSlide kit (Trevigen) according to the manufacturer's protocol. Briefly, cells were treated with the indicated amount of drug, trypsinized, and resuspended in the provided melted agarose mixture. The mixture was pipetted onto slides and allowed to harden, after which the slides were submerged in lysis solution and then run in a standard electrophoresis chamber, incubated with SYBR green, and visualized via epifluorescence. The percentage of DNA in the tail was analyzed with Comet Assay IV software (Perceptive Instruments).
RESULTS
Loss-of-function screen to identify senescence regulators.
To identify genes that regulate the entry of human cells into replicative senescence, we performed a pooled-format loss-of-function screen (Fig. 1A) in late-passage BJ human foreskin fibroblasts using a portion of the TRC shRNA library composed of 5 pools containing a total of 13,799 shRNA constructs targeting 2,939 genes (see Table S1 in the supplemental material). We selected BJ fibroblasts for this screen, since prior work had shown that suppression of p53 extends the life span of these cells and that constitutive expression of hTERT alone suffices to immortalize the cells (3, 4, 55). Infection of 1 × 105 cells was performed at a multiplicity of infection of 0.5 to ensure that the majority of the cells in the culture were infected with a single lentivirus-delivered shRNA. After selection with puromycin to purify cells that integrated a lentiviral vector, we propagated each of the pools for several weeks to identify cells that had bypassed replicative senescence. Most of these pools displayed significant proliferation past that of control cells, suggesting that each shRNA pool harbored shRNAs that gave the desired phenotype (Fig. 1A). After this period of extended proliferation, DNA was isolated from the pooled cultures, and the shRNAs present within the extended-proliferation populations were identified by PCR followed by Sanger sequencing. We identified 17 different genes by this method, 10 of which were identified multiple times (Table 1). In addition, we included two genes identified only once for further evaluation based upon prior reports implicating these genes as tumor suppressors (FBXW7 and RUNX3 [36, 38]).
Fig 1.

Pooled shRNA screen to identify genes affecting senescence. (A) Flow chart of the experimental design used in the study. BJ cells were infected with individual pools of shRNAs and passaged when nearly confluent until control cultures had entered replicative senescence. The graph shows the cumulative population doublings of the pools during the course of the experiment. DNA was isolated from these six pools at the final time point, and shRNAs were amplified by PCR, cloned, and sequenced to determine the identities of individual shRNAs in each pool. gDNA, genomic DNA. (B) Suppression of CHEK2 expression. One control shRNA (shGFP no. 3) and four shRNAs targeting CHEK2 (sh1 to sh4) were introduced into BJ cells at PD 45, and knockdown was measured by qRT-PCR. The error bars represent standard deviations (SD) from triplicate experiments. (C) Proliferation of cells upon suppression of CHEK2. Cells expressing two shRNAs that suppress CHEK2 (red data points, corresponding to the red bars in panel B) were grown in serial passages, together with cells expressing an shRNA targeting p53 (green data points) and two control cell lines expressing shGFP (blue data points). On the indicated days, the number of PD achieved by each culture was measured.
Table 1.
Identities of cloned shRNAs
| Gene namea | Gene product | No. of shRNAsb |
|---|---|---|
| ZNF193 | Zinc finger protein 193 | 7 |
| EHBP1 | EH domain binding protein 1 | 7 |
| NEK4 | NIMA-related kinase 4 | 6 |
| PRKACB | Cyclic AMP (cAMP)-dependent protein kinase catalytic subunit | 5 |
| CHEK2 | Checkpoint kinase 2 | 3 |
| WDR79 | WD repeat domain 79 | 3 |
| RAB3B | Ras family member | 2 |
| TCF20 | Transcription factor 20 | 2 |
| RUNX3 | Runt-related transcription factor 3 | 1 |
| CHRM3 | Cholinergic receptor, muscarinic 3 | 1 |
| CDC2 | Cell division cycle 2 | 3 |
| CSNK1E | Casein kinase 1 epsilon | 2 |
| MCTP1 | Multiple C2 domains, transmembrane 1 | 1 |
| SNF1LK | SNF1-like kinase | 1 |
| FBXW7 | Fbox and WD40 domain protein 7 | 1 |
| KLF11 | Kruppel-like factor 11 | 1 |
| RAB14 | Ras family member | 1 |
Boldface, eliminated from follow-up due to low frequency of identification or previous experience with the genes or shRNAs.
The DNA obtained from pooled cultures was amplified by PCR and sequenced in bulk. For three of the five experimental pools (italics), a single shRNA was identified by this bulk-sequencing method. Amplified shRNAs from these pools were also cloned and sequenced, and the number of shRNAs identified for each gene is reported.
For these genes, we obtained 3 to 6 additional shRNAs from the TRC collection and introduced the shRNAs individually into independently propagated presenescent BJ cells to determine whether the shRNAs suppressed their target genes, as well as whether they permitted BJ cells to bypass senescence. Although the shRNAs identified in the screen suppressed the expression of their target genes for 7 of these 10 genes (see Fig. S1A in the supplemental material), for 2 genes, we reproducibly found that introduction of multiple shRNAs led to an extension of the time to senescence (Fig. 1B and C and 2).
Fig 2.

Suppression of Nek4 increases the time to replicative senescence in human foreskin fibroblasts. (A) Suppression of NEK4 in BJ, IMR-90, and WI-38 cells and HFF. Two control shRNAs (shGFP no. 1 and no. 2) and two shRNAs targeting NEK4 (shN4 no. 1 and no. 2) were introduced into BJ, IMR-90, or WI-38 cells or HFF at 10 to 15 PD preceding the expected onset of replicative senescence, and Nek4 suppression was measured by immunoblotting. Actin is shown as a loading control. Immunoblots to confirm SV40 LT expression in IMR-90 and WI-38 cells are also included. Lanes X, untransfected cells. (B) Proliferation of NEK4-suppressed fibroblasts. The number of PD achieved in BJ, IMR-90, or WI-38 fibroblasts or HFF was measured at the time points indicated and reported as the average number in triplicate experiments of cumulative PD for each line. *, P < 0.05; **, P < 0.005; ***, P < 0.0005 compared to control shGFP cells.
The two genes identified by this approach were the checkpoint control kinase gene, CHEK2, and the NIMA-related kinase gene, NEK4. Suppression of CHEK2 expression extended the time to senescence by 8 to 10 PD, depending on the suppression efficiency of the shRNA used (Fig. 1B and C). Since prior work had implicated CHEK2 as a gene that regulates entry into senescence (24), this observation validated our approach. Of note, two shRNAs targeting CHEK2 potently suppressed Chek2 expression (97 to 99%) and also markedly inhibited cell proliferation so that a proliferation curve could not be generated, suggesting that CHEK2 is an essential gene in this cell type (Fig. 1B and C).
Suppression of NEK4 expression extends the time to senescence.
In addition to CHEK2, we identified several shRNAs specific for NEK4, also known as STK2 (34). Nek4 is a serine/threonine kinase related to the Aspergillus G2/M cell cycle regulator NIMA. Although more extensive studies have been performed on other Nek family kinases, the biological functions of Nek4 are relatively uncharacterized (reviewed in reference 44). To validate the extension of time to senescence phenotype observed when we suppressed NEK4 expression, we generated cell lines that stably expressed either of two distinct shRNAs targeting NEK4 (BJ shN4 no. 1 and shN4 no. 2) or control shRNAs targeting the green fluorescent protein gene (GFP) (BJ shGFP no. 1 and shGFP no. 2) in BJ cells at PD 50. Expression of the shRNAs targeting NEK4 suppressed Nek4 protein expression, as determined by immunoblotting with an anti-human C-terminal Nek4 rabbit polyclonal antibody (Fig. 2A). This antibody recognizes two predicted splice variants of Nek4 (Fig. 2A) in the same proportions as their mRNA precursors, as determined by quantitative RT-PCR (data not shown).
Long-term culture of BJ cells in which NEK4 expression is suppressed (BJ shN4) revealed an extension of the time to replicative senescence of 10 to 20 PD compared to BJ shGFP cells (Fig. 2B). The upper limit of this range attained by BJ shN4 no. 2 cells is similar to the extension of proliferative capacity seen upon expression of SV40 LT, although we note that BJ shN4 no. 2 cells entered senescence at PD 90 while cells expressing LT entered crisis at PD 90 (Fig. 2 and data not shown). These observations demonstrate that suppression of NEK4 extended the proliferative limit of BJ fibroblasts, similar to that induced by elimination of known replicative-senescence checkpoints. To confirm that NEK4 plays a similar role in other human fibroblast cell lines, we introduced the NEK4-specific or control GFP-specific shRNAs into independently isolated HFF, as well as IMR90 and WI-38 fibroblasts, and found that NEK4 expression was suppressed (Fig. 2A) and that suppression of NEK4 resulted in an extension of the time to senescence by 5 to 17 PD (Fig. 2B). Thus, suppression of NEK4 delays the initiation or enforcement of replicative senescence in human fibroblasts.
Effects of NEK4 suppression on telomeres and telomerase.
Because primary BJ fibroblasts and HFF can be immortalized by constitutive expression of hTERT, we examined whether NEK4-suppressed cells display an extended life span due to changes in telomeres or telomerase. To determine if alterations in NEK4 expression affected telomere length or telomerase activity, we manipulated NEK4 expression in BJ and HT-1080 cells, which show intermediate telomere length and telomerase activity and therefore can be monitored for alterations in telomere length in the setting of constitutive expression of telomerase (54), either by introducing shRNAs targeting GFP or NEK4 (shGFP no. 3, shN4 no. 1, or shN4 no. 2) or by introducing an expression vector to overexpress NEK4, and confirmed the effects of these manipulations on Nek4 protein levels (Fig. 3A). As indicated by a telomere repeat amplification protocol (TRAP) assay, suppression or overexpression of NEK4 failed to induce telomerase activity in BJ fibroblasts (Fig. 3B and data not shown) and failed to alter telomerase activity in the HT-1080 cells (Fig. 3B).
Fig 3.

Effects of NEK4 suppression on telomerase and the telomere. (A) Nek4 overexpression and suppression in HT-1080 cells. HT-1080 cells were infected with either a control vector, a Nek4 overexpression construct, a control shRNA (shGFP no. 3), or each of two NEK4-specific shRNAs (shN4 no. 1 and shN4 no. 2), as indicated. Nek4 levels were measured by immunoblotting. β-Actin is shown as a loading control. (B) Telomerase activity in the presence of Nek4 overexpression or suppression. The telomere repeat amplification protocol was utilized to measure telomerase activity from 2 μg of lysates from HT-1080 cells, shown in panel A, or BJ cells, as indicated. An internal control (IC) for PCR inhibition is shown (bottom), as is a negative control for heat-inactivated lysate (Boil). (C) Telomere length Southern blot of HT-1080 cells. HT-1080 cells, shown in panel A, were cultured for 5, 14, or 29 PD postselection. DNAs collected from these cultures were subjected to TRF Southern blotting, and the intensity of hybridization is shown. Size markers in kb are shown on the left. (D) Telomere length Southern blot of BJ cells. BJ cells expressing control or NEK4-specific shRNAs were cultured for 10, 20, or 30 PD postselection, and the DNA collected was subjected to TRF Southern blotting as in panel C.
Because the TRAP assay is not quantitative, subtle changes in telomerase activity or telomerase activity-independent telomere length effects may not be detectable by the assay. Therefore, to determine if alteration of NEK4 expression could affect telomere length in long-term culture, we collected DNA from these BJ and HT-1080 cells after extended passage in culture and subjected the DNA to Southern blotting using telomere restriction fragments (TRF). Manipulating NEK4 expression in BJ or HT-1080 cells failed to induce a measurable change in TRF length over 29 to 30 PD after stable expression of the indicated shRNAs (initiated at approximately PD 20) (Fig. 3C and D). These observations suggest that Nek4 does not directly affect telomerase activity or telomere length.
Suppression of NEK4 leads to an increased proliferation rate and reduced transcription of p21.
Although there was no detectable affect on telomere length or telomerase activity upon suppression of NEK4, we observed that cells expressing shRNAs specific for NEK4 displayed an increased proliferation rate. At late passage (PD 55 to 60), BJ shN4 cells proliferated at nearly twice the rate of BJ shGFP cells (P < 0.003) (Fig. 4A). To gain insight into the increased proliferation rate of BJ shN4 cells, we examined the steady-state protein levels of various cell cycle regulators. The levels of p53, cyclin E, cyclin D1, CDK2, and p27KIP1 (p27) appear similar in BJ shGFP and BJ shN4 cells (Fig. 4B). As determined by qRT-PCR, the mRNA levels of p16 also appear similar in BJ shGFP and BJ shN4 cells (data not shown).
Fig 4.

Suppression of NEK4 alters the proliferation rate and p21 expression. (A) Effect of NEK4 suppression on doubling time. Proliferation was measured in BJ cells over 15 days and used to extrapolate the doubling time. The error bars represent SD from triplicate experiments. *, P < 0.008 compared to shGFP no. 1 control. (B) Immunoblotting of cell cycle regulators in control BJ shGFP no. 1 and no. 2 or BJ shN4 no. 1 and shN4 no. 2 cells. Actin is shown as a loading control. (C) qRT-PCR analysis of NEK4, p53, and p21 mRNA levels in BJ shN4 no. 1 and shN4 no. 2 cells compared to control BJ shGFP no. 1 cells. *, P < 0.0005 compared to shGFP no. 1 control. (D) Luciferase assay in transiently transfected 293T cells to assess transcription off the p21 promoter in the presence of control shGFP no. 1 or shN4 no. 1 and 2 (left) and in the presence of empty vector, ectopic Nek4, or ectopic p53 (right). *, P < 0.04, and **, P < 0.0006 compared to shGFP no. 1 control (left); *, P < 0.0006 compared to empty-vector control (right).
However, we found that the levels of cyclin-dependent kinase inhibitor 1A, p21CIP1 (referred to below as p21), were reduced in BJ shN4 cells compared to BJ shGFP cells (Fig. 4B). To determine whether the decrease in p21 levels was due to decreased transcription rather than a posttranscriptional alteration, we performed quantitative RT-PCR and found that BJ shN4 cells exhibit decreased transcription of p21, as evidenced by reduced p21 mRNA levels (Fig. 4C). Suppression of NEK4 by shN4 no. 1 resulted in a decrease in p21 mRNA levels smaller than that observed upon expression of shN4 no. 2 (0.81- and 0.27-fold, respectively), but we note that the levels of p21 mRNA and protein were still reduced compared to those in BJ shGFP cells (Fig. 4B and C). To confirm the observed decrease in p21 transcription, we utilized a firefly luciferase construct in which luciferase expression is driven by the p21 promoter (22). Similar to what we observed using qRT-PCR, transient cotransfection of the luciferase construct, together with shN4 no. 1 and no. 2, led to decreased expression of luciferase (0.58- and 0.38-fold, respectively) compared to cotransfection with shGFP no. 1 (P < 0.4) (Fig. 4D, left), indicating that suppression of NEK4 decreases transcription from the p21 promoter. Overexpression of NEK4 did not alter luciferase expression, while overexpression of p53 led to dramatic upregulation of luciferase expression, as expected (6.09-fold compared to the control vector) (Fig. 4D, right). However, we note that NEK4-suppressed cells are not refractory to p53 expression, since overexpression of a Flag epitope-tagged p53 in BJ shGFP no. 1, BJ shN4 no. 1, or BJ shN4 no. 2 cells (Fig. 5A) resulted in comparable levels of cell cycle arrest, as assessed from decreased BrdU incorporation (Fig. 5B). These observations indicate that the p21 promoter is still responsive to p53 in these cells and suggest that Nek4 functions upstream of p53-mediated p21 transcription.
Fig 5.
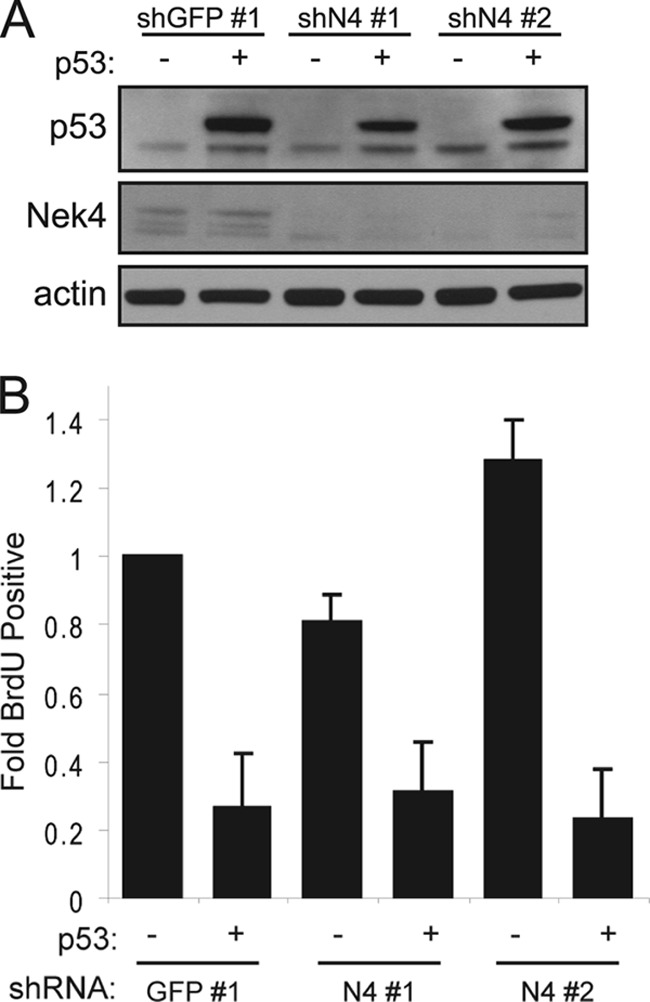
Response of NEK4-suppressed cells to p53 overexpression. (A) Overexpression of p53 in BJ cells. BJ shGFP no. 1 or BJ shN4 no. 1 and no. 2 cells were infected with a control vector (−) or Flag-tagged p53 (+). The cell lines were assessed for NEK4 suppression and p53 overexpression by immunoblotting. Actin is shown as a loading control. (B) BrdU incorporation assays to assess cell cycle arrest. BJ cells, characterized in panel A, were incubated with BrdU for 18 h postselection to assess the percentage of cells undergoing proliferation. At least 100 cells were scored for each triplicate experiment, and the error bars represent SD.
Consistent with p21 being a primary mediator of p53-dependent cell cycle arrest, loss of p21 has previously been linked to an extension of the cell life span (21, 57). We characterized BJ shN4 cells during the extended life span period by assessing the status of previously described markers of senescence in BJ shGFP and BJ shN4 cells at a time when the control cells (BJ shGFP) had begun to enter senescence and the BJ shN4 cells continued to proliferate (PD 64). Similar to what has been described for late-passage p21−/− human diploid fibroblasts (HDFs) (21, 57), we found that senescent BJ shGFP and proliferating BJ shN4 cells exhibited similar levels of SA β-Gal activity (Fig. 6A). Furthermore, we observed that the levels of p21 gradually increase over progressive population doublings, coincident with the eventual proliferative arrest that the BJ shN4 cells undergo (Fig. 6B). NEK4, however, remains suppressed in late-passage and senescing BJ shN4 cells (Fig. 6B). Thus, our observations indicate that the extended life span of BJ shN4 cells is due to decreased transcription of p21.
Fig 6.

Characterization of extended-life-span NEK4-suppressed cells. (A) (Left) SA β-Gal staining. BJ shGFP no. 1 and no. 2 and BJ shN4 no. 1 and no. 2 cells were fixed and stained with X-Gal to identify the percentages of cells with SA β-Gal activity at the indicated population doublings. At least 100 cells were scored for each time point. (Right) Parallel BrdU incorporation assays. Cells were also incubated with BrdU for 48 h to assess the percentages of proliferating cells at the indicated population doublings. At least 100 cells were scored for each time point. (B) Immunoblotting (IB) of markers of senescence. Steady-state levels of p53, p21, Nek4, and actin were assessed via immunoblotting in BJ shGFP no. 1 or BJ shN4 no. 1 and no. 2 cells at the indicated PD. Control cells senesced prior to PD 88, as can be seen in panel A. The last time points for shN4 no. 1 and no. 2, PD 92 and 101, respectively, were assessed together in the bottom row.
Cell cycle arrest, but not acute senescence, is altered by suppression of NEK4.
Because we observed decreased levels of p21 in BJ shN4 cells, we were interested in determining whether acute senescence and/or stimulus-induced cell cycle arrest are globally defective in these cells. An acute senescence response has been demonstrated upon oncogene activation, such as the expression of an oncogenic allele of H-RAS (H-RasV12). To investigate if suppression of NEK4 renders cells resistant to the acute oncogene-induced senescence response, we expressed H-RasV12 in BJ shGFP or shN4 cells (Fig. 7A). Expression of H-RasV12 in BJ shGFP cells induced senescence, as determined from a dramatic decrease in BrdU incorporation, and suppression of NEK4 failed to alter the percentage of BrdU-incorporating cells in response to H-RasV12 expression (Fig. 7B). Thus, suppression of NEK4 is not sufficient for cells to escape from acute senescence triggered by expression of H-RasV12.
Fig 7.
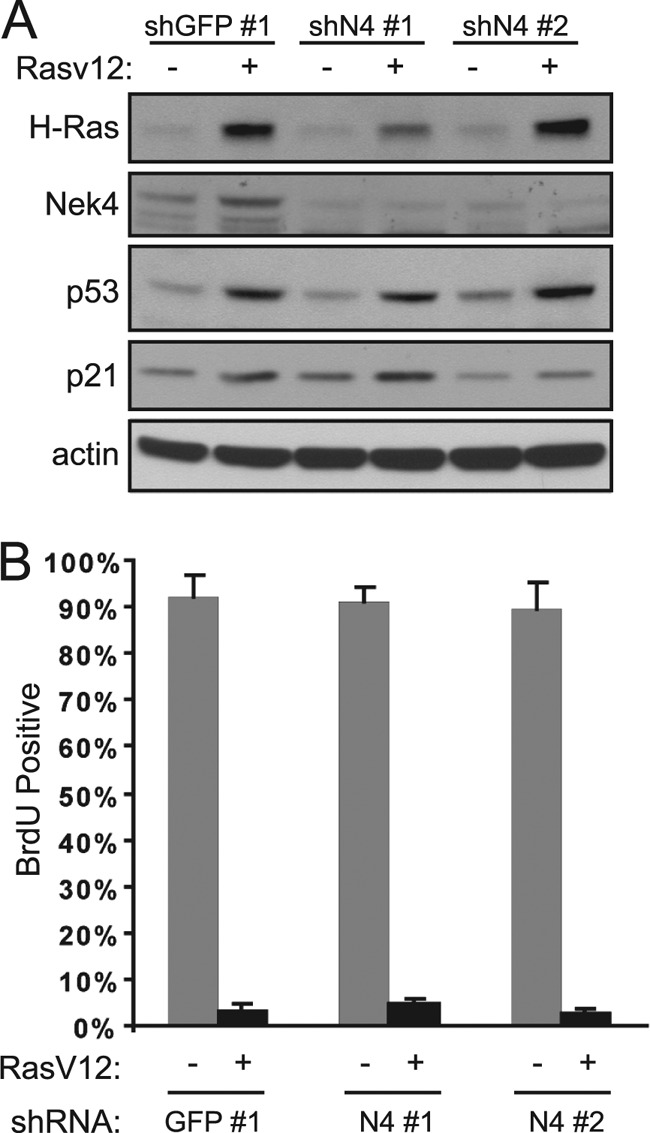
Effect of NEK4 suppression on oncogene-induced senescence. (A) Suppression of NEK4 in BJ cells plus H-RasV12 overexpression. BJ cells were infected with a control shRNA (GFP no. 1) or each of two shRNAs targeting NEK4 (shN4 no. 1 and shN4 no. 2), followed by infection with a control vector (−) or H-RasV12 (+). These cell lines were assessed for NEK4 suppression and H-RasV12 expression, p53 levels, and p21 levels by immunoblotting. β-Actin is shown as a loading control. (B) BrdU incorporation as a measure of proliferation. Five days postinfection with H-RasV12, the cells shown in panel A were incubated with BrdU for 24 h to assess the percentage of proliferating cells. At least 100 cells were scored for each triplicate experiment, and the error bars represent SD.
We also determined whether reversible cell cycle arrest, such as that induced by DNA damage, was altered in these cells. Colony formation assays were performed on BJ shGFP and BJ shN4 cells that were treated with vehicle control or increasing concentrations of the DNA-damaging agents etoposide and mitomycin C (MMC). Etoposide inhibits DNA topoisomerase II, leading primarily to double-stranded DNA breaks, while MMC is a DNA cross-linker. BJ shN4 cells displayed decreased sensitivity to etoposide treatment (Fig. 8A, top) (P < 0.015), but not to treatment with MMC (Fig. 8A, middle), suggesting that NEK4 suppression interferes with the arrest induced by double-stranded DNA damage. To explore this finding further, we performed colony formation assays using other double-stranded DNA-damaging drugs, bleomycin and neocarzinostatin, and observed similar decreases in sensitivity (see Fig. S2 in the supplemental material). Moreover, following treatment with increasing doses of gamma irradiation, BJ shN4 no. 2 cells again displayed decreased sensitivity compared to BJ shGFP no. 1 cells (P < 0.015); BJ shN4 no. 1 cells displayed sensitivity intermediate between those of BJ shGFP no. 1 and BJ shN4 no. 2 cells, consistent with the intermediate phenotypes observed throughout other assays (Fig. 8A, bottom).
Fig 8.

Effect of NEK4 suppression on DNA damage-mediated cell cycle arrest. (A) Colony formation assays to examine the sensitivity of BJ shGFP no. 1 or BJ shN4 no. 1 and no. 2 fibroblasts to treatment with etoposide (top), mitomycin C (middle), or gamma irradiation (bottom). Survival is expressed as the percentage of colonies formed following treatment compared to mock-treated cells. The error bars indicate the standard deviations of triplicate experiments. *, P < 0.015 compared to shGFP no. 1 control. (B) BrdU incorporation assays to assess cell cycle arrest of cells mentioned in part A in response to treatment with etoposide (top), mitomycin C (middle), or gamma irradiation (bottom). The bars indicate the percentages of BrdU-positive cells following treatment with the indicated drug or irradiation, normalized to the percentage of BrdU-positive mock-treated cells. The error bars represent the standard deviations of triplicate experiments. *, P < 0.03; **, P < 0.004; †, P < 0.016; ‡, P < 0.0003 compared to the shGFP no. 1 control. (C) Reexpression of Nek4 restores etoposide-induced cell cycle arrest. A BrdU incorporation assay was performed as shown in panel B using BJ shGFP no. 1 or BJ shN4 no. 1 cells stably transfected with empty vector or Flag-tagged Nek4. Below the bar graph is an immunoblot confirming overexpression of Flag-tagged Nek4 in the appropriate cell lines; actin is shown as a loading control. *, P < 0.024 compared to shN4 no. 1 cells expressing the empty vector.
To confirm that the observed changes in sensitivity were due to impaired cell cycle arrest, we performed short-term BrdU incorporation assays. Specifically, BJ shGFP and BJ shN4 cells were treated with vehicle control and 5 μM etoposide or 0.05 μg/ml MMC in the presence of BrdU for 20 h. In parallel, BJ shGFP and BJ shN4 cells were treated with 1 Gy of irradiation and cultured with BrdU for 20 h. As expected, the proliferation of BJ shGFP no. 1 cells was arrested or decreased upon treatment with all of the DNA-damaging agents (Fig. 8B, top). However, even in the continuous presence of etoposide, approximately 40 to 50% of BJ shN4 cells continued to proliferate (P < 0.03 compared to BJ shGFP no. 1), further suggesting that NEK4-suppressed cells do not arrest as expected in response to double-stranded DNA damage. Similar results were observed in IMR-90 cells expressing shGFP or shN4 and treated with etoposide, showing that the phenotype is not specific to BJ fibroblasts (see Fig. S3 in the supplemental material). No appreciable difference was seen in BrdU incorporation rates upon treatment of the cells with MMC, in agreement with the colony formation assays (Fig. 8B, middle). In response to gamma irradiation, BJ shN4 cells exhibited a higher BrdU incorporation rate than control BJ shGFP cells (approximately 10 to 30% higher; P < 0.016), again in accordance with a defective cell cycle arrest in response to double-stranded DNA damage (Fig. 8B, bottom).
To confirm that the decreased level of Nek4 is specifically responsible for the observed impairment in DNA damage-induced cell cycle arrest, Nek4 was ectopically expressed in BJ shGFP and BJ shN4 no. 1 cells (Fig. 8C). Upon expression of Nek4 in these cells, we found no appreciable change in basal BrdU incorporation compared to cells containing a control vector. When treated with etoposide, however, reintroduction of Nek4 into BJ shN4 no. 1 cells restores the cell cycle arrest induced by DNA damage to levels similar to those observed in empty-vector- or Nek4-overexpressing BJ shGFP cells (Fig. 8C). Together, these observations show that suppression of NEK4 does not interfere with acute senescence responses but does disrupt double-stranded DNA damage-mediated cell cycle arrest.
Identification of Nek4-interacting proteins.
To gain insight into Nek4 function, we isolated Nek4 immune complexes and identified putative Nek4-interacting proteins by mass spectrometric analysis. We expressed the full-length NEK4 cDNA as a Flag epitope-tagged protein (F-Nek4) in 293T cells, isolated immune complexes with Flag M2-conjugated agarose beads, and eluted bound proteins with excess Flag peptide. Upon SDS-PAGE, we readily detected a band corresponding to the expected size of Nek4 by silver staining in lysates derived from 293T cells expressing F-Nek4, but not in lysates derived from control transfected 293T cells (Fig. 9A). Furthermore, differentially displayed bands corresponding to potential interacting proteins were seen in the F-Nek4 immune complexes (Fig. 9A). We excised these bands from a replicate colloidal blue-stained gel and performed mass spectrometry analysis. We cross referenced the list of putative interacting proteins with a list of common contaminants of Flag-M2 IPs from 293T cells (11) and, based upon this information, compiled a list of putative Nek4-interacting proteins (Table 2).
Fig 9.

Identification of Nek4-interacting proteins. (A) IP of Nek4 complexes. Representative silver-stained SDS-PAGE gel containing Flag peptide eluates of mock (−)- or Flag-Nek4 (F-Nek4)-transfected 293T cell lysates immunoprecipitated for the Flag epitope tag. Size markers (in kilodaltons) are indicated on the left. F-Nek4 is indicated on the right. (B) Validation of Nek4 interactions by endogenous co-IPs. Immune complexes were immunoprecipitated using antibodies specific for endogenous Nek4 or Ku80 and were subjected to SDS-PAGE, together with whole-cell lysate (WCL). The antibody used for subsequent immunoblotting is indicated. (C) IP of Nek4 complexes in the presence of 0.1 mg/ml ethidium bromide (EtBr), as indicated. Immunoblots were performed as described in the legend to panel B. (D) IP of Nek4 complexes in mock- or 50 μM etoposide-treated cells, as indicated. Immunoblots were performed as described in the legend to panel B.
Table 2.
Mass spectrometry resultsa
| Interacting protein | Peptide coverage (%) |
|---|---|
| BCAS2 | 25.3 |
| CDC2 | 43.1 |
| CDK2 | 23.8 |
| DNA-PK(cs) | 10.4 |
| GNL3 | 29.7 |
| Ku70 | 54.9 |
| Ku80 | 29.5 |
| MCM7 | 8.3 |
| NAT10 | 13.3 |
| PCNA | 26.1 |
Bands excised from a replicate of the gel depicted in Fig. 9A were submitted to mass spectrometry. After comparison with known contaminants, the list of interacting proteins and the peptide coverage of each was compiled.
The putative interactions with DNA-PK(cs), Ku70, and Ku80 were of particular interest to us, since we had found a role for Nek4 in cell cycle arrest in response to DNA damage. Together, these proteins make up the active DNA-PK kinase, a key mediator of nonhomologous end joining (NHEJ) and, thus, the recognition and repair of double-stranded DNA breaks (reviewed in reference 42). To confirm that these proteins interact with Nek4, we isolated immune complexes using antibodies specific for either endogenous Nek4 or Ku80 in BJ fibroblasts (Fig. 9B). Nek4 and Ku80 immune complexes isolated from BJ cells and resolved via SDS-PAGE were immunoblotted with antibodies specific for endogenous DNA-PK(cs), Ku70, and Ku80. We detected each of these proteins in complex with Nek4 (Fig. 9B). To eliminate the possibility that the interaction of the Nek4 and Ku proteins was mediated by DNA, we isolated Nek4 immune complexes in the presence of ethidium bromide and found that interaction between Nek4 and Ku70 was intact (Fig. 9C). Furthermore, we also confirmed that this interaction remains intact following etoposide treatment (Fig. 9D). Together, these observations indicate that Nek4 associates with the NHEJ proteins DNA-PK(cs), Ku70, and Ku80.
Impairment of DNA-PK upon suppression of NEK4.
Since our observations implicated Nek4 in the response to double-stranded DNA damage, we investigated the effects of suppressing NEK4 on the DNA-PK complex. We examined the steady-state levels of DNA-PK(cs), Ku70, and Ku80 in asynchronous BJ shGFP and BJ shN4 cells but did not observe any notable differences (Fig. 10A). Furthermore, the overall nuclear localization of these proteins was not altered upon suppression of NEK4 (data not shown), and the interaction between Ku70 and Ku80 is not impaired in BJ shN4 cells (Fig. 10B).
Fig 10.

Effect of NEK4 suppression upon DNA-PK activity. (A) Immunoblotting of Nek4, DNA-PK(cs), Ku80, Ku70, and actin (loading control) in BJshGFP no. 1 or BJshN4 no. 1 and 2 cells. (B) IP of Ku70 complexes in BJ shGFP no. 1, BJ shN4 no. 1, and BJ shN4 no. 2 cells to examine the interaction between Ku70 and Ku80. The antibody used for subsequent immunoblotting is indicated. (C) Immunoblotting of cellular fractionation assays. The soluble nuclear fractions (10%) and chromatin fractions (10%) of dimethyl sulfoxide (DMSO)- or etoposide-treated BJ shGFP no. 1 and BJ shN4 no. 1 and no. 2 cells were separated by SDS-PAGE and immunoblotted to examine levels of DNA-PK(cs), Ku70, XRCC4, and H2A. (D) Flow cytometric analysis of chromatin-bound DNA-PK(cs) levels. The bars indicate the percentages of DNA-PK(cs) bound to chromatin following etoposide treatment normalized to DMSO-treated levels. The error bars represent SD from triplicate experiments. *, P < 0.0005, and **, P < 0.002 compared to shGFP no. 1 control.
Normally, DNA-PK(cs) is recruited to sites of DNA damage via a Ku70/Ku80 heterodimer that assembles at the site, and the activated kinase subsequently signals to and recruits downstream factors necessary for DNA repair (reviewed in reference 42). To investigate whether these functions are altered in NEK4-suppressed cells, BJ shGFP and BJ shN4 cells were treated with 50 μM etoposide and fractionated into cytoplasmic, soluble nuclear, and insoluble nuclear (chromatin) lysates, as previously described (52). The nuclear fractions were separated by SDS-PAGE and immunoblotted with antibodies specific for DNA-PK(cs), Ku70, and histone variant H2A as a loading control. Upon treatment with etoposide, we failed to identify changes in the proportion of soluble or insoluble chromatin-bound Ku70 in BJ shGFP or BJ shN4 cells (Fig. 10C). However, although the chromatin-bound fraction of DNA-PK(cs) increased upon etoposide treatment in BJ shGFP cells, as expected, we found that this increase was diminished in BJ shN4 cells (Fig. 10C). To assess this in a quantitative manner, we adapted previously described assays in which soluble nuclear DNA-PK(cs) is extracted from cells prior to fixation in order to analyze the percentage of insoluble DNA-PK(cs) that remains bound to DNA (2, 20, 35). BJ shGFP and BJ shN4 cells were treated with vehicle control or 50 μM etoposide and fixed either prior to or following extraction. These cells were incubated with an antibody specific for DNA-PK(cs) and a fluorescent secondary antibody to analyze the amount of bound, insoluble DNA-PK(cs) using flow cytometry. Using this method, we found that, following double-stranded DNA damage, the observed percentage of DNA-PK(cs) that is bound to DNA was decreased in BJ shN4 cells compared to control BJ shGFP cells (61.1% in BJ shN4 no. 1 cells and 77.8% in BJ shN4 no. 2 cells; P < 0.002) (Fig. 10D). These observations are in agreement with our immunoblot analyses and, together, indicate that NEK4 suppression interferes with recruitment of DNA-PK(cs) to damaged DNA.
To examine the consequences of impaired recruitment of DNA-PK to sites of DNA damage, we examined the activation of p53 in NEK4-suppressed cells. Following 50 μM etoposide treatment, phosphorylation of p53 at serine 15 was reduced in BJ shN4 no. 1 and no. 2 cells compared to BJ shGFP cells (Fig. 11A). In addition, we found decreased levels of HDM2 mRNA, a transcriptional target of p53, in BJ shN4 no. 1 and no. 2 cells compared to BJ shGFP cells, following etoposide treatment (P < 0.045) (Fig. 11B). We also examined another primary target of DNA-PK, the histone variant H2AX. In untreated asynchronous BJ shGFP or BJ shN4 cells, we failed to observe a notable difference in the numbers of γ-H2AX foci (see Fig. S4 in the supplemental material). However, when BJ shGFP and BJ shN4 cells were treated with 50 μM etoposide, a decreased level of phosphorylated H2AX (γ-H2AX) was present in treated BJ shN4 cells compared to control BJ shGFP cells, as evidenced via immunofluorescence (Fig. 11C). To examine this more quantitatively, we analyzed the cells using flow cytometry and found that shN4 no. 1 cells and shN4 no. 2 cells contain only 72.8% and 77.7% of the amount of γ-H2AX observed in BJ shGFP cells, respectively (P < 0.03) (Fig. 11D). Moreover, these differences occur despite the fact that the control and shN4 cells acquire similar levels of DNA damage as assessed via a traditional comet assay (Fig. 11E). Additionally, ATM, another key regulator of the DNA damage response (DDR), is similarly activated in BJ shGFP and BJ shN4 cells upon treatment with etoposide, as measured by autophosphorylation at serine 1981 (Fig. 11F). Together, these data support the notion that recognition of double-stranded DNA damage by DNA-PK and the consequent downstream signaling are impaired in NEK4-suppressed cells.
Fig 11.

Effect of NEK4 suppression on events downstream of DNA-PK. (A) Immunoblotting of phosphoserine 15 (pSer15) p53, total p53, and actin (loading control) in BJ shGFP no. 1, BJ shN4 no. 1, and BJ shN4 no. 2 cells treated with vehicle control or 50 μM etoposide. (B) qRT-PCR analysis of HDM2 mRNA levels in BJ shGFP no. 1 and BJ shN4 no. 1 and no. 2 cells treated with 50 μM etoposide. *, P < 0.045 compared to shGFP control. The fold change is shown. The error bars indicate SD. (C) Immunofluorescence staining of γ-H2AX foci in BJ shGFP no. 1 or BJ shN4 no. 1 and no. 2 fibroblasts following 50 μM etoposide treatment. The arrows indicate cells that display reduced γ-H2AX levels compared to control shGFP no. 1 cells. (D) Flow cytometric analysis of γ-H2AX levels. The bars indicate the percentages of γ-H2AX fluorescence levels in etoposide-treated BJ shGFP no. 1 or BJ shN4 no. 1 and no. 2 cells normalized to DMSO-treated cells. *, P < 0.03, and **, P < 0.009 compared to shGFP control. (E) Comet assay to assess the amount of DNA damage acquired. (Left) Fluorescence images of representative control and etoposide-treated cells after electrophoresis. (Right) The bars indicate the percentages of DNA found in the comet tail of DMSO or etoposide-treated BJ shGFP no. 1 and BJ shN4 no. 1 and no. 2 cells. At least 100 cells were analyzed per condition for each triplicate experiment. (F) Immunoblot analysis of ATM activation. Whole-cell lysates from DMSO- or etoposide-treated BJ shGFP no. 1 and BJ shN4 no. 1 and no. 2 cells were separated by SDS-PAGE and immunoblotted to examine levels of phosphoserine 1981 (pSer1981) ATM, total ATM, and actin as a loading control.
DISCUSSION
NEK4 and replicative senescence.
Entry into replicative senescence occurs with extended passage of explanted human cells derived from normal tissues (29, 58). While the p53 and pRB pathways play key roles in enforcing the senescent state, the mechanism for triggering replicative senescence is poorly understood. Here, we used a loss-of-function approach to identify genes involved in regulating entry into replicative senescence and identified the NIMA-related kinase gene, NEK4, as a gene whose expression is required for timely entry into replicative senescence. Further characterization of NEK4-suppressed cells revealed a reduction in the levels of p21, a protein well described as an important mediator of cell cycle arrest and replicative senescence (reviewed in reference 30). The extension of replicative capacity beyond the normal replicative barrier has been described for suppression of the primary p21 trans activator, p53 (5, 6, 23, 25, 49, 59), and for direct suppression of p21 itself (9). Notably, extended-life-span BJ shN4 cells phenocopy late-passage p21−/− and p21+/− cells (21, 57), strongly supporting the idea that reduced p21 levels are responsible for the extended life span of BJ shN4 cells.
Suppression of NEK4 expression did not affect the acute senescence response induced by oncogenic H-RasV12 expression, although we observed an effect on cell cycle arrest induced by various DNA-damaging drugs. We believe this discrepancy may be due to the possibility that senescence-inducing phenotypes, such as oncogenic Ras expression, might be more potent and therefore more difficult to mitigate than temporary cell cycle arrest or even replicative senescence, in which reduction of the proliferative rate is more gradual. In other words, NEK4-suppressed cells are not refractory to p53- or p21-mediated proliferative arrest, but in cases where the induction of p21 is low level or gradual, the decreased basal p21 levels observed in NEK4-suppressed cells cause the cell cycle arrest to be delayed or dampened, requiring higher activation of p21 than usual. Another explanation for this observation is that temporary cell cycle arrest and replicative senescence are fundamentally different than acute senescence, in a way that uniquely requires the presence of Nek4. Indeed our results suggest that Nek4 may play a role specifically in the double-stranded DNA damage response, which is likely not primarily responsible for oncogenic Ras-induced senescence.
The role of Nek4 in the response to DNA damage.
Mass spectrometry analysis of Nek4-interacting proteins identified the mutually interacting proteins DNA-PK(cs), Ku70, and Ku80. The Ku70/Ku80 heterodimer localizes to sites of double-stranded DNA breaks and recruits DNA-PK(cs), becoming the active DNA-PK kinase complex that is responsible for the cascade of events necessary to repair the breaks (reviewed in reference 42). The observed association between Nek4 and all components of the DNA-PK complex suggests that this interaction may occur at sites of DNA damage; however, it also remains possible that Nek4 may interact with each protein individually. Nevertheless, we note that in NEK4-suppressed cells, we observed a defect in the recruitment of DNA-PK(cs) to chromatin upon the induction of double-stranded DNA damage. We thus hypothesize that Nek4 plays a role in this recruitment or in the stability of the interaction between DNA-PK(cs) and Ku70/Ku80. Although we observed only a partial disruption in DNA-PK(cs) recruitment, this may be due to incomplete suppression of NEK4 in these fibroblasts. Therefore, the remaining amounts of Nek4 may be able to mediate DNA-PK complex formation, albeit at reduced efficiency. Alternatively, it is possible that Nek4 regulates a subpopulation of DNA-PK or plays a role in complex formation in specific contexts.
In line with the decreased recruitment of DNA-PK(cs) to DNA, fibroblasts containing shRNAs specific for NEK4 also display decreased activation of p53 and decreased levels of γ-H2AX upon treatment with etoposide. Although several kinases are known to phosphorylate H2AX, it has been reported that DNA-PK is the primary kinase responsible for H2AX phosphorylation in the context of double-stranded DNA damage (1). These results correlate with the observation that NEK4 suppression decreases the sensitivity of fibroblasts to double-stranded DNA-damaging drugs, but not to a DNA cross-linker, mitomycin C. We also showed that the decreased sensitivity is a result of defective cell cycle arrest in NEK4-suppressed cells, likely linked to defective signaling by DNA-PK. We thus believe that Nek4 is a novel mediator of the response to double-stranded DNA damage.
Several lines of evidence implicate DNA damage as a significant contributor to the induction of replicative senescence. In agreement with this, we identified Chek2, a key substrate of the DNA damage kinase ataxia-telangiectasia mutated (ATM), as a regulator of replicative senescence, as has been previously described (24). Moreover, it was shown that inactivation of Chek2 also resulted in decreased p21 expression in human fibroblasts due to defective p53 activation (24). It was previously shown that DNA-PK-deficient scid mouse embryonic fibroblasts display delayed and attenuated induction of p21 expression in response to ionizing radiation (31). Thus, our findings that Nek4 associates with DNA-PK and is involved in the response to double-stranded DNA damage is consistent with the extension of life span acquired upon NEK4 suppression. We believe that inefficient DNA-PK signaling, which may otherwise be occurring sporadically or at low levels in normal cells, is likely responsible for the observed decrease in p21 levels of NEK4-suppressed cells, leading to delayed replicative senescence.
Interestingly, other NIMA-related kinases have been implicated in cell cycle arrest in response to DNA damage (reviewed in reference 44). Nek1 appears to be activated in response to ionizing radiation and plays a role in the consequent cell cycle arrest (12, 13). Nek10 is a mediator of the G2/M cell cycle arrest that occurs in response to UV irradiation (45). Nek11 is implicated in the infrared (IR)-induced degradation of CDC25A and G2/M arrest (40).
Relevance of Nek4 to cancer.
Both replicative and oncogene-induced senescence have been implicated as tumor-suppressive mechanisms (reviewed in reference 37), and bypass of senescence may be an important step in tumorigenesis. Furthermore, it is clear that defective DNA damage repair facilitates the accumulation of tumor-promoting mutations. Therefore, one would expect that a gene required for timely entry into replicative senescence and for the proper recognition and repair of double-stranded DNA breaks might be suppressed or lost in human cancers. Indeed, a survey of lung cancer samples showed that the genomic region containing NEK4, chromosome 3p, is frequently deleted (53). Although clearly recurrently deleted in many cancers, a key tumor suppressor gene located in this region has not yet been identified. Based on the data that we provide here, NEK4 may be one target of this region of recurrent deletion.
Since NEK4-suppressed cells eventually enter replicative senescence and do not proliferate indefinitely, it is likely that loss of NEK4 requires the concomitant alteration of other oncogenes and tumor suppressors to initiate transformation. While Nek4 may be involved in the efficient recognition and repair of DNA damage, loss of Nek4 should not necessarily result in a hypermutagenic state. Nevertheless, loss of Nek4 could decrease the efficiency of repair when DNA damage occurs through other means, and extension of the time to replicative senescence might allow the prolonged expansion of premalignant cells, further allowing the possibility of subsequent mutations and selection for abnormal variants.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Hahn and Cichowski laboratories for assistance and helpful comments and Matthew Meyerson, Kumiko Tanaka, Jordi Barretina, Heidi Greulich, Hongbin Ji, and Kwok-Kin Wong for assistance with lung cancer cell line data and reagents. We thank Daosong Xu and Martin Hemler for the HT-1080 cell line.
C.L.N. was supported by an American Cancer Society postdoctoral fellowship (PF-09-117-01-CCG), and R.P was supported by a Howard Hughes predoctoral fellowship. This work was supported in part by NIH/NIA grant R01 AG023145 and by the H. L. Snyder Medical Foundation.
Footnotes
Published ahead of print 30 July 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. An J, et al. 2010. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol. Biol. 11: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balajee AS, Geard CR. 2001. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 29: 1341–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beausejour CM, et al. 2003. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 22: 4212–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bodnar AG, et al. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352 [DOI] [PubMed] [Google Scholar]
- 5. Bond JA, et al. 1995. Mutant p53 rescues human diploid cells from senescence without inhibiting the induction of SDI1/WAF1. Cancer Res. 55: 2404–2409 [PubMed] [Google Scholar]
- 6. Bond JA, et al. 1999. Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol. 19: 3103–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bond JA, Wyllie FS, Wynford-Thomas D. 1994. Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene 9: 1885–1889 [PubMed] [Google Scholar]
- 8. Braig M, et al. 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665 [DOI] [PubMed] [Google Scholar]
- 9. Brown JP, Wei W, Sedivy JM. 1997. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277: 831–834 [DOI] [PubMed] [Google Scholar]
- 10. Campisi J, d'Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8: 729–740 [DOI] [PubMed] [Google Scholar]
- 11. Chen GI, Gingras AC. 2007. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 42: 298–305 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Chen CF, Riley DJ, Chen PL. 2011. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 10: 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, Chen PL, Chen CF, Jiang X, Riley DJ. 2008. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 7: 3194–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Z, et al. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collado M, et al. 2005. Tumour biology: senescence in premalignant tumours. Nature 436: 642. [DOI] [PubMed] [Google Scholar]
- 16. Courtois-Cox S, et al. 2006. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 10: 459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dickson MA, et al. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20: 1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dimri GP, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DiRenzo J, et al. 2002. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer Res. 62: 89–98 [PubMed] [Google Scholar]
- 20. Drouet J, et al. 2005. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J. Biol. Chem. 280: 7060–7069 [DOI] [PubMed] [Google Scholar]
- 21. Dulic V, Beney GE, Frebourg G, Drullinger LF, Stein GH. 2000. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol. Cell. Biol. 20: 6741–6754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. el-Deiry WS, et al. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- 23. Gallimore PH, et al. 1997. Adenovirus type 12 early region 1B 54K protein significantly extends the life span of normal mammalian cells in culture. J. Virol. 71: 6629–6640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gire V, Roux P, Wynford-Thomas D, Brondello JM, Dulic V. 2004. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 23: 2554–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gire V, Wynford-Thomas D. 1998. Reinitiation of DNA synthesis and cell division in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol. Cell. Biol. 18: 1611–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hahn WC. 2002. Immortalization and transformation of human cells. Mol. Cells 13: 351–361 [PubMed] [Google Scholar]
- 27. Hahn WC, et al. 1999. Creation of human tumour cells with defined genetic elements. Nature 400: 464–468 [DOI] [PubMed] [Google Scholar]
- 28. Harley VR, et al. 1990. Vaccinia virus expression and sequence of an avian influenza nucleoprotein gene: potential use in diagnosis. Arch. Virol. 113: 133–141 [DOI] [PubMed] [Google Scholar]
- 29. Hayflick L, Moorhead PS. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25: 585–621 [DOI] [PubMed] [Google Scholar]
- 30. Herbig U, Sedivy JM. 2006. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech. Ageing Dev. 127: 16–24 [DOI] [PubMed] [Google Scholar]
- 31. Kachnic LA, et al. 1999. The ability of p53 to activate downstream genes p21(WAF1/cip1) and HDM2, and cell cycle arrest following DNA damage is delayed and attenuated in scid cells deficient in the DNA-dependent protein kinase. J. Biol. Chem. 274: 13111–13117 [DOI] [PubMed] [Google Scholar]
- 32. Kim NW, et al. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266: 2011–2015 [DOI] [PubMed] [Google Scholar]
- 33. Kiyono T, et al. 1998. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396: 84–88 [DOI] [PubMed] [Google Scholar]
- 34. Levedakou EN, et al. 1994. Two novel human serine/threonine kinases with homologies to the cell cycle regulating Xenopus MO15, and NIMA kinases: cloning and characterization of their expression pattern. Oncogene 9: 1977–1988 [PubMed] [Google Scholar]
- 35. Li J, Stern DF. 2005. DNA damage regulates Chk2 association with chromatin. J. Biol. Chem. 280: 37948–37956 [DOI] [PubMed] [Google Scholar]
- 36. Li QL, et al. 2002. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109: 113–124 [DOI] [PubMed] [Google Scholar]
- 37. Lundberg AS, Hahn WC, Gupta P, Weinberg RA. 2000. Genes involved in senescence and immortalization. Curr. Opin. Cell Biol. 12: 705–709 [DOI] [PubMed] [Google Scholar]
- 38. Mao JH, et al. 2004. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432: 775–779 [DOI] [PubMed] [Google Scholar]
- 39. McConnell BB, Starborg M, Brookes S, Peters G. 1998. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 8: 351–354 [DOI] [PubMed] [Google Scholar]
- 40. Melixetian M, Klein DK, Sorensen CS, Helin K. 2009. NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat. Cell Biol. 11: 1247–1253 [DOI] [PubMed] [Google Scholar]
- 41. Michaloglou C, et al. 2005. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436: 720–724 [DOI] [PubMed] [Google Scholar]
- 42. Misteli T, Soutoglou E. 2009. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 10: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moffat J, et al. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- 44. Moniz L, Dutt P, Haider N, Stambolic V. 2011. Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div. 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moniz LS, Stambolic V. 2011. Nek10 mediates G2/M cell cycle arrest and MEK autoactivation in response to UV irradiation. Mol. Cell. Biol. 31: 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18: 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nguyen CL, Eichwald C, Nibert ML, Munger K. 2007. Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J. Virol. 81: 13533–13543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ramirez RD, et al. 2001. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 15: 398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rogan EM, et al. 1995. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell. Biol. 15: 4745–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- 51. Shay JW, Wright WE, Werbin H. 1991. Defining the molecular mechanisms of human cell immortalization. Biochim. Biophys. Acta 1072: 1–7 [DOI] [PubMed] [Google Scholar]
- 52. Spardy N, et al. 2007. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J. Virol. 81: 13265–13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tonon G, et al. 2005. High-resolution genomic profiles of human lung cancer. Proc. Natl. Acad. Sci. U. S. A. 102: 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Steensel B, de Lange T. 1997. Control of telomere length by the human telomeric protein TRF1. Nature 385: 740–743 [DOI] [PubMed] [Google Scholar]
- 55. Vaziri H, Benchimol S. 1998. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 8: 279–282 [DOI] [PubMed] [Google Scholar]
- 56. Voorhoeve PM, Agami R. 2003. The tumor-suppressive functions of the human INK4A locus. Cancer Cell 4: 311–319 [DOI] [PubMed] [Google Scholar]
- 57. Wei W, Sedivy JM. 1999. Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp. Cell Res. 253: 519–522 [DOI] [PubMed] [Google Scholar]
- 58. Wright WE, Shay JW. 1992. The two-stage mechanism controlling cellular senescence and immortalization. Exp. Gerontol. 27: 383–389 [DOI] [PubMed] [Google Scholar]
- 59. Yan Y, Ouellette MM, Shay JW, Wright WE. 1996. Age-dependent alterations of c-fos and growth regulation in human fibroblasts expressing the HPV16 E6 protein. Mol. Biol. Cell 7: 975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.