

Abstract
During translation initiation in Saccharomyces cerevisiae, an Arg- and Ser-rich segment (RS1 domain) of eukaryotic translation initiation factor 4G (eIF4G) and the Lys-rich segment (K-boxes) of eIF2β bind three common partners, eIF5, eIF1, and mRNA. Here, we report that both of these segments are involved in mRNA recruitment and AUG recognition by distinct mechanisms. First, the eIF4G-RS1 interaction with the eIF5 C-terminal domain (eIF5-CTD) directly links eIF4G to the preinitiation complex (PIC) and enhances mRNA binding. Second, eIF2β-K-boxes increase mRNA binding to the 40S subunit in vitro in a manner reversed by the eIF5-CTD. Third, mutations altering eIF4G-RS1, eIF2β-K-boxes, and eIF5-CTD restore the accuracy of start codon selection impaired by an eIF2β mutation in vivo, suggesting that the mutual interactions of the eIF segments within the PIC prime the ribosome for initiation in response to start codon selection. We propose that the rearrangement of interactions involving the eIF5-CTD promotes mRNA recruitment through mRNA binding by eIF4G and eIF2β and assists the start codon-induced release of eIF1, the major antagonist of establishing tRNAiMet:mRNA binding to the P site.
INTRODUCTION
In eukaryotic translation, initiation factors (eIFs) promote dissociation of vacant 80S (canonical initiation) or posttermination (recycling) ribosomes and assist binding of Met-tRNAiMet and 5′-capped mRNA to the 40S subunit to form 43S and 48S preinitiation complexes (PICs), respectively (for reviews, see references 15 and 17). eIF2 binding of Met-tRNAiMet is dependent on GTP bound to its γ subunit, delivering it to the 40S subunit. The 43S complex additionally contains eIF1A, eIF1, eIF3, and eIF5, the latter three forming a multifactor complex (MFC) with the eIF2/GTP/Met-tRNAiMet ternary complex (TC) (3, 39). Except for eIF3, which binds the solvent side of the 40S subunit (38), these factors bind the decoding site of the 40S subunit and together play a role in positioning Met-tRNAiMet in the P site. The cytoplasmic mRNA cap-binding complex eIF4F is made of three subunits, the major scaffold eIF4G, m7G-cap-binding subunit eIF4E, and the mRNA helicase, eIF4A. eIF4F primes the 5′-proximal area of the m7G-capped mRNA by the action of eIF4A and recruits the 43S complex to the primed region of the mRNA to form the 48S PIC. According to the scanning model, the 5′-proximal 48S complex migrates downstream along the mRNA, searching for the start codon.
Currently, much effort is devoted to understanding the mechanism by which these initiation factors coordinately regulate the state of the 48S PIC. During scanning, the PIC is presumed to be in the scanning-competent open state, and once a start codon is reached, it is shifted to the closed state, establishing tRNAiMet:mRNA binding in the P site. Thus, the PIC in the closed state does not scan; instead, it tightly binds the selected start codon to promote formation of the 40S initiation complex (IC) (6, 22). eIF1 is present in the PIC in the open state and plays a major role in antagonizing stable tRNAiMet:mRNA binding to the P site, thus suppressing transition to the closed state (27, 30). The release of eIF1 in response to AUG selection is considered to be the key regulatory event during the ribosome response to a start codon (10). eIF5 promotes GTP hydrolysis of eIF2-GTP upon or subsequent to PIC formation (GTPase activation protein or GAP function), but the GDP and inorganic phosphate resulting from the hydrolysis remain bound to eIF2. Inorganic phosphate is released after eIF1 release, allowing eIF2-GDP to be ejected from the ribosome (2).
While eIF1 release is considered primarily as a ribosomal response to start codon recognition, the contribution of other factors in retaining eIF1 in the open complex and facilitating its release on start codon selection remains elusive. Recently, a novel role for eIF5 in antagonizing eIF1 function by promoting its release has been proposed (27), but the mechanism by which eIF5 promotes eIF1 release is unknown. Another open question is how mRNA binding to the PIC is stabilized with the mRNA entry channel open (30).
eIF4G is the major scaffolding subunit of eIF4F, linking eIF4A to the m7G-cap of the mRNA and thereby recruiting mRNA to the PIC (24, 43). Mammalian and most other metazoan eIF4G contain three HEAT domains, the first two of which serve as eIF4A-binding sites and the last as the binding site for Mnk eIF4E kinases (17). Yeast eIF4G lacks the last two HEAT domains but retains the first HEAT domain, which is sufficient to bind eIF4A. Despite this structural difference, the functions of mammalian and yeast eIF4G are very similar, most notably in their ability to bind eIF4A, eIF4E, and the poly(A)-binding protein (Pab). In addition, conserved RS domains of eIF4G, here termed RS1 and RS2, are located N and C terminally to the MIF4G HEAT domain (26). While the RS1 domain of mammalian eIF4G1 is shown to promote scanning for a start codon under the control of the internal ribosome entry site (31), the precise role of these disordered segments is unknown.
The N-terminal tail (NTT) of eIF2β contains three lysine-rich segments called K-boxes (4). eIF2β-NTT is the common binding site for eIF5 and eIF2Bε, promoting eIF2 TC recruitment to the ribosome (3) and the reactivation of eIF2 by guanine nucleotide exchange (4), respectively. Interestingly, eIF2β-NTT also binds mRNA via its K-boxes, suggesting a role for eIF2β in mRNA recruitment (20). Moreover, depletion of eIF4G in yeast does not compromise mRNA binding to the 40S subunit in vivo, suggesting that additional factors, including eIF2 and eIF3, are involved in stably anchoring mRNA to the 40S subunit (19). Furthermore, alanine substitutions altering K-boxes increase the accuracy of start codon selection, suggesting that eIF2β is also involved in a subsequent step in AUG recognition (20). The precise roles for eIF2β K-boxes in contributing to mRNA binding to the PIC and start codon selection have not been defined.
To answer the questions mentioned above, we have studied the positively charged, disordered segments of eIF4G and eIF2β, the RS1 domain of eIF4G, and the K-boxes of eIF2β, which we find bind eIF1, eIF5, and mRNA (7, 14, 25). Our data indicate that eIF4G-RS1 interaction with eIF5 is directly responsible for eIF4G-mediated mRNA binding to the 43S complex, and that the eIF2β-K-boxes stabilize mRNA binding to the 40S subunit.
In selecting AUG start codons, we find that the mutation of eIF4G-RS1 or eIF5 increases the accuracy of the start codon. Our data support a model whereby the eIF2β-K-boxes and eIF4G-RS1, along with the eIF5 C-terminal domain (eIF5-CTD, or eIF5-C), are involved in shifting the ribosome to a closed state after mRNA binding. Based on previous studies and the data presented here, we propose a detailed protein-protein interaction model explaining how the rearrangement of interactions involving eIF2β-NTT and eIF4G-RS1 leads to eIF1 release in response to start codon recognition.
MATERIALS AND METHODS
Yeast strains and methods.
Plasmids used in this study (listed in Table S1 in the supplemental material) were generated as described in the supplemental text using oligodeoxyribonucleotides listed in Table S2 in the supplemental material. The yeast Saccharomyces cerevisiae genome encodes two copies of eIF4G, eIF4G1 (encoded by TIF4631) and eIF4G2 (encoded by TIF4632). Two series of S. cerevisiae tif4632 strains were used in this study and are listed in Table S3 in the supplemental material. One is derived from the GCN2+ strain YAS1955 (see Table S3, column 5) (41), which was employed to study the Gcn2p-dependent general control response to 3AT-induced histidine starvation. The other is derived from KAY220 (see Table S3, column 6) (43), carrying his4-303 with the start codon altered to AUU, a phenotypic reporter for stringency of start codon selection. They were generated by introducing wild-type or mutant HA-TIF4632 TRP1 plasmid to YAS1948 (41) and KAY173 (43), respectively, and evicting the resident TIF4632 URA3 plasmid by the growth of the resulting transformants in the presence of fluoroorotic acid (FOA) (plasmid shuffling).
KAY976 (TIF5 his4-306 ura3), KAY978 (tif5-AN1 his4-306 ura3), KAY979 (tif5-AN1_ΔE396 his4-306 ura3), KAY977 (tif5-BN1-FL his4-306 ura3), and KAY936 (ssu2-1-FL his4-303 ura3) were constructed similarly by FOA-mediated plasmid shuffling with the parental strain EY920 [MATa ura3-52 leu2-3 leu2-112 trp1Δ-63 his4-306(UUG) tif5Δ::hisG p(scURA3 TIF5)] (36) and the single-copy LEU2 plasmids YCpL-TIF5-FL (4), YCpL-tif5-AN1, YCpL-tif5-AN1_ΔE396, YCpL-tif5-BN1-FL (44), and YCpL-tif5-G62S (K. Asano, personal collection).
Standard yeast molecular biology methods, including growth and β-galactosidase assays, were used throughout (21).
Biochemical methods.
Glutathione S-transferase (GST) pulldown assays of 35S-labeled proteins (35), 32P-β-globin mRNA (44), and the purified 40S subunit (28), as well as the 32P-MFA2 mRNA recruitment assay in yeast cell extracts (5), were conducted as described previously.
eIF2β-dependent mRNA recruitment to the 40S subunit was assayed as follows. 32P-MFA2 mRNA (24,500 cpm) prepared as described previously (5), purified 40S subunit (0.144 A260 U or 240 pmol) (1), and appropriate recombinant eIF fragments (10 μg or 260 pmol of GST-eIF2β or 8 μg or 400 pmol of His-eIF5-B6) were incubated in 50 μl binding buffer [20 mM HEPES-KOH, 75 mM potassium acetate (KOAc), 2 mM Mg(OAc)2, 1 mM EDTA, 1 mM dithiothreitol] at 4°C for 90 min. The samples then were fixed with 1% HCHO on ice for 5 min, quenched with 0.1 M glycine, and layered on a 5 to 40% sucrose gradient containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 1 mM MgCl2. After ultracentrifugation at 39,000 rpm for 4.5 h, the gradient sample was fractionated by an Isco gradient fractionator. Half of the fractions were used for scintillation counting, and the other half were used for immunoblotting with anti-SUI3 or anti-RpsO antibodies.
RESULTS
Yeast model to study eIF4G.
Like mammals, yeast genomes encode two copies of eIF4G, eIF4G1 and eIF4G2, which are highly homologous to each other. The deletion of both TIF4631 (encoding eIF4G1) and TIF4632 (encoding eIF4G2) is lethal (13), and either copy can complement the lethal effect of the deletion, provided that the copy is expressed from the stronger tif4631 promoter (40). Both eIF4G1 and eIF4G2 are able to bind eIF1 and eIF5 (14, 43). Since we narrowed down the minimal eIF1- and eIF5-binding sites in eIF4G2 (14), we have chosen to study eIF4G2 in this communication and further dissect its interaction with eIF1 and eIF5. Accordingly, we used a tif4631 tif4632 double deletion strain carrying a plasmid expressing wild-type or mutant HA-tagged eIF4G2 under the tif4631 promoter. The bottom four lines of Fig. 1A depict eIF4G2 alleles used in this study.
Fig 1.

Minimal binding domains of eIF4G2 (Tif4632p) for eIF1, eIF4A, eIF5, and mRNA. (A) Primary structure of yeast eIF4G2 mutations used in this study with defined binding sites for Pab1p, eIF4A, and eIF4E (gray boxes). Boldface underlining denotes RNA-binding segments. Dark boxes indicate the RS domains for RNA binding (43). Light gray boxes are homologous to RNA1, an RNA-binding domain in yeast eIF4G1 (9), and box 3, displaying high homology among fungal eIF4G (b3) (30). Tables to the left summarize the results of interaction assays with RNA and Pab1p. NT, not tested; ΔX, deletion of aa 847 to 914; ΔN, deletion of aa 1 to 237. (B, C, E, and F) GST-eIF4G2 fusion proteins (2 to 5 μg of full-length products, indicated by arrowheads in the top gel), listed across the top, were bound to 35S-labeled proteins synthesized in rabbit reticulocyte lysate in 200 μl binding buffer. GST fusion-containing complexes together with 20% input amounts of the reaction mixtures were analyzed by SDS-PAGE followed by Coomassie staining (top) and autoradiography (bottom). Horizontal arrows indicate the full-length products. The downward arrow indicates a reproduced decrease in the interaction compared to the control (n = 3). (B) Binary interactions of GST or the indicated GST-eIF4G2 segments with 35S-eIF4A, eIF1, and eIF5-C. (C) The effect of RS1 mutation tif4632-8* (8*) or tif4632-7R (7R) (as defined in Table S3 in the supplemental material) on eIF4G2 binding to eIF1 and eIF5. W, wild type. (D) The effect of tif4632-7R on GST-eIF4G-C binding to 32P-β-globin mRNA, as examined by Northwestern blotting (8). Left, Coomassie staining of proteins used. Right, autoradiography of proteins bound to 32P-β-globin mRNA. Arrowheads, locations of full-length products. W, wild type. (E and F) Indicated GST-eIF4G2 proteins were bound to 35S-eIF5-C in the presence of 20 μg His-eIF1 (E) or 35S-eIF1 in the presence of 20 μg His-eIF5-C (F). Arrowheads indicate locations of bands representing His-eIF1 or His-eIF5-C bound. (G) GST-eIF1 and its M4 and M5 mutant derivatives were allowed to bind 35S-eIF4G2-A, and the 35S proteins bound were analyzed together with 20% input amounts (lane 1).
The eIF4G2 RS1 domain binds eIF1, eIF5, and mRNA.
Optimal interaction of yeast eIF4G2 with eIF1 and eIF5 requires (i) its intact HEAT domain (amino acids [aa] 557 to 812; Fig. 1A shows its location) and (ii) its Arg- and Ser-rich segment (RS1) located N terminal to the HEAT domain (14). To better characterize the RS1 segment of eIF4G2, we determined the minimal eIF1- and eIF5-binding segment of RS1 by truncation analysis and crucial amino acids therein by point mutations. Figure 1A lists truncated versions of eIF4G2 used here for interaction assays; the alphabetical designation of the fragments is used throughout the text (see Fig. 4A for additional constructs). eIF4G2_fragment B (frgB; eIF4G2439-577) was the previously determined minimal binding segment for eIF1 and eIF5, but it still contained a part of the HEAT domain, making it difficult to draw a definitive conclusion about the contribution of RS1 to eIF1 and eIF5 binding (Fig. 1B, lane 5). As shown in Fig. 1B, a GST-eIF4G2 fusion carrying only the RS1 domain (frgC; eIF4G2439-513) was sufficient for binding to eIF1 and eIF5-CTD (eIF5-C), which is minimally required for eIF4G2 binding. In contrast, eIF4G2-frgC did not interact with the mRNA helicase eIF4A (Fig. 1B, autoradiography-labeled 35S-eIF4A), confirming the specificity for the interaction involving frgC.
Fig 4.

eIF5 regulates mRNA binding by eIF4G2 and eIF2β in vitro. (A) Primary structure of eIF4G2 with defined binding sites for the indicated initiation factors. Thick lines below the structure represent the portion of eIF4G2 polypeptide found in each construct (see Fig. S3 in the supplemental material for experimental design). The chart on the right summarizes the fold change in mRNA binding caused by eIF5 in a logarithmic scale, with P values listed in the box to the right. A positive value indicates fold increase, while a negative value indicates fold decrease. Bars indicate standard deviations (SD). GST-eIF2β-Ν was also tested, as shown at the bottom. (B) Interaction of eIF2β-NTT with the 40S subunit. GST (5 μg), GST-eIF2β-N (10 μg), and GST-eIF1 (5 μg) were bound to the 40S subunit purified from the wild-type (WT) or A1193U strain with (+mRNA) or without (blank) nonradiolabeled β-globin mRNA (10 μg). The bound 40S subunit was detected by anti-Rps0 antibodies (bottom gels). Top gel, Coomassie staining of proteins used in this study. In, 25% input amount of the 40S subunit. N.T., not tested; however, the same preparation of the mutant 40S subunit used here did not bind to GST-eIF1 in a different experiment (28). (C) Summary of percentages of the 40S subunit bound to GST-eIF2β-N calculated from lanes 1 and 3 in panel B. Parentheses indicate values from an independent experiment.
eIF4G2-frgC (positions 439 to 513) is a hydrophilic segment enriched in Arg (17%), Ser (17%), Lys (8%), Pro (8%), and Asp (9%). To determine which amino acids in RS1 are responsible for its interaction with eIF1 and eIF5, we started by examining the effect of mutations altering the most prominent category of amino acids, positively charged Arg and Lys. Thus, we created tif4632-8*, altering R473, K491, and R501 to Ile, Arg, and Ser, respectively, and tif4632-7R, altering R487, R492, R493, R496, R501, R502, and R505 all to glutamine, a similarly sized polar amino acid. The latter mutation of the RS1 domain altering the positively charged cluster, but not the former mutation, abolished its binding to eIF1 and eIF5-CTD (Fig. 1C, lanes 3 to 5). However, the same mutation constructed in the context of the entire C-terminal portion of eIF4G2 (eIF4G2-frgA) did not abolish but did reduce the binding to these factors (Fig. 1C, lanes 6 and 7). The latter observation supports the idea that eIF4G2 has more than one region that participates in eIF1 and eIF5 binding that is dependent on the integrity of the HEAT domain (14).
Since eIF4G2-frgC also binds RNA (43), we tested the effect of tif3632-7R on mRNA binding by this fragment. Interestingly, tif4632-7R also abolished 32P-labeled β-globin mRNA binding to eIF4G2-frgC, which was immobilized onto a nitrocellulose membrane (Northwestern assay) (Fig. 1D, lanes 4 to 6). Together, these results indicate that the Arg-rich motif in the RS1 domain is responsible at least in part for its binding to eIF1, eIF5-CTD, and mRNA.
eIF1 and eIF5-CTD compete for eIF4G2-RS1.
Competition for binding by two distinct proteins forms the basis of remodeling protein complexes and potentially complex functions via changes in protein-protein interactions. We previously showed that eIF1 and eIF5 compete for binding to the C-terminal half of eIF4G2 (eIF4G2-frgA) (14), suggesting that these factors are involved in such protein-mediated remodeling. However, this experiment used a GST-eIF4G2 construct produced in a low yield, which bound only ∼1% of the 35S-eIF5-C added (see Fig. 3D in reference 14). Therefore, we were unable to quantitatively evaluate the competition between eIF5-C and eIF1. To overcome this problem, we tested competition with a GST-GB (streptococcal protein G B1 domain) double fusion form of eIF4G2 that is better expressed and purifies to produce better yields of GST-eIF4G2 (43). As shown in Fig. 1E, lanes 4 and 5, GST-GB-eIF4G2-frgA binding to eIF5-C was strongly inhibited by His-eIF1. Similarly, GST-GB-eIF4G2-frgA binding to eIF1 was inhibited by His-eIF5-C, albeit more weakly (Fig. 1F, lanes 3 and 4). Importantly, eIF5-C and eIF1 also competed for binding to the minimal segment, eIF4G2-frgC (Fig. 1E, lanes 6 and 7, and F, lanes 5 and 6), but the competition appeared to be weaker than that for eIF4G2-frgA. Together, these results confirm that the interaction of eIF4G2 with eIF1 and eIF5 are mutually competitive and also suggests that efficient competition between eIF1 and eIF5 requires the HEAT and RS2 domains.
Fig 3.

Interaction of eIF4G2-RS1 with eIF5 mediates mRNA recruitment to the 43S complex. (A and B) Transformants bearing the indicated mutations (WT, YAS1955; 7R, KAY874; and gcn2Δ, KAY24) were diluted and spotted onto SC-His plates without (−3AT) or with (+3AT) the indicated concentrations of 3AT, and the plates were incubated for 3 days (−3AT) and 5 days (+3AT), respectively. In row 5 of panel B are immunoblots of 5 μg whole-cell extracts from KAY24 transformants (rows 7 and 9) with antibodies against the proteins labeled to the right. (C) eIF4G interaction with eIF5 promotes mRNA recruitment to the 40S subunit in vitro. Cell-free translation extracts prepared from strains with the indicated mutations (lower box) were incubated with 32P-labeled poly(A)-tailed MFA2 mRNA with or without FLAG-eIF5 and fractionated by sucrose gradient velocity sedimentation. The assay was analyzed and presented as described for Fig. 2E, except the reaction volume was 200 μl and 80 μg of eIF5 was added.
Mutational studies indicated that the conserved acidic and basic faces of the eIF5-CTD interact with eIF4G2-frgA (44). To learn which part of eIF1 binds eIF4G2, we performed GST pulldown assays with eIF1 mutant proteins bearing altered Lys-rich hydrophobic surfaces (KH), Lys- and Arg-rich (KR) surfaces of the globular domain, or NTT of eIF1 (see Fig. S1A and B in the supplemental material). Mutations altering several amino acids in the KH and KR surfaces abolished GST-eIF1 binding to eIF4G2-frgA (Fig. 1G, M4 and M5). Conversely, GST-eIF4G2-frgA binding to eIF1 was decreased significantly by the same mutations and less profoundly by M1, M2, and M3 altering eIF1-NTT (see Fig. S1C). Thus, the KH and KR regions of eIF1 are the major eIF1 binding surfaces for eIF4G, similar to a previous report indicating that the KH and KR regions bind to eIF5-CTD and eIF3c (33).
Three unstructured segments of yeast eIF4G2 are capable of binding RNA and are involved in mRNA recruitment to the 40S subunit.
To contrast the role of eIF4G2-RS1 in translation initiation with that of other RNA-binding domains of eIF4G2, we sought to identify additional RNA-binding domains in eIF4G2. We recently showed that eIF4G2-frgC, encompassing RS1 and the C-terminal segment of eIF4G2 (see Fig. 4A, frgH) containing RS2, binds 32P-β-globin mRNA in vitro (43). Of the GST-eIF4G2 fragments tested, we identified two additional fragments of eIF4G2 that influenced β-globin mRNA binding, eIF4G2-frgG (eIF4G21-176) and a longer segment, eIF4G2-frgF (eIF4G21-239), both of which contain a distinct RNA-binding domain located in the N-terminal region (see Fig. S2A, lanes 2 and 3, in the supplemental material). These results localize three RNA-binding domains within eIF4G2, similar to yeast eIF4G1 (9).
To examine the in vivo function of individual RNA-binding domains, we generated yeast expressing eIF4G2 mutants lacking these sites as the sole copy of eIF4G. Because tif4632-ΔA encoding eIF4G lacking the entire C-terminal tail did not express in yeast (R. Watanabe and K. Asano, unpublished data), we generated a strain bearing tif4632-ΔX, lacking half of RS2 to the C-terminal end of the protein (Fig. 1A). In addition, we generated strains expressing tif4632-ΔN lacking the N-terminal RNA-binding domain and tif4632-7R, which alters 7 Arg amino acids that are required for binding to RNA, eIF1, and eIF5 (Fig. 1). All three alleles expressed the eIF4G2 mutants at a level equivalent to or higher than that of wild-type eIF4G2 (Fig. 2A). All strains expressing eIF4G2 mutants grew normally at a permissive temperature of 30°C, but they also all grew slowly at a more restrictive temperature of 37°C (Fig. 2B, sections 1 to 3, shows tif4632-7R; data not shown for others). Thus, we concluded that all RNA-binding domains of eIF4G2 play important roles in maintaining cell viability at 37°C.
Fig 2.

Yeast phenotypes of mutants with altered RNA-binding sites of eIF4G2. (A) The expression of eIF4G2 mutants used in this study (YAS1955 derivatives) (Fig. 1A; also see Table S3 in the supplemental material). Twenty and 40 μg total protein from whole-cell extracts of the indicated strains were used for immunoblotting with anti-HA (to detect HA-eIF4G2) and antitubulin antibodies (loading control). (B) Temperature- and 3AT-sensitive growth of tif4632-7R was assayed by spotting 5 μl of 0.15 A600 U culture (columns 1) and 10-fold serial dilutions (columns 10−1 and 10−2) onto SC-His plates without (−) and with (+) 10 mM 3AT and incubated at the indicated temperatures for 3 days. (C) eIF4G2 mutations impair mRNA recruitment to the 40S subunit in vitro. Cell-free translation mixture with 32P-labeled poly(A)-tailed MFA2 mRNA was fractionated by sucrose gradient velocity sedimentation. 32P counts in relevant fractions are shown with an arrow indicating the fraction containing free 40S subunit. (D) Effect of tif4632-7R on GCN4-lacZ expression from p180. The graph summarizes β-galactosidase activity of YAS1955 (WT) and KAY901 (tif4632-7R [7R]) transformants carrying p180 in the presence (+) or absence (−) of 10 mM 3AT. The P value for differences between the values in columns 1 and 3 is presented.
Cell extracts were prepared from eIF4G2 mutant strains to test whether 32P-labeled model mRNA [poly(A)-tailed MFA2 mRNA] is as efficiently recruited to 40S subunits by mutant eIF4G2 proteins. The addition of the nonhydrolyzable GTP analog GMPPNP to block hydrolysis of nucleotide bound to eIF2 allowed us to evaluate assembly of 48S complexes prior to start codon selection. All three mutants displayed a defect in stable mRNA binding to the 40S subunit (Fig. 2C; also see Fig. S3A in the supplemental material for statistical analysis of binding). Minor growth defects of yeast caused by the tif4632 mutations and reduced mRNA binding to 40S ribosomal subunits in extracts containing mutant eIF4G2 proteins suggest there is redundancy built into eIF4G2 RNA binding for recruitment of mRNA to the 40S subunit.
We observed that the strongest mRNA-binding defect was caused by tif4632-ΔN (see Fig. S3A in the supplemental material), and we considered that this may be due to alteration of the Pab1p-binding site (Fig. 1A). In agreement with this idea, recent studies on yeast eIF4G1 showed that box 3, which is conserved among fungal eIF4G N termini, is responsible for Pab1p binding (29). In eIF4G2, box 3 is located within the region deleted by tif4632-ΔN (labeled b3 in Fig. 1A). To test if box 3 is required for Pab1p binding to eIF4G2, we performed a pulldown assay with GST-eIF4G2 fragments and 35S-Pab1p. eIF4G2-frgF (eIF4G21-239) or a still longer construct, eIF4G2-frgE (eIF4G21-514), but not eIF4G2-frgG (eIF4G21-172), bound Pab1p (see Fig. S2B in the supplemental material), confirming that a region including box 3 is the Pab1p-binding site. Thus, the N-terminal RNA- and Pab1p-binding sites are functionally conserved between yeast eIF4G1 and eIF4G2.
The eIF4G2-RS1 interaction with eIF5 mediates mRNA recruitment to the 43S complex.
During the characterization of the tif4632-7R mutant, we found that it showed sensitivity to 3-aminotriazole (3AT), the His3p inhibitor, at the semirestrictive temperature of 34°C (Fig. 2B, compare sections 2 and 4). Histidine starvation caused by 3AT activates Gcn2p eIF2α kinase, which in turn promotes translation of Gcn4p, the transcriptional activator of amino acid biosynthesis enzymes, including His3p. GCN4-lacZ reporter assays indicated, however, that GCN4 is normally induced by 3AT in tif4632-7R cells (Fig. 2D, columns 2 and 4), although the basal GCN4 expression in the absence of 3AT was increased in these cells (columns 1 and 3) (this unexpected finding is discussed below). Because tif4632-7R impairs mRNA binding to the ribosome (Fig. 2C), we presumed that the 3AT sensitivity of tif4632-7R would be a result of a general translation defect; a strong eIF4G function would be required to conduct general translation in the presence of 3AT, which inhibits Met-tRNAiMet binding via eIF2 phosphorylation. Thus, an impaired eIF4G function would result in slower growth in the presence of 3AT. If this is the case, we reasoned that 3AT sensitivity would be suppressed by overexpression of critical binding partners of eIF4G2-RS1, eIF1, eIF5, and potentially eIF4A due to mass action effects. As shown in Fig. 3A, the overexpression of eIF5, but not eIF1 or eIF4A, suppressed the 3AT sensitivity of tif4632-7R. This is not due simply to a general change in growth of yeast overexpressing eIF5, since overexpression of eIF5 does not affect the growth of wild-type cells in the presence of 3AT (Fig. 3B, row 2). Thus, the eIF4G2-RS1 interaction with eIF5 mediates the critical function impaired by the tif4632-7R mutation.
A caveat to this interpretation was the previous observation that eIF5 overexpression can induce GCN4 translation by inhibiting eIF2B-catalyzed eIF2 recycling by guanine nucleotide exchange, thereby converting 3AT-sensitive gcn2Δ strains to 3AT resistance (Gcd− phenotype) (Fig. 3B, rows 7 and 8) (37). Thus, eIF5 overexpression in tif4632-7R cells could increase 3AT resistance by constitutively expressing GCN4 without restoring defective eIF4G2 function. To rule out this possibility, we examined the effect of tif5-Δ20, a deletion of a 20-amino-acid-long segment of eIF5 that was recently identified as a segment inhibiting GDP release from eIF2 (GDI activity) (18). The GDI activity of eIF5 antagonizes eIF2B-mediated eIF2 reactivation; thus, deletion of this domain eliminates the ability of eIF5 to decrease ternary complex (TC) abundance and hence its ability to confer 3AT resistance (Fig. 3B, rows 8 and 9) without changing the eIF5 expression level (Fig. 3B, section 5). The overexpression of eIF5-Δ20 still suppressed the 3AT sensitivity by tif4632-7R (Fig. 3B, rows 5 and 6), in support of the idea that the phenotype was suppressed by eIF5 mass action but not by enhancing GCN4 induction through TC levels.
To directly test if the eIF4G2-RS1 interaction with eIF5 mediates mRNA recruitment to the ribosome, we examined the effect of eIF5 addition on the 32P-MFA2 mRNA recruitment defect observed in eIF4G2-7R cell extracts. The addition of eIF5 restored the mRNA binding defect that was observed in the mutant extracts (Fig. 3C; also see Fig. S3B in the supplemental material for statistical analysis), demonstrating that the eIF4G2-RS1 interaction with eIF5 mediates mRNA recruitment to the ribosome. The restoration of mRNA recruitment under these conditions is specific to eIF5, because addition of eIF1 did not restore mRNA binding (see Fig. S4A in the supplemental material), and addition of eIF5-C did not restore the mRNA binding defect (see Fig. S4B), indicating that the eIF5-CTD is not sufficient to mediate stable complex formation with mRNA.
eIF5 stimulates mRNA binding by eIF4G in vitro.
In an effort to delineate the mechanism by which eIF4G interaction with eIF5 promotes mRNA binding by the 43S complex, it was found that the addition of full-length eIF5 or eIF5-C (CTD) increases 32P-β-globin mRNA binding to eIF4G2-frgA by 2-fold (Fig. 4A, rows 1 and 2). GST pulldown assays were used with recombinant GST-eIF4G2 and 32P-β-globin mRNA to tease apart the contributions provided by eIF5 and eIF4G2 to mRNA binding to the ribosome. Each of the recombinant GST-eIF4G2 constructs used here bound 10 to 20% of the 32P-mRNA in the absence of eIF5 (see Fig. S5A in the supplemental material). To ensure accurate comparison, the glutathione resin bound to GST fusion proteins was divided equally into two tubes, eIF5 proteins (10 μg) were added to one tube, and then the pulldown assay was performed for both tubes (see Fig. S5B). We column purified the longer GST-eIF4G2 species to eliminate perturbation from contaminating shorter species (see Fig. S5C). Because eIF5-C can bind eIF4G2-frgC (RS1 alone) (Fig. 1) but does not stimulate its binding to 32P-mRNA (Fig. 4A, row 3), the increased mRNA binding by eIF4G2-frgA is not due to eIF5 interaction with the RS1 domain per se but presumably involves the HEAT and RS2 domains. 32P-mRNA binding to eIF4G2-frgH (RS2 alone) or eIF4G2-frgD (HEAT plus RS2) was not affected by eIF5-C (Fig. 4A, rows 4 and 5), consistent with their inability to bind eIF5 (14). Interestingly, partial or complete elimination of RS2 in eIF4G2-frgI or eIF4G2-frgJ, respectively, not only abolished the ability of eIF5 to stimulate mRNA binding to eIF4G2 but also rendered eIF5 inhibitory to mRNA binding by eIF4G2 (Fig. 4A, rows 6 and 7). Since eIF5 did not inhibit mRNA binding by eIF4G2-frgC, the eIF5 inhibition of mRNA binding by eIF4G-frgI and -frgJ appears to involve the HEAT domain. These results indicate that the RS2 domain is required for the eIF5-stimulated mRNA binding by eIF4G2 and also suggest that this process involves eIF5 interaction with the HEAT domain.
eIF5 antagonizes eIF2β-dependent mRNA binding to the 40S subunit.
In addition to the eIF4G2 RS domains, the eIF2β K-box segment alone can bind mRNA, and its role in mRNA recruitment to the ribosome was postulated (20). In contrast to its effect on mRNA binding by eIF4G2, eIF5-C reduced the efficiency of eIF2β binding to 32P-mRNA by 2-fold (Fig. 4A, row 7). To address whether this observation relates to the ability of eIF2β to mediate mRNA binding to the ribosome, we first tested if eIF2β-NTT can bind directly to the 40S subunit. As shown in Fig. 3B, row 3, middle gel, GST-eIF2β-N (eIF2β1-140) carrying all three K-boxes bound the 40S subunit purified from wild-type yeast. As a control, GST-eIF1 bound the 40S subunit (lane 5) as shown previously (28). This K-box–40S subunit interaction was not impeded by an excess amount of nonradiolabeled β-globin mRNA (Fig. 4B), ruling out that the interaction is due to general RNA-binding activity of the K-boxes. More importantly, this interaction was significantly decreased by the 18S rRNA A1193U mutation (28) altering the 40S subunit P site (Fig. 4B and C). Thus, the eIF2β K-box segment alone interacts with the 40S subunit, and this interaction specifically depends on the intact structure of the 40S subunit decoding site.
Having observed that the eIF2β segment containing K-boxes bind mRNA and the 40S subunit and that the mRNA in excess did not inhibit eIF2β binding to the 40S subunit, it was conceivable that the eIF2β segment alone can serve as a bridge between mRNA and the 40S subunit. To test this idea, we incubated the 40S subunit and 32P-MFA2 mRNA in the absence or presence of GST-eIF2β-N and examined if the eIF2β protein increases mRNA bound to the ribosome in a sucrose gradient-velocity sedimentation analysis (see Materials and Methods). Immunoblot analysis of the gradient fractions indicated that GST-eIF2β-N comigrates with the 40S subunit, confirming the mutual interaction between eIF2β-N and the 40S subunit (Fig. 5A, section 2, bottom gel). Scintillation counting of the gradient fractions showed that GST-eIF2β-N increases 32P-MFA2 mRNA binding to the 40S subunit (Fig. 5B and C). Importantly, the addition of eIF5-C to the preformed complexes significantly decreased eIF2β-stimulated 32P-mRNA recruitment (Fig. 5B and C) without affecting the amount of GST-eIF2β-N comigrating with the 40S subunit (Fig. 5A, section 3). Thus, eIF5-C specifically inhibits mRNA binding by eIF2β but not 40S subunit binding by eIF2β (Fig. 5D). These results suggest that the eIF2β-K-box segment can promote mRNA recruitment to the 40S subunit, and that this recruitment is favored in the absence of eIF5-CTD–eIF2β-K-box interaction (see Discussion).
Fig 5.
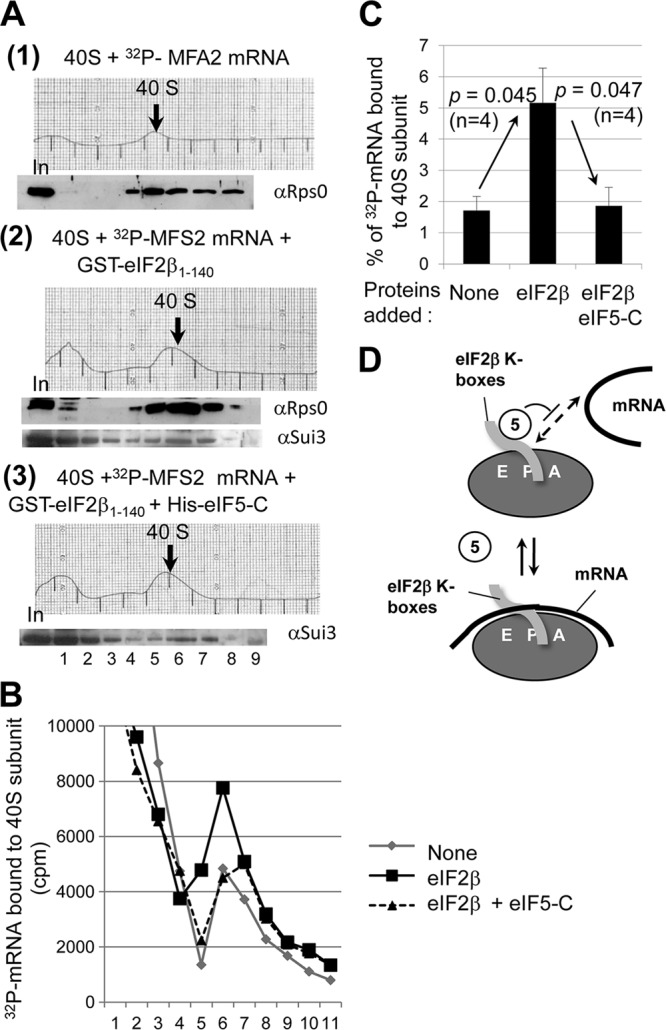
eIF5-CTD inhibits eIF2β-mediated 32P-MFA2 mRNA binding to the 40S subunit in vitro. (A) Purified wild-type 40S subunit was incubated in the reaction buffer at 4°C for 90 min with 32P-MFA2 mRNA in the absence (section 1) or presence of GST-eIF2β-N (section 2) or in the presence of GST-eIF2β-N and eIF5-C (section 3). The binding reaction was terminated by cross-linking with formaldehyde (5 min on ice), followed by glycine treatment and then fractionation by sucrose gradient velocity sedimentation; the A254 profile of gradient fractions is shown. The relevant 40S subunit-containing fractions are shown with a thick arrow. Proteins from one-half of the gradient samples were precipitated by ethanol, followed by immunoblot analysis with antibodies raised against proteins listed to the right. In, 10% input amount. The resulting immunoblot patterns were shown under the corresponding A254 profile. (B) Scintillation count of the 32P-MFA2 mRNA present in the other half of the gradient samples from the experiments shown in panel A. Gray line, section 1; black solid line, section 2; black dotted line, section 3. (C) Amount of 32P-MFA2 mRNA found in 40S subunit fractions in the absence or presence of GST-eIF2β-N (eIF2β) or GST-eIF2β-N and eIF5-CTD (eIF2β eIF5-C). Bars indicate standard errors. (D) Model showing that eIF5-C (empty oval) competes with mRNA (thick line) binding without impeding eIF2β K-box (gray tube) binding to the 40S subunit (gray oval with E P A denoting the decoding site).
eIF4G2-RS1 is involved in the 48S PIC commitment to initiation in response to start codon selection.
Our GCN4-lacZ reporter assay showed that tif4632-7R significantly increased GCN4 expression in the absence of starvation signals (Fig. 2D). Translation of GCN4 mRNA is normally blocked by reinitiation of translation at upstream open reading frame (uORF) elements in the 5′-leader region, which leads to ribosome dissociation (15). The starvation-induced eIF2 phosphorylation inhibits eIF2 reactivation and decreases eIF2/GTP/Met-tRNAiMet TC abundance, thereby allowing the 40S ribosome to bypass the inhibitory uORFs and reinitiate at the GCN4 AUG. Mutations impairing TC loading or retention on the 40S subunit mimic the effect of eIF2 phosphorylation, thereby increasing GCN4 expression in the absence of inducing signals (general control derepressed, or Gcd−, phenotype) (6). Therefore, our findings indicate that eIF4G2-7R confers a Gcd− phenotype, suggesting that this mutation destabilizes TC binding to the 40S subunit.
To get more insights into the role of eIF4G2-RS1 during the PIC function in vivo, we examined translation from UUG, a noncanonical start codon. If an eIF mutation relaxes the stringency of start codon selection, it allows translation initiation of a reporter gene where the start codon has been switched from AUG to UUG, the so-called suppressor of initiation codon mutation phenotype (Sui−). If the mutation increases the accuracy of translation initiation, it suppresses the relaxed stringency of initiation caused by a Sui− mutation (suppressor of Sui, or Ssu−, phenotype) (6, 12, 34). Recent studies indicate that mutations impairing the open state of the PIC tend to relax stringent AUG selection by allowing the PIC to achieve the closed state at a UUG codon (Sui− phenotype), while mutations impairing the closed state tend to increase the stringency of initiation by inhibiting formation of the PIC to the closed state at a UUG codon (Ssu− phenotype) (34). Since tif4632-7R conferred a Gcd− phenotype, suggesting destabilization of the scanning ribosomal complex, it was anticipated that this mutation also displays the Sui− or Ssu− phenotype. To test whether tif4632-7R confers the Sui− or Ssu− phenotype, we introduced this allele into yeast carrying a his4-303 allele in which the start codon was altered to AUU. As shown in Fig. 6A, row 3, tif4632-7R did not promote His4p expression and hence was judged to be Sui+ (16). In contrast, we found that tif4632-7R suppressed the Sui− phenotype caused by SUI3-2 (Fig. 6A, row 3; Ssu− phenotype).
Fig 6.

Mutations in eIF4G2-RS1 and eIF5-CTD suppress relaxed start codon selection caused by eIF2β mutation (SUI3-2). (A) Five microliters of 0.15 and 0.015 A600 U cultures of transformants of KAY220 (WT) and KAY862 (7R) carrying SUI3 or SUI3-2 URA3 plasmids (see Table S1 in the supplemental material) were spotted onto SC-Ura (+His) and SC-Ura-His (−His) plates and incubated at 30°C for 3 and 7 days, respectively. (B) Translation initiation from UUG codons in tif4632-7R. Double transformants of YAS1955 (WT) and KAY901 (7R) with a HIS4-lacZ reporter plasmid, p367 with its natural start codon, HIS4AUG-lacZ, p400 with HIS4UUG-lacZ, and with SUI3 or SUI3-2 ADE2 plasmids (see Table S1) were grown in SC-Ura-Ade medium and assayed for β-galactosidase activity. Shown are the averages and standard errors from 6 independent measurements. (C) Bars indicate the percentage of the HIS4UUG-lacZ values relative to HIS4AUG-lacZ values shown in panel B, calculated as UUG suppression activity. (D) Five microliters of 0.15 A600 U culture and its 10-fold serial dilutions of transformants of KAY976 (WT) and tif5 derivatives were assayed as described for panel A, except +His and −His plates were incubated for the times indicated above the images.
To confirm that the Ssu− phenotype is due to altered translation from his4-303, we transformed the tif4632-7R mutant with plasmids carrying HIS4-lacZ reporters, either with its natural start codon (HIS4AUG-lacZ) or with an AUU start codon in his4-303 (HIS4AUU-lacZ). tif4632-7R significantly increased expression from HIS4AUG-lacZ compared to the wild type (P = 0.015) (Fig. 6B, columns 6 and 8). This is consistent with increased GCN4 expression or Gcd− phenotype of this mutant without inducing amino acid starvation (Fig. 2D); because Gcn4p is an activator of HIS4 transcription, the reporter expression was likely due to increased transcription. Similar observations were made with Gcd− mutations altering eIF2γ (11). With tif4632-7R decreasing the relative frequency of translation initiation from the UUG codon (UUG/AUG ratio in Fig. 6C, columns 3 and 4), we conclude that eIF4G2-RS1 is important for 48S PIC commitment to initiation at the noncognate start codon.
eIF2β-, eIF4G-, and eIF3c-binding faces of eIF5-CTD are also involved in the 48S PIC commitment to initiation at a UUG codon.
The Ssu− phenotype observed with tif4632-7R is akin to the phenotype caused by a K-box mutation that alters eIF2β-N-terminal domain (20). The common protein partners of these protein segments are eIF1 and eIF5. Many mutations altering eIF1 cause increased tolerance of noncognate start codon usage, suggesting that the interactions of eIF1 and other translation initiation factors contribute to stabilizing the open state of a PIC-phase ribosome (6). We wished to examine more closely how abolishing eIF5-CTD interactions with its known binding partners affects the formation of the closed state of the PIC. tif5-AN1 alters the acidic area I of eIF5-CTD, and ΔE396 eliminates E396 and the acidic C-terminal tail. The former mutation reduces the eIF5 interaction with eIF2β, while the double mutation AN1_ΔE396 does so to a larger extent (44). tif5-AN1 also reduces the interaction with eIF4G2 (44). tif5-BN1 changes the basic area II of eIF5-CTD, disrupting the interaction with eIF3c and eIF1 (44). As a control, tif5-G62S, which decreases the eIF5 GAP activity (7), displayed a Ssu− phenotype in our assay (Fig. 6D, sections 2 and 3, rows 1 and 9). As shown in Fig. 6D, sections 2 and 3, rows 3, 5, and 7, AN1 weakly, the double mutant AN1_ΔE396 more strongly, and BN1 still more strongly suppressed the His+ phenotype caused by the eIF2β mutation SUI3-2 (Ssu− phenotype). Thus, similar to eIF2β-K-box (20) and eIF4G2-RS1 (Fig. 6A to C) mutations, mutations at the acidic faces of eIF5-CTD interacting with these proteins displayed the Ssu− phenotype. Unexpectedly, we also found that the basic face of eIF5-CTD involved in eIF3c and eIF1 binding also displayed the Ssu− phenotype (see Discussion). These results support the model that the mutual interactions of eIF5-CTD with eIF2β-K-boxes and eIF4G2-RS1 are involved in the 48S PIC commitment to initiation, presumably driving the PIC to the closed state.
DISCUSSION
In this communication, we report that yeast eIF4G2 contains three distinct RNA-binding sites (Fig. 1A), and that the RS1 domain plays a unique role in mRNA recruitment to the ribosome. The eIF4G2-RS1-mediated mRNA recruitment involves interaction with eIF5 (Fig. 1 and 3). tif4632-7R altering eIF4G2-RS1 reduced 32P-mRNA binding to the 40S subunit in cell extracts, which was reversed by adding excess eIF5 (Fig. 3C), demonstrating that a weakened interaction with eIF5 is responsible for the mRNA binding defect. eIF5 is a component of 43S PIC (3), and here we show that it increases mRNA binding to eIF4G2 containing RS1, HEAT, and RS2 domains (Fig. 4A). Our data demonstrate that eIF5 stabilizes the 48S PIC by enhancing eIF4G interaction with the mRNA, in addition to serving as a bridge to eIF4F-mRNA complexes. The eIF5-stimulated RNA binding by eIF4G2 requires the RS2 domain, which is located C terminally to the RS1 and HEAT domains. Thus, eIF5 binding through the RS1 and HEAT domains regulates RNA binding activity of RS2 that is located at a distance from RS1 in terms of the primary structure.
Of note was the finding that tif4632-7R reverses the relaxed stringency of AUG selection caused by eIF2β-S254Y (Ssu− phenotype). Ssu− phenotypes are thought to be caused by mutations impairing transition of PIC to the closed state (12). eIF1 is proposed to be a key element in repressing transition of the PIC from the open to closed state (10). Because the eIF4G-binding face of eIF1 (Fig. 1G; also see Fig. S2 in the supplemental material) overlaps its ribosome-binding face (KR area including K60) (32), a mechanism to explain the Ssu− phenotype of eIF4G-RS1 mutation is that, in response to AUG recognition, eIF4G-RS1 (together with the HEAT domain) excludes eIF1 from the P site by competitively inhibiting its binding to the ribosome. Because eIF1 appears to inhibit eIF4G2-frgA (RS1-HEAT-RS2) binding to the eIF5-CTD more strongly than it did for eIF4G2-frgC (RS1 only) (Fig. 1E), the ability to exclude eIF1 would be stronger with the intact eIF4G containing the HEAT domain. Prior to AUG recognition, eIF4G-RS1 is likely to be bound to eIF5-CTD, enhancing a stable anchoring of mRNA to the 40S subunit decoding site. Thus, it is reasonable to propose that switching eIF4G interaction with eIF5-CTD to eIF1 is an important step in promoting eIF1 release.
In strong support for a model where factor remodeling facilitates eIF1 release, in collaboration we recently showed that an eIF5-CTD mutation disrupting binding to eIF2β impaired PIC transition to the closed state in vitro, and it also displayed a Ssu− phenotype in combination with eIF2β-S264Y, eIF1-93-97, or eIF1-K60E in vivo (23). It was postulated that eIF2β-NTT is not bound to eIF5-CTD in the open state, in agreement with a model that eIF2β can stabilize mRNA binding at this stage (Fig. 5). Start codon recognition is proposed to promote eIF2β-NTT binding of eIF5-CTD, which disrupts a network of factor interactions that anchor eIF1 to the 40S subunit and thereby drives PIC to the closed state by facilitating eIF1 release. The Ssu− phenotypes caused by mutations altering eIF2β-K-boxes (20), as well as eIF5-CTD area I and the acidic tail (eIF2β-binding site) (Fig. 6D, rows 3 to 5), support this model. The in vitro reconstitution system used in this study does not include eIF4G and eIF3. However, Ssu− phenotypes caused by eIF4G-RS1 (this study) or eIF3c-NTT (42) suggest strongly that factor remodeling involving disordered segments of these factors can facilitate eIF1 release, as proposed above for eIF4G-RS1.
Similar to eIF4G-RS1, the NTT of the c subunit of eIF3 (eIF3c/Nip1p) also interacts with the KR area of eIF1. The eIF3c-NTT contains three distinct 10-aa-long elements, box 2, box 6, and box 12 (42). Of these, box 6 and box 12 are likely eIF1-binding sites, since alteration of these 10 aa strongly reduced eIF1 binding by each one (12- and 5-fold, respectively) (42). The interaction of eIF1 with these motifs presumably helps to dissociate eIF1 from the ribosomal P site by competitive inhibition. In support of this model, a mutation altering box 6 restores the stringency caused by a Sui− mutation to favor AUG selection (Ssu−) (42). Box 12 mutation shows a Sui− phenotype, suggesting that eIF1 binding to this element helps anchor eIF1 within the open PIC. Finally, mutational studies also suggest that box 2 and box 6 are eIF5-binding sites (42). Together with the Ssu− phenotype caused by tif5-BN1 (Fig. 6D) disrupting eIF5 binding to eIF1 and eIF3c (44), these findings suggest that eIF5-eIF3c interaction is required for the shift to the PIC closed state.
In conclusion, we propose that the myriad of eIF interactions, as depicted in Fig. 7a, can be sorted into two distinct sets of interactions which characterize the open and closed states of the PIC. In the closed PIC, the key interaction is eIF1-KR binding to eIF3c-NTT and eIF4G-RS1, which together help to exclude eIF1 from the ribosome (dotted blue lines in the bottom of Fig. 7c). Because eIF1 can bind simultaneously to eIF3c and eIF4G (14), eIF1 may dissociate from the ribosome in complex with eIF3 and eIF4G. Alternatively, eIF4G-RS1 may help eIF1 to dissociate from eIF3 after eIF1 release. The interaction between eIFs found in the open PIC is proposed to prevent these interactions while helping eIF1 anchored to the PIC (dotted purple lines in Fig. 7b). The data presented in this report indicate that the positively charged segments, eIF4G-RS1, eIF4G-RS2 (not shown in Fig. 7b), and eIF2β-NTT (K-boxes), stabilize mRNA binding to the ribosome (orange arrows in Fig. 7b). We propose that eIF5-CTD is bound to eIF3c-NTT at this stage, positioning the latter in proximity to eIF1. In this way, eIF5-CTD–eIF3c-NTT interaction would promote eIF1 release (dotted blue lines in Fig. 7b), which would be initiated through eIF2β-NTT binding to the eIF5-CTD (Fig. 7c). The evidence for the involvement of eIF4G-RS1 interaction with eIF3c-NTT is indirect; validating this model will require further work.
Fig 7.

Hypothetical model of eIF assembly rearrangement within the PIC. (a) Summary of interaction between eIF1 (1), eIF2β-NTT (2β-NTT), eIF3c-NTT (3c-NTT), eIF5-CTD (5-CTD), and eIF4G-CTD (4G-CTD), as well as that between eIF1 (with K60, the ribosome contact site) and the 40S subunit (gray oval with E P A denoting the decoding site). Boxes indicate unstructured charged segments, K-boxes in eIF2β, RS1 in eIF4G, and glutamate (E)- and lysine (K)-rich segments (boxes 2, 6, and 12) in eIF3c. Circles indicate the folded domains of these eIFs, with charged amino acids (E or K) in designated surface areas. A dotted line or arrow indicates proposed interaction between the indicated amino acids or surface area (a question mark indicates that the interaction surface has not been determined). Orange line, interaction important for mRNA recruitment to or stabilization within the PIC identified in this study. Purple line, mutational disruption of this interaction causes the Sui− phenotype (10, 14, 23, 33, 42). Blue line, mutational disruption of this interaction causes the Ssu− phenotype (20, 23, 42, and this study). (b and c) The network of interactions stabilizing the open and closed states of the PIC, respectively. The interactions whose mutations cause the Sui− phenotype are proposed to assist anchoring eIF1 to the 40S subunit shown in panel b. The interactions whose mutations cause the Ssu− phenotype (except for eIF5-eIF3c interactions [see the text]) are proposed to promote eIF1 release in panel c.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alan Hinnebusch for anti-SUI3 antibodies and stimulating discussions, Leos Valasek for anti-RpsO antibodies, Tom Donahue for anti-eIF5 antibodies, Stefan Rothenberg, Chris Fraser, and John Hershey for comments on the manuscript, and Gerhard Wagner, Alan Hinnebusch, and Jon Lorsch for discussion and sharing results prior to publication. We are also greatly indebted to James Anderson for carefully editing the manuscript.
This work was supported by NIH GM64781 and the K-State Terry Johnson Cancer Center (K.A.).
Footnotes
Published ahead of print 30 July 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR. 2007. Reconstitution of yeast translation initiation. Methods Enzymol. 430: 111– 145 [DOI] [PubMed] [Google Scholar]
- 2. Algire MA, Maag D, Lorsch JR. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20: 251– 262 [DOI] [PubMed] [Google Scholar]
- 3. Asano K, Clayton J, Shalev A, Hinnebusch AG. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14: 2534– 2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asano K, Krishnamoorthy T, Phan L, Pavitt GD, Hinnebusch AG. 1999. Conserved bipartite motifs in yeast eIF5 and eIF2Bε, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 18: 1673– 1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Asano K, et al. 2002. Analysis and reconstitution of translation initiation in vitro. Methods Enzymol. 351: 221– 247 [DOI] [PubMed] [Google Scholar]
- 6. Asano K, Sachs MS. 2007. Translation factor control of ribosome conformation during start codon selection. Genes Dev. 21: 1280– 1287 [DOI] [PubMed] [Google Scholar]
- 7. Asano K, et al. 2001. Multiple roles for the carboxyl terminal domain of eIF5 in translation initiation complex assembly and GTPase activation. EMBO J. 20: 2326– 2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Asano K, et al. 1997. Structure of cDNAs encoding human eukaryotic initiation factor 3 subunits: possible roles in RNA binding and macromolecular assembly. J. Biol. Chem. 272: 27042– 27052 [DOI] [PubMed] [Google Scholar]
- 9. Berset C, Zurbriggen A, Djafarzadeh S, Altmann M, Trachsel H. 2003. RNA-binding activity of translation initiation factor eIF4G1 from Saccharomyces cerevisiae. RNA 9: 871– 880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung Y-N, et al. 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 21: 1217– 1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dorris DR, Erickson FL, Hannig EM. 1995. Mutations in GCD11, the structural gene for eIF-2γ in yeast, alter translational regulation of GCN4 and the selection of the start site for protein synthesis. EMBO J. 14: 2239– 2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fekete CA, et al. 2007. N- and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO J. 26: 1602– 1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goyer C, Altmann M, Trachsel H, Sonenberg N. 1989. Identification and characterization of cap binding proteins from yeast. J. Biol. Chem. 264: 7603– 7610 [PubMed] [Google Scholar]
- 14. He H, et al. 2003. The yeast eIF4G HEAT domain interacts with eIF1 and eIF5 and is involved in stringent AUG selection. Mol. Cell. Biol. 23: 5431– 5445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinnebusch AG, Dever TE, Asano K. 2007. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, p 225–268 In Mathews MB, Sonenberg N, Hershey JWB. (ed), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16. Huang H, Yoon H, Hannig EM, Donahue TF. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 11: 2396– 2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jackson RJ, Hellen CUT, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 10: 113– 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jennings MD, Pavitt GD. 2010. eIF5 has GDI activity necessary for translational control by eIF2 phosphorylation. Nature 465: 378– 381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jivotovskaya AL, Valasek L, Hinnebusch AG, Nielsen KH. 2006. eIF3 and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol. Cell. Biol. 26: 1355– 1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laurino JP, Thompson GM, Pacheco E, Castilho BA. 1999. The β subunit of eukaryotic translation initiation factor 2 binds mRNA through the lysine repeats and a region comprising the C2-C2 motif. Mol. Cell. Biol. 19: 173– 181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee B, Udagawa T, Singh CS, Asano K. 2007. Yeast phenotypic assays on translational control. Methods Enzymol. 429: 105– 137 [DOI] [PubMed] [Google Scholar]
- 22. Lorsch JR, Dever TE. 2010. Molecular view of 43S complex formation and start site selection in eukaryotic translation initiation. J. Biol. Chem. 285: 21203– 21207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luna RE, et al. 2012. The C-terminal domain of eukaryotic initiation factor 5 promotes start codon recognition by its dynamic interplay with eIF1 and eIF2β. Cell Rep. 1: 689– 702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marintchev A, et al. 2009. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 136: 447– 460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mitchell SF, et al. 2010. The 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell 39: 950– 962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morley SJ, Curtis PS, Pain VM. 1997. eIF4G: translation's mystery factor begins to yield its secrets. RNA 3: 1085– 1104 [PMC free article] [PubMed] [Google Scholar]
- 27. Nanda JS, et al. 2009. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 394: 268– 285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nemoto N, et al. 2010. Yeast 18S rRNA is directly involved in the ribosomal response to stringent AUG selection during translation initiation. J. Biol. Chem. 285: 32200– 32212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park EH, et al. 2011. Multiple elements in the eIF4G1 N-terminus promote assembly of eIF4G1 · PABP mRNPs in vivo. EMBO J. 30: 302– 316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Passmore LA, et al. 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26: 41– 50 [DOI] [PubMed] [Google Scholar]
- 31. Prevot D, et al. 2003. Characterization of a novel RNA-binding region of eIF4GI critical for ribosomal scanning. EMBO J. 22: 1909– 1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331: 730– 736 [DOI] [PubMed] [Google Scholar]
- 33. Reibarkh M, et al. 2008. Eukaryotic initiation factor (eIF) 1 carries two distinct eIF5-binding faces important for multifactor assembly and AUG selection J. Biol. Chem. 283: 1094– 1103 [DOI] [PubMed] [Google Scholar]
- 34. Saini AK, Nanda JS, Lorsch JR, Hinnebusch AG. 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNAiMet binding to the ribosome. Genes Dev. 24: 97– 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh CR, Asano K. 2007. Localization and characterization of protein-protein interaction sites. Methods Enzymol. 429: 139– 161 [DOI] [PubMed] [Google Scholar]
- 36. Singh CR, et al. 2005. eIF5 is critical for the integrity of the scanning preinitiation complex and accurate control of GCN4 translation. Mol. Cell. Biol. 25: 5480– 5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh CR, et al. 2006. An eIF5/eIF2 complex antagonizes guanine nucleotide exchange by eIF2B during translation initiation. EMBO J. 25: 4537– 4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siridechadilok B, Fraser CS, Hall RJ, Doudna JA, Nogales E. 2005. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science 310: 1513– 1515 [DOI] [PubMed] [Google Scholar]
- 39. Sokabe M, Fraser CS, Hershey JW. 2012. The human translation initiation multi-factor complex promotes methionyl-tRNAi binding to the 40S ribosomal subunit. Nucleic Acids Res. 40: 905– 913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tarun SZ, Sachs AB. 1996. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 15: 7168– 7177 [PMC free article] [PubMed] [Google Scholar]
- 41. Tarun SZ, Sachs AB. 1997. Binding of eukaryotic translation initiation factor 4E (eIF4E) to eIF4G represses translation of uncapped mRNA. Mol. Cell. Biol. 17: 6876– 6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Valásek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG. 2004. Interaction of eIF3 subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 24: 9437– 9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watanabe R, et al. 2010. The eIF4G HEAT domain promotes translation re-initiation in yeast both dependent on and independent of eIF4A mRNA helicase. J. Biol. Chem. 285: 21922– 21933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamamoto Y, et al. 2005. The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3 and eIF4G. Proc. Natl. Acad. Sci. U. S. A. 102: 16164– 16169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.