

Abstract
Interleukin-17 (IL-17) is critically involved in the pathogenesis of various inflammatory disorders. IL-17 receptor (IL-17R)-proximal signaling complex (IL-17R-Act1-TRAF6) is essential for IL-17-mediated NF-κB activation, while IL-17-mediated mRNA stability is TRAF6 independent. Recently, inducible IκB kinase (IKKi) has been shown to phosphorylate Act1 on Ser 311 to mediate IL-17-induced mRNA stability. Here we show that TANK binding kinase 1 (TBK1), the other IKK-related kinase, directly phosphorylated Act1 on three other Ser sites to suppress IL-17R-mediated NF-κB activation. IL-17 stimulation activated TBK1 and induced its association with Act1. IKKi also phosphorylated Act1 on the three serine sites and played a redundant role with TBK1 in suppressing IL-17-induced NF-κB activation. Act1 phosphorylation on the three sites inhibited its association with TRAF6 and consequently NF-κB activation in IL-17R signaling. Interestingly, TRAF6, but not TRAF3, which is the upstream adaptor of the IKK-related kinases in antiviral signaling, was critical for IL-17-induced Act1 phosphorylation. TRAF6 was essential for IL-17-induced TBK1 activation, its association with Act1, and consequent Act1 phosphorylation. Our findings define a new role for the IKK-related kinases in suppressing IL-17-mediated NF-κB activation through TRAF6-dependent Act1 phosphorylation.
INTRODUCTION
Interleukin-17 (IL-17A or IL-17) was originally identified as a characteristic cytokine secreted by Th17 cells (19). More IL-17-producing sources have now been found, especially innate immune cells (6, 23). IL-17 belongs to a cytokine family containing six members (IL-17A to IL-17F) which are structurally different from other cytokines (7, 8, 16, 26, 40). IL-17 is the best-characterized member of the family. IL-17 is a proinflammatory cytokine and acts on local tissue cells to induce production of genes for proinflammatory proteins, including cytokines, chemokines, and matrix metalloproteases, to amplify inflammation (8, 16, 26). The IL-17 level is elevated in a variety of inflammatory and autoimmune disorders, while IL-17 deficiency or its functional blockage reduces the development and pathogenesis of autoimmune diseases, suggesting that IL-17 is a promising target for various inflammatory disorders (16, 26).
While the IL-17 family can induce common signaling pathways, including NF-κB and mitogen-activated protein kinases (MAPKs) (P38, Jun N-terminal protein kinase [JNK], and extracellular signal-regulated kinase [ERK]) (8, 33), IL-17 receptor (IL-17R)-proximal signaling is being discovered and shown to be unique and tightly regulated (2, 3, 22, 27–29, 31, 32, 38). Act1 is identified as a key adaptor to recruit TRAF6 to IL-17R to mediate IL-17-induced NF-κB activation, while TRAF3 and TRAF4 are found to interfere with the complex formation of IL-17R-Act1-TRAF6 (3, 27, 37, 38). Both Act1 and TRAF6 function as E3 ubiquitin ligases in IL-17R signaling (22). Act1-mediated polyubiquitination of TRAF6 leads to TRAF6 autoubiquitination, which is required for IL-17-induced NF-κB activation and production of genes for proinflammatory proteins (22).
IL-17 has also been shown to stabilize mRNAs such as the chemokine CXCL1 induced by tumor necrosis factor (TNF) (11). IL-17-induced mRNA stability is Act1 dependent but TRAF6 independent (12). Very recently, two studies reported that inducible IκB kinase (IKKi; also known as IKKξ), TRAF2, and TRAF5 are important for IL-17-induced mRNA stability (1, 34). IKKi belongs to the IKK-related kinases, which contain another kinase, TANK binding kinase 1 (TBK1) (9, 17). The IKK-related kinases have been shown to have similar as well as differential roles in antiviral signaling, autophagy, and oncogenesis (5, 13, 15, 25, 30, 35, 36).
While IKKi has been found as a kinase to phosphorylate Act1 on Ser 311 to regulate TRAF6-independent function in IL-17R signaling in a mouse system (1), it still remains to be determined whether there are other kinases to phosphorylate Act1 and whether Act1 can be phosphorylated on other sites in addition to Ser 311. Here we report that TBK1 is another direct kinase for Act1 and phosphorylates Act1 on three other serine sites in both human and mouse systems. IKKi also redundantly phosphorylates Act1 on these three sites in a mouse system. Both TBK1 and IKKi suppressed IL-17-induced NF-κB activation through phosphorylating Act1 on the three sites. Mechanistic studies showed that TRAF6 was crucial for IL-17-induced TBK1 activation and its association with Act1 for the consequent Act1 phosphorylation. Act1 phosphorylation on the three sites interfered with its interaction with TRAF6 to suppress continuous inflammatory signaling. Our findings identify a new role for the IKK-related kinases in suppressing IL-17-mediated NF-κB activation and provide a molecular mechanism of how Act1 gets phosphorylated by these kinases for the negative-feedback regulation of IL-17R signaling.
MATERIALS AND METHODS
Reagents, cell lines, and constructs.
Recombinant human and mouse IL-17 were from R&D Systems. Anti-M2 (Flag) and antiactin were from Sigma-Aldrich. Antihemagglutinin (anti-HA) was from Covance. Anti-p-IκBα, anti-p-JNK, anti-JNK, anti-p-P38, and p-ERK were from Cell Signaling Technology. Anti-Act1, anti-IKKi, anti-TRAF6, anti-TRAF3, and normal mouse IgG were from Santa Cruz Biotechnology. Anti-TBK1 was from Imgenex. Anti-p-TBK1 (S172) was from BD Bioscience. Shrimp alkaline phosphatase (SAP) was from Fermentas.
HEK293T cells, HeLa cells, human astrocytes (U87-MG), wild-type mouse embryonic fibroblasts (MEFs), Act1−/− MEFs, TBK1−/− IKKi−/− MEFs, TBK1−/− MEFs, Traf6−/− MEFs, and Traf3−/− MEFs were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; HyClone), penicillin G (100 μg/ml), and streptomycin (100 μg/ml) as previously described (10, 13, 22, 25, 27, 38).
The cDNAs encoding HA-tagged IKKα, IKKβ, and NIK were constructed into pcDNA3.1. The cDNAs encoding HA or Flag-tagged TBK1 and IKKi were cloned into pcDNA3. The plasmid for Flag-tagged Act1 was previously described (31). The cDNAs encoding mouse Act1 and its point mutants, including the S147A, S209A, S222A, S147/209A, and S147/209/222A (3SA) mutants, as well as mouse Flag-tagged TRAF6 and its mutants (Flag-TRAF6-K124R, Flag-TRAF6-C70A, and Flag-TRAF6-ΔRing with residues 1 to 108 deleted), were constructed into the retroviral vector pMSCV-IRES-GFP. The cDNAs for human or mouse HA-tagged TBK1, IKKi, DNIKKi (K38A), and DNTBK1 (K38A) were subcloned into pMSCV-IRES-GFP.
Coimmunoprecipitations and immunoblot analysis.
Coimmunoprecipitations and immunoblot analysis were performed as previously described (38). HEK293T cells were transfected with plasmids by a calcium phosphate transfection method. Cells left untreated or treated with IL-17 at different time points were harvested by washing with ice-cold PBS and then lysed with Co-IP buffer (0.5% Triton X-100, 20 mM HEPES [pH 7.6], 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 1 mM Na3VO4) freshly supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Roche). Cell extracts were incubated with 0.5 μg appropriate antibody and 20 μl protein A-Sepharose beads (GE Healthcare). After overnight incubation, beads were washed four times with Co-IP buffer and boiled in protein loading buffer for 10 min to release and denature immunoprecipitated proteins before separation on SDS-PAGE.
Cells were directly lysed in Co-IP buffer as described above and separated by SDS-PAGE. Immunoblot analysis was performed by initial transfer of proteins onto polyvinylidene fluoride filters using Mini Trans-Blot (Bio-Rad Laboratories) and followed by a blocking step using Tris-buffered saline with 0.1% Tween 20 plus 5% nonfat dried milk for 1 h at room temperature. The blots were then incubated with primary antibody overnight and subsequently washed, followed by incubation with a secondary antibody conjugated to horseradish peroxidase (HRP) for 1 h at room temperature. After extensive washing of the blots, signals were visualized with chemiluminescent HRP substrate (Millipore).
Expression of recombinant Act1-GST fusion fragments.
The cDNAs encoding the deletion mutants or point mutants of human Act1 (amino acids [aa] 1 to 565) or mouse Act1 (aa 1 to 555), as shown in Fig. 6 and Fig. 7, were cloned into the pGEX 4T-1 vector. All constructs were verified by sequencing. Glutathione S-transferase (GST) fusion proteins were expressed in Escherichia coli BL21(DE3) and purified with glutathione-Sepharose (GE Healthcare) according to the manufacturer's instructions.
Fig 6.

The IKK-related kinases phosphorylate human Act1 at Ser 162, Ser 220, and Ser 233. (A) Schematic representation of human Act1 protein. The major isoform of human Act1 contains 565 amino acids. The indicated regions of Act1 were expressed as recombinant GST fusion proteins in E. coli and purified with glutathione-Sepharose. The fusion proteins for in vitro kinase assay in panels B and C were subjected to Coomassie blue staining (CB). *, target protein. Molecular mass is indicated in kilodaltons next to the gel blots. (B and C) IKKi (B) or TBK1 (C) phosphorylates Act1 at the regions of aa 121 to 191 and 191 to 330. Flag-tagged IKKi (B) or TBK1 (C) was expressed and immunoprecipitated with anti-M2 from HEK293T cells and incubated with the recombinant proteins of human Act1 (hAct1) truncations as indicated in panel A. The incorporated radioactivity was detected by autoradiography. Arrows indicate IKKi or TBK1 autophosphorylation. Stars indicate phosphorylated Act1. (D and E) Conserved serine sites are showed by amino acid alignment of Act1 protein sequences. Alignment of Act1 protein sequences in different species was performed by the multiple-sequence alignment program ClustalW2. S143, S146, S149, S156, S159, and S162 residues in the human Act1 region of aa 121 to 191 (D) and S201, S220, S233, and S256 residues in the human Act1 region of aa 191 to 330 (E) are indicated by arrows. (F) IKKi or TBK1 phosphorylates Ser 162 of Act1 in the region of aa 121 to 191. The indicated point mutants of the GST-hAct1 region of aa 121 to 191 were used as substrates for Flag-tagged IKKi (top) or TBK1 (middle) in an in vitro kinase assay as in panels B and C. The expression of the GST fusion protein mutants was checked by Coomassie blue staining (CB). (G and H) IKKi phosphorylates Ser 220 of Act1, while TBK1 phosphorylates Ser 220 and Ser 233 of Act1 in the region of aa 191 to 330. In vitro kinase assays were performed with the indicated point mutants of the GST-hAct1 region of aa 191 to 330 as in panel F. 4SA, four-serine mutant (S201A, S220A, S233A, and S256A). Arrows in panels F to H indicate IKKi or TBK1 autophosphorylation. Stars indicate phosphorylated Act1.
Fig 7.

The IKK-related kinases phosphorylate mouse Act1 at Ser 147, Ser 209, and Ser 222. Flag-tagged mTBK1 (top) or mIKKi (middle) were expressed and immunoprecipitated from HEK293T cells and incubated with recombinant mouse Act1 (mAct1) deletion mutants or point mutants with changes of the mAct1 region of aa 121 to 390 as indicated. The incorporated radioactivity was detected by autoradiography. The recombinant GST fusion proteins of Act1 mutants were checked by Coomassie blue staining (CB) (bottom). The arrows indicate the autophosphorylation bands of IKK-related kinases. Stars indicate phosphorylated Act1. Asterisks indicate target protein.
In vitro kinase assay.
HEK293T cells were transfected with plasmids encoding Flag-tagged TBK1 or Flag-tagged IKKi by calcium phosphate precipitation. MEFs infected with retrovirus for Flag-IKKi were stimulated with IL-17 (50 ng/ml) for various times. Cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed using Co-IP buffer for 30 min on ice. Cell lysates were incubated with 0.5 μg anti-M2 and 20 μl protein A-Sepharose beads overnight at 4°C. The beads were washed twice with Co-IP buffer followed by two additional washes with kinase buffer (20 mM HEPES [pH 7.6], 10 mM MgCl2, 10 mM NaCl, 12.5 mM β-glycerophosphate, 10 mM sodium orthovanadate, 2 mM dithiothreitol [DTT], and 20 μM ATP). Washed beads were incubated with 30 μl kinase buffer including 1μCi of [γ-32P]ATP and with or without recombinant GST fusion proteins of Act1 (as indicated below) for 30 min at 30°C. Reactions were stopped by boiling in protein loading buffer and subjected to SDS-PAGE. After electrophoresis, gels were dried and the incorporated radioactivity was detected by autoradiography.
Retroviral transduction.
Retrovirus vectors and helper vectors were transfected into 293FT cells (Invitrogen) by calcium phosphate precipitation for viral packaging. At 60 h after transfection, virus was collected for infection of target cells in the presence of Polybrene (10 mg/ml; Sigma). At day 4 after infection, cells were used for experiments.
RNA interference (RNAi) and transfection.
HeLa cells were transfected with small interfering RNA (siRNA) oligonucleotides and Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). The siRNA sequence for human TRAF6 gene knockdown is 5′-GAUCCAGGGAUAUGAUGUAdTdT-3′; that for human Act1 gene knockdown is 5′-GCUUCAGAACACUCAUGUCUAdTdT-3′. The scrambled control siRNA sequence is 5′-UUCUCCGAACGUGUCACGUdTdT-3′. Seventy-two hours after transfection, cells were collected for protein isolation.
RNA isolation and real-time quantitative PCR.
Total RNA was extracted from cells with TRIzol reagent according to the manufacturer's instructions (Invitrogen). For cDNA synthesis, RNA was reverse transcribed with a PrimeScript RT reagent kit (TaKaRa). The expression of the genes encoding Ccl20, Ccl2, KC, Cxcl2, IL-6, granulocyte colony-stimulating factor (G-CSF), TNF-α, and IL-8 was quantified by real-time PCR with SYBR Premix Ex Taq kit (TaKaRa). All gene expression results were normalized to expression of housekeeping gene Rpl13a. Amplification of cDNA was performed on an ABI Prism 7900 HT cycler (Applied Biosystems) and with the following primers: 5′-GCGAATCAGAAGCAGCAAG-3′ and 5′-CGTGTGAAGCCCACAATAAA-3′ for human Ccl20, 5′-AACTGGGTGAAAAGGGCTGT-3′ and 5′-GTCCAATTCCATCCCAAAAA-3′ for mouse Ccl20, 5′-TTCTGTGCCTGCTGCTCAT-3′ and 5′-GGGGCATTGATTGCATCT-3′ for human Ccl2, 5′-AGGTCCCTGTCATGCTTCTG-3′ and 5′-TCTGGACCCATTCCTTCTTG-3′ for mouse Ccl2, 5′-AGACAGCAGAGCACACAAGC-3′ and 5′-ATGGTTCCTTCCGGTGGT-3′ for human IL-8, 5′-TCCTGCATCCCCCATAGTTA-3′ and 5′-CTTCAGGAACAGCCACCAGT-3′ for human KC, 5′-AGACTCCAGCCACACTCCAA-3′ and 5′-TGACAGCGCAGCTCATTG-3′ for mouse KC, 5′-CATCGAAAAGATGCTGAAAAATG-3′ and 5′-TTCAGGAACAGCCACCAATA-3′ for human Cxcl2, 5′-CCTGGTTCAGAAAATCATCCA-3′ and 5′-CTTCCGTTGAGGGACAGC-3′ for mouse Cxcl2, 5′-GATGAGTACAAAAGTCCTGATCCA-3′ and 5′-CTGCAGCCACTGGTTCTGT-3′ for human IL-6, 5′-GATGGATGCTACCAAACTGGAT-3′ and 5′-CCAGGTAGCTATGGTACTCCAGA-3′ for mouse IL-6, 5′-CCTGGAGCAAGTGAGGAAGA-3′ and 5′-CAGCTTGTAGGTGGCACACA-3′ for mouse G-CSF, 5′-CAGCCTCTTCTCCTTCCTGAT-3′ and 5′-GCCAGAGGGCTGATTAGAGA-3′ for human TNF-α, 5′-CGAGGTTGGCTGGAAGTACC-3′ and 5′-CTTCTCGGCCTGTTTCCGTAG-3′ for human Rpl13a, and 5′-GGGCAGGTTCTGGTATTGGAT-3′ and 5′-GGCTCGGAAATGGTAGGGG-3′ for mouse Rpl13a.
Statistics.
Data are presented as means; error bars indicate standard errors of the means (SEM). A two-tailed Student's t test was used for analysis of differences between the groups. A one-way analysis of variance was initially done to determine whether an overall statistically significant change existed before analysis with a two-tailed paired or unpaired Student t test. P values of less than 0.05 were considered statistically significant.
RESULTS
Both TBK1 and IKKi are required for Act1 phosphorylation in IL-17R signaling.
Act1 has been shown to be phosphorylated upon IL-17 stimulation (1, 14, 31). Act1 has also been shown to interact with IKK complex (20, 21). We tested if IKK or related kinases were involved in Act1 phosphorylation. Interestingly, overexpression of TBK1 or IKKi, the IKK-related kinases, resulted in Act1 mobility shift, while overexpression of the dominant negative form of IKKi (the kinase dead mutant) did not, suggesting that these two kinases are likely responsible for Act1 phosphorylation (Fig. 1A). In contrast, overexpression of IKKα, IKKβ, or NIK did not lead to Act1 mobility shift, indicating that Act1 was specifically phosphorylated by the IKK-related kinases (Fig. 1A). To confirm that the Act1 shift induced by the IKK-related kinases was due to phosphorylation, the cell lysates were treated with shrimp alkaline phosphatase (SAP) to remove kinase-mediated protein phosphorylation. The SAP treatment indeed removed the Act1 mobility shift by overexpression of TBK1 or IKKi, suggesting that the IKK-related kinases phosphorylated Act1 (Fig. 1B). To see whether the IKK-related kinases are responsible for IL-17-induced Act1 phosphorylation, we made use of the TBK1−/− IKKi−/− mouse embryonic fibroblasts (DKO MEFs). The IL-17-induced Act1 phosphorylation was severely reduced in the doubly deficient MEFs compared to that in wild-type cells (Fig. 1C). To determine whether both kinases contributed to Act1 phosphorylation, the doubly deficient MEFs were infected with retrovirus encoding TBK1 or IKKi and then stimulated with IL-17. Restoration of either TBK1 or IKKi increased IL-17-induced Act1 phosphorylation (Fig. 1D, top). TBK1 and IKKi together showed additive effects on Act1 phosphorylation (Fig. 1D, top). We further found that the kinase-dead mutants of TBK1 or IKKi did not restore IL-17-induced Act1 phosphorylation in the DKO MEFs (Fig. 1D, bottom), indicating that the kinase activity of the IKK-related kinases was required for Act1 phosphorylation in IL-17R signaling. Similarly, we found that dominant negative forms of human IKKi or mouse TBK1 suppressed IL-17-induced Act1 phosphorylation in human U87-MG cells or wild-type MEFs, respectively (data available on request). Although IL-17 has been shown to induce IKKi activation for Act1 phosphorylation (1), here we show that TBK1 is also required for Act1 phosphorylation. So we next checked whether TBK1 can be activated by IL-17 stimulation. Indeed, IL-17 stimulation resulted in TBK1 phosphorylation at Ser 172, phosphorylation of which is necessary for its kinase activity (18), in both human U87-MG cells (Fig. 1E) and MEFs (Fig. 1F). Together, our data suggest that the kinase activities of both TBK1 and IKKi are specifically and redundantly required for Act1 phosphorylation in IL-17R signaling.
Fig 1.

The IKK-related kinases specifically phosphorylate Act1 in IL-17R signaling. (A) Overexpression of the IKK-related kinases results in Act1 mobility shift. HEK293T cells were transfected with the indicated HA-tagged expression plasmids or empty vector (EV), and lysates were analyzed by immunoblotting with antibodies against Act1, actin, and HA. (B) The IKK-related kinases phosphorylate Act1. Lysates from HEK293T cells transfected with plasmids for M2-Act1, M2-Act1 plus HA-IKKi, and M2-Act1 plus HA-TBK1 were left untreated or treated with SAP (30 min at 37°C), followed by immunoblot analysis with antibodies against M2, actin, and HA. (C) The IKK-related kinases are required for IL-17-induced Act1 phosphorylation. Wild-type (WT) and TBK1−/− IKKi−/− (DKO) MEFs were stimulated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with antibodies against Act1 and actin. Levels of expression of TBK1, IKKi, and Act1 were measured by immunoblotting with lysates from WT and DKO MEFs (right). (D) The IKK-related kinases redundantly phosphorylate Act1. DKO MEFs infected with control virus (EV) or retrovirus encoding TBK1, IKKi, or IKKi plus TBK1 (top) or TBK1, DNTBK1 (K38A), or DNIKKi (K38A) (bottom) were left untreated or treated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with antibodies against Act1, actin, TBK1, and IKKi. (E and F) IL-17 stimulation results in TBK1 phosphorylation. Lysates from U87-MG cells (E) or MEFs (F) treated with IL-17 (50 ng/ml) for the indicated times were immunoblotted with antibodies against p-TBK1, TBK1, p-IκBα, and actin. Arrowheads in panels C and D indicate the shifted (phosphorylated) Act1 band. L.E., long exposure; S.E., short exposure.
Act1 associates with the IKK-related kinases in an IL-17 signal-dependent manner.
As kinases are supposed to associate with their substrates, we sought to determine whether the IKK-related kinases really interact with Act1. We found that Act1 associated with TBK1 or IKKi in an overexpression system (Fig. 2A). We then checked if Act1 bound to the kinases in an IL-17-dependent manner. HeLa cells were infected with retrovirus encoding HA-tagged TBK1 or HA-tagged IKKi and then stimulated with IL-17. Endogenous Act1 was indeed associated with the tagged TBK1 (Fig. 2B) or IKKi (Fig. 2C) in a signal-dependent manner. We further confirmed that IL-17 stimulation led to association of endogenous Act1 with endogenous TBK1 or IKKi in both human HeLa cells (Fig. 2D) and U87-MG cells (Fig. 2E). Since IKKi has been shown to interact with Act1 in MEFs, we further investigated the association of Act1 with TBK1 in MEFs. We found that IL-17 stimulation resulted in Act1 binding to HA-tagged TBK1 as well as phosphorylated TBK1 (Fig. 2F). The IL-17-induced association of Act1 with phosphorylated TBK1 was also confirmed in an endogenous system (Fig. 2G). Domain deletion analyses showed that domain 182 to 282 of human Act1 mediated its association with TBK1 or IKKi (data available on request). Together, our results firmly establish that the IKK-related kinases talk to Act1 in IL-17R signaling in both human and mouse cells.
Fig 2.
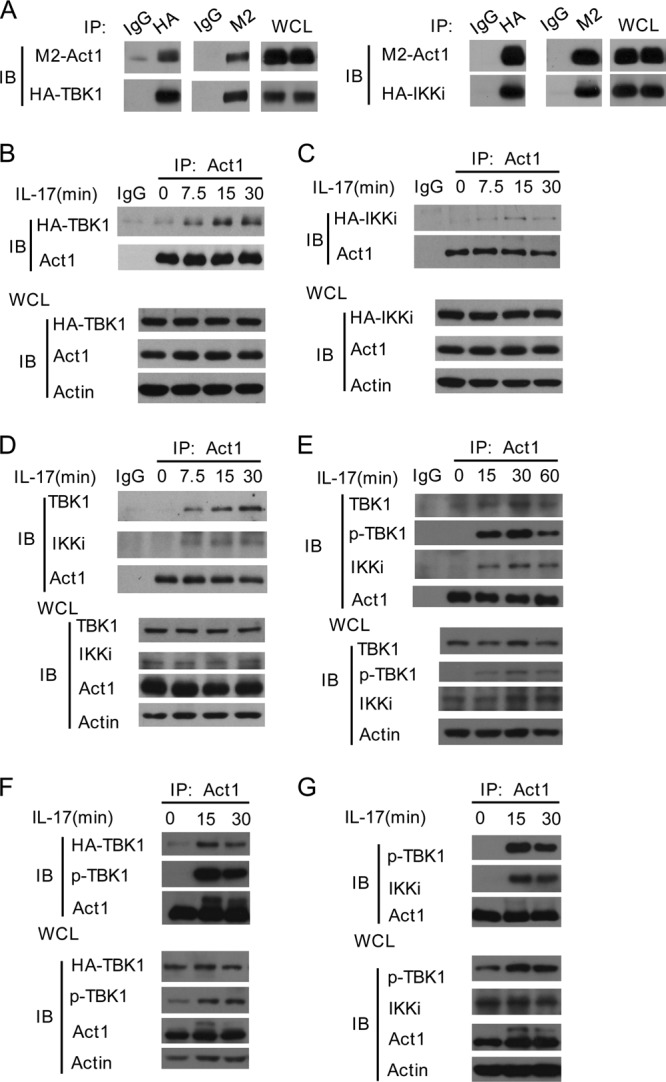
Act1 associates with the IKK-related kinases in an IL-17 stimulation-dependent manner. (A) Act1 associates with the IKK-related kinases in an overexpression system. HEK293T cells transfected with plasmids for M2-Act1 and HA-TBK1 (left) or HA-IKKi (right) were immunoprecipitated (IP) with anti-M2 or control IgG and anti-HA or control IgG, followed by immunoblotting with antibodies against M2 or HA. WCL, whole-cell lysates. (B and C) Endogenous Act1 is associated with the HA-tagged TBK1 (B) or IKKi (C) in an IL-17 signal-dependent manner. HeLa cells infected with retrovirus encoding HA-tagged TBK1 (B) or IKKi (C) were stimulated with IL-17 for the indicated times. Lysates were immunoprecipitated (IP) with anti-Act1 or IgG, followed by immunoblot analysis with antibodies against HA, Act1, or actin. (D and E) Endogenous Act1 is associated with the endogenous IKK-related kinases in an IL-17 signal-dependent manner. Lysates from HeLa cells (D) or U87-MG cells (E) treated with IL-17 (50 ng/ml) for the indicated times were immunoprecipitated with anti-Act1 or IgG, followed by immunoblot analysis with the indicated antibodies. (F and G) IL-17 stimulation results in Act1 binding to the IKK-related kinases in mouse cells. MEFs infected with retrovirus for HA-tagged TBK1 (F) or left uninfected (G) were treated with IL-17 for 0, 15, or 30 min, and then cell lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis with the indicated antibodies.
The IKK-related kinases suppress IL-17-induced NF-κB activation.
To determine the potential role of the IKK-related kinases in IL-17-mediated signaling, we utilized the dominant negative forms of IKKi or TBK1 which inhibited IL-17-induced Act1 phosphorylation. HeLa cells were first infected with retrovirus encoding these dominant negative forms or their wild-type controls and then stimulated with IL-17. Either dominant negative TBK1 or dominant negative IKKi greatly enhanced while wild-type TBK1 or IKKi suppressed IL-17-induced NF-κB activation (p-IκBα) in human HeLa cells (Fig. 3A and B). Similarly, these dominant negative forms increased while their wild-type controls suppressed IL-17 induced NF-κB activation in human U87-MG cells (Fig. 3C and D). These results suggest that the IKK-related kinases suppress IL-17-induced NF-κB activation. Consistently, the dominant negative form of IKKi significantly increased IL-17-induced downstream gene production in both HeLa cells (Fig. 3E) and U87-MG cells (Fig. 3F). As it is reported that IKKi does not affect IL-17-induced NF-κB activation in mouse airway epithelial cells (1), we reasoned that the dominant negative form of IKKi does not just affect IKKi-mediated function but may also affect TBK1-mediated function. We indeed found that overexpression of the dominant negative IKKi interfered with the association of TBK1 with Act1 in an IL-17-dependent manner, probably because of the competitive binding of the dominant negative form to Act1 (data not shown).
Fig 3.

The IKK-related kinases suppress IL-17-induced NF-κB activation in the human system. (A to D) The IKK-related kinases repress IL-17-induced NF-κB activation. (A and B) HeLa cells infected with control virus (EV) or retrovirus encoding HA-tagged dominant negative TBK1 (DNTBK1) or wild-type TBK1 (WT-TBK1) (A) or HA-tagged DNIKKi or WT-IKKi (B) were treated with IL-17 for the indicated times. Lysates were analyzed by immunoblotting with antibodies against p-IκBα, HA, and actin. (C and D) U87-MG cells infected with retroviral control or retrovirus for HA-tagged DNTBK1 or WT-TBK1 (C) or HA-tagged DNIKKi or WT-IKKi (D) were left untreated or treated with IL-17 for the indicated times. Lysates were analyzed by immunoblotting with antibodies against p-IκBα, HA, and actin. (E and F) The dominant negative form of IKKi significantly increases IL-17-induced gene production. HeLa cells (E) or U87-MG cells (F) were infected with retroviral control or virus for HA-tagged DNIKKi and then left untreated or treated with IL-17 for 2 h. The induction of Ccl20, Ccl2, IL-8, Cxcl2, IL-6, TNF, and KC mRNA was measured by real-time PCR. Graphs in panels E and F show means and SEM; n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test).
We then checked the function of the IKK-related kinases in the mouse system through restoration of IKKi or TBK1 to the DKO MEFs. Restoration of either IKKi or TBK1 suppressed IL-17-induced NF-κB activation (Fig. 4A). Similarly, restoration of IKKi, TBK1, or both significantly inhibited IL-17-induced gene production, while expression of the dominant negative form of TBK1 (the kinase-dead mutant) had no suppressive effect in the DKO cells (Fig. 4B). It is reported that IKKi expression is low in MEFs (25). Similar to the results with DKO MEFs, we observed that restoration of TBK1 to TBK1-deficient MEFs suppressed IL-17-mediated activation of NF-κB and JNK (Fig. 5A). Consistently, restoration of TBK1 inhibited IL-17-induced downstream gene production (Fig. 5B). Although IKKi is shown to be required for IL-17-induced mRNA stability of KC (1), we found that TBK1 was not required for this pathway (data available on request), suggesting that IKKi and TBK1 have differential roles in IL-17R-mediated signaling. Together, our results from both human and mouse systems suggest that the IKK-related kinases play a suppressive and likely redundant role in IL-17-induced NF-κB activation.
Fig 4.
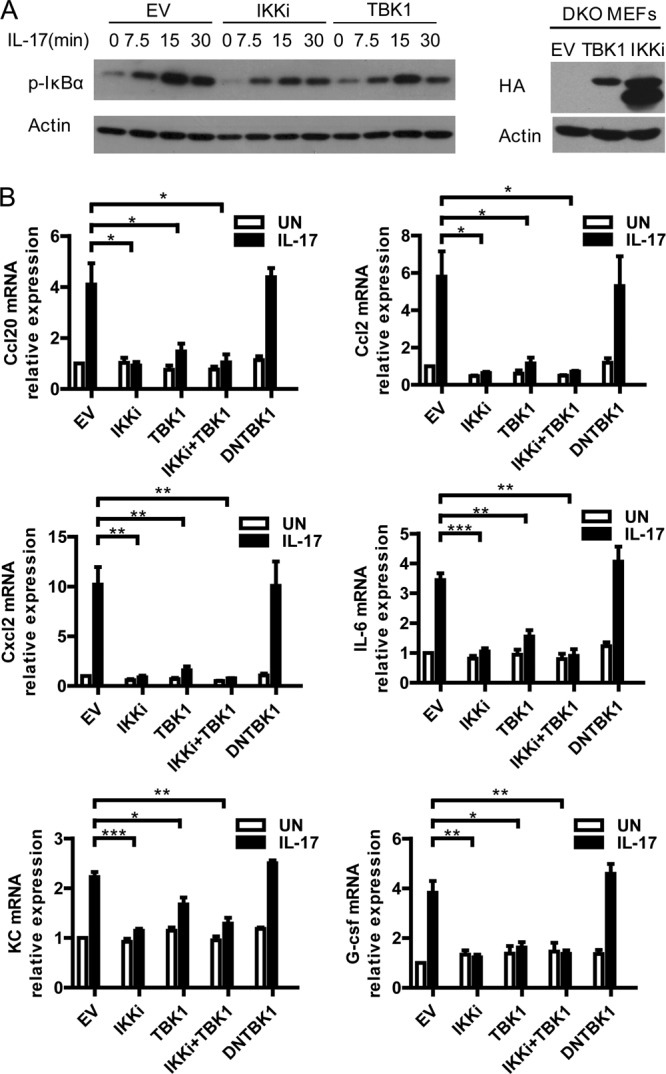
The IKK-related kinases suppress IL-17-induced NF-κB activation in the mouse system. (A) Restoration of the IKK-related kinases to DKO MEFs greatly suppresses IL-17-induced NF-κB activation. DKO MEFs infected with retroviral control or virus for HA-tagged IKKi or HA-tagged TBK1 were left untreated (UN) or treated with IL-17 for the indicated times. Lysates were analyzed by immunoblotting with antibodies against p-IκBα and actin. Levels of expression of HA-TBK1 and HA-IKKi in DKO MEFs are shown on the right. (B) Restoration of the IKK-related kinases to DKO MEFs significantly inhibits IL-17-induced gene production. DKO MEFs infected with retroviral control or virus for HA-tagged IKKi, TBK1, TBK1 plus IKKi, or DNTBK1 were left untreated or treated with IL-17 for 1 h. The induction of Ccl20, Ccl2, Cxcl2, IL-6, KC, and G-CSF mRNA was analyzed by real-time PCR. Graphs show means and SEM; n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test).
Fig 5.

TBK1 suppresses IL-17-induced NF-κB activation. (A) TBK1−/− MEFs infected with retrovirus encoding empty vector (EV) or HA-tagged mouse TBK1 (mTBK1) were treated with IL-17 (50 ng/ml) for 0, 7.5, 15, or 30 min. Lysates were analyzed by immunoblotting with the indicated antibodies. Arrowhead indicates the shifted (phosphorylated) Act1 band. (B) TBK1−/− MEFs reconstituted with HA-tagged mTBK1 or empty vector (EV) were left untreated or treated with IL-17 for 1 h. The induction of Ccl20, Ccl2, Cxcl2, and KC mRNA was measured by real-time PCR. Graphs show means and SEM; n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test).
The IKK-related kinases redundantly phosphorylate Act1 at three serine sites.
To look for the phosphorylation sites in Act1 by the IKK-related kinases, we constructed different GST-tagged deletion mutants of human Act1 (Fig. 6A). These GST fusion proteins were expressed in E. coli and purified with glutathione-Sepharose (Fig. 6A). We then constructed Flag-tagged human TBK1 or Flag-tagged human IKKi. The Flag-tagged proteins were expressed in HEK293T cells and immunoprecipitated by anti-M2 (Flag) antibodies for in vitro kinase assay. IKKi phosphorylated Act1 at the region of aa 1 to 400 but not at the C-terminal region of aa 390 to 565 (Fig. 6B). Further deletion analyses showed IKKi phosphorylated Act1 at the region of aa 121 to 191 as well as aa 191 to 330 (Fig. 6B). TBK1 phosphorylated Act1 at the same regions as IKKi (Fig. 6C). As the IKK-related kinases have been shown to phosphorylate their substrates at serine sites, we then compared Act1 sequences from different species for conserved serine sites in the two regions (Fig. 6D and E). The closely located and conserved serine sites (sites 143, 146, and 149 or sites 156, 159, and 162) in the region of aa 121 to 191 were mutated at the same time (Fig. 6D). The in vitro kinase assays showed that combined mutation of three serine sites (sites 156, 159, and 162) blocked TBK1- or IKKi-mediated phosphorylation of the Act1 region (Fig. 6F). Further single serine mutation showed that Ser 162 was the site of phosphorylation by TBK1 or IKKi in the Act1 region of aa 121 to 191 (Fig. 6F). We then mutated the four conserved serine sites at the region of aa 191 to 330 all together (Fig. 6E) and found that the mutation blocked either TBK1- or IKKi-mediated phosphorylation of the Act1 region (Fig. 6G). Combined mutation of three serine sites (Ser 201, Ser 220, and Ser 233) showed the same blockage of phosphorylation (Fig. 6G). We then mutated the serine sites either singly or with different combinations of two sites and found that IKKi phosphorylated Ser 220 while TBK1 phosphorylated Ser 220 and Ser 233 (Fig. 6G and H). Thus, in the human system, IKKi phosphorylates Act1 at two sites (Ser 162 and Ser 220), while TBK1 phosphorylates Act1 at three sites (Ser 162, Ser 220, and Ser 233). However, all three equivalent serine sites (Ser 147, Ser 209, and Ser222) in mouse Act1 are utilized by both mouse TBK1 and mouse IKKi (Fig. 7).
To see whether the three serine sites in Act1 phosphorylated by the IKK-related kinases in the in vitro assays are utilized in IL-17R signaling, we restored wild-type Act1 or individual serine point mutants of Act1 into Act1-deficient MEFs and then checked IL-17-induced Act1 phosphorylation. Although the levels of IL-17-induced Act1 phosphorylation were not obviously altered in the three single mutants (S147A, S209A, or S222A mutant) compared to wild-type Act1, their mobility shifts appear lower than that of wild-type Act1, indicating that IL-17 induced phosphorylation of all the sites (Fig. 8A). A similar result was found for the Act1 double-site mutant with changes at both Ser 147 and Ser 209 (Fig. 8B). However, when all the three serine sites (3SA) were mutated, IL-17-induced Act1 phosphorylation was severely reduced (Fig. 8C), indicating that the three serine sites are the major ones of Act1 phosphorylation in IL-17R signaling.
Fig 8.

Mouse Act1 is phosphorylated at Ser 147, Ser 209, and Ser 222 upon IL-17 stimulation. Act1−/− MEFs were infected with retrovirus for wild-type Act1 (WT), Act1 single point mutants (S147A, S209A, and S222A) (A), the Act1 double point mutant (S147A and S209A) (B), or the Act1 triple point mutant (3SA: S147A, S209A, and S222A) (C). The MEFs were then left untreated or treated with IL-17 (50 ng/ml) for 15 or 30 min. Lysates were immunoblotted with anti-Act1 and antiactin. Arrowheads indicate the shifted (phosphorylated) Act1 band.
Act1 phosphorylation at the three serine sites inhibits IL-17-mediated NF-κB activation.
Our results showed that the IKK-related kinases were required for IL-17-induced Act1 phosphorylation and suppressed IL-17-induced NF-κB activation (Fig. 1, 3, and 4). As the mouse IKK-related kinases phosphorylated Act1 at the same sites (Ser 147, Ser 209, and Ser 222), we sought to determine whether Act1 phosphorylation at these sites plays a negative role in IL-17-induced NF-κB activation. Consistent with the suppressive role of the IKK-related kinases, mutation of the three Ser sites of Act1 enhanced IL-17-induced NF-κB activation and JNK activation (Fig. 9A). Similarly, mutation of these sites increased IL-17-induced downstream gene production (Fig. 9B). As Act1 association with TRAF6 is essential for IL-17-induced NF-κB activation, we asked whether Act1 phosphorylation at these serine sites by the IKK-related kinases might affect the interaction of Act1 with TRAF6 to exert a suppressive role. The IL-17-induced association of Act1 with TRAF6 was indeed increased in the MEFs doubly deficient in TBK1 and IKKi compared to that in wild-type MEFs (Fig. 9C). Similarly, mutation of the three Ser sites of Act1 enhanced IL-17-induced binding of Act1 to TRAF6 (Fig. 9D). Our results suggest that upon IL-17 stimulation, the IKK-related kinases redundantly phosphorylate Act1 on the three Ser sites, and the Act1 phosphorylation may lead to Act1 conformational change to interfere with its association with TRAF6 for NF-κB and JNK activation.
Fig 9.

Act1 phosphorylation by the IKK-related kinases suppresses IL-17-mediated NF-κB activation. (A) Mutation of the three Act1 phosphorylation sites enhances IL-17-induced NF-κB. Act1−/− MEFs infected with retrovirus encoding wild-type Act1 (WT) or the Act1 mutant (3SA) were left untreated or treated with IL-17 (50 ng/ml) for 15 or 30 min. Lysates were analyzed by immunoblotting with the indicated antibodies. Arrowhead indicates the shifted (phosphorylated) Act1 band. (B) Mutation of the three Act1 phosphorylation sites increases IL-17-induced gene induction. Shown is real-time PCR analysis of Ccl20, Ccl2, Cxcl2, and KC mRNA in Act1−/− MEFs infected as described for panel A followed by treatment with IL-17 (50 ng/ml) for 0 or 1 h. Graphs show means and SEM; n = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t test). (C) IL-17-induced association of Act1 with TRAF6 is increased in DKO MEFs. Wild-type (WT) and IKKi−/− TBK1−/− (DKO) MEFs were left untreated or treated with IL-17 (50 ng/ml) for 7.5 or 15 min. Lysates were immunoprecipitated with anti-TRAF6 and subjected to immunoblot analysis with antibodies against TRAF6 and Act1. (D) Mutation of the three Act1 phosphorylation sites enhances IL-17-induced binding of Act1 to TRAF6. Act1−/− MEFs were infected with retrovirus encoding wild-type Act1 (WT) or the Act1 triple point mutant (3SA) and then left untreated or treated with IL-17A (50 ng/ml) for 7.5 or 15 min. Lysates were immunoprecipitated with anti-TRAF6 and subjected to immunoblot analysis with antibodies against TRAF6 and Act1.
TRAF6 is required for IL-17-induced Act1 phosphorylation.
TRAF3 is essential for the IKK-related kinase-mediated IRF3 phosphorylation in Toll-like receptor (TLR) or RIG-I-like receptor (RLR)-mediated antiviral signaling (17). We have previously shown that TRAF3 suppresses IL-17R-mediated NF-κB activation (38). We reasoned that TRAF3 might be involved in Act1 phosphorylation in IL-17R signaling. We unexpectedly found that TRAF3 deficiency did not affect IL-17-induced Act1 phosphorylation (Fig. 10A). Surprisingly, we found that TRAF6 was required for IL-17-induced Act1 phosphorylation (Fig. 10B), although TRAF6 has been reported to function downstream of Act1 to mediate IL-17-induced NF-κB activation (22, 27). As primary MEFs may vary in their responses to IL-17, we restored TRAF6 to TRAF6-deficient MEFs and found that TRAF6 reconstituted IL-17-induced Act1 phosphorylation (Fig. 10C), confirming that TRAF6 was critical for Act1 phosphorylation. It is worth noting that there was still TRAF6-independent phosphorylation of Act1, as IL-17-induced Act1 phosphorylation was not totally blocked, although it was severely reduced in TRAF6-deficient MEFs (Fig. 10B and C). TRAF6 is a ring-type E3 ubiquitin ligase, and its E3 activity is important for NF-κB activation induced by proinflammatory cytokines like IL-1 and IL-17 (4, 22). We found that the TRAF6 E3 deletion mutation (ΔRing) or E3 point mutation (C70A) did not obviously restore IL-17-induced Act1 phosphorylation, while wild-type TRAF6 did (Fig. 10D), indicating that TRAF6 E3 ligase activity was required for Act1 phosphorylation. While TRAF6 autoubiquitination is essential for its downstream NF-κB activation (4, 22), we found that TRAF6 autoubiquitination was not important for Act1 phosphorylation, as TRAF6 autoubiquitination site mutation (K124R) still restored IL-17-induced Act1 phosphorylation in TRAF6-deficient MEFs (Fig. 10E). TRAF6 activates NF-κB through TAK1 complex in IL-17R signaling (39). We then checked if TAK1 is involved in Act1 phosphorylation and found that TAK1 was not required for IL-17-induced Act1 phosphorylation (data not shown). Thus, our findings show TRAF6 is specifically and uniquely required for Act1 phosphorylation in IL-17R signaling.
Fig 10.

TRAF6 but not TRAF3 is critical for Act1 phosphorylation. (A) Traf3-deficient (Traf3 KO) and wild-type (Traf3 WT) MEFs were treated with IL-17 (50 ng/ml) for 0, 15 or 30 min. Lysates were analyzed by immunoblotting with antibodies against Act1, actin, and TRAF3. (B) TRAF6 is required for IL-17-induced Act1 phosphorylation. WT and Traf6−/− MEFs were stimulated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with antibodies against Act1, TRAF6, p-IκBα, and actin. (C) TRAF6 restores IL-17-induced Act1 phosphorylation in Traf6-deficient MEFs. Traf6−/− MEFs infected with control virus (EV) or retrovirus for Flag-TRAF6 were stimulated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with antibodies against Act1, M2, p-IκBα, and actin. (D) TRAF6 E3 ligase activity is required for Act1 phosphorylation. Traf6−/− MEFs infected with control virus (EV) or retrovirus for Flag-tagged TRAF6 (WT) or its E3 mutants (Flag-Traf6-ΔRing and Flag-Traf6-C70A, respectively) were left untreated or treated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with antibodies against Act1, M2, p-IκBα, and actin. (E) TRAF6 autoubiquitination is not important for Act1 phosphorylation. Traf6−/− MEFs infected with retrovirus for wild-type Flag-TRAF6 (WT) or its autoubiquitination point mutant (Flag-TRAF6-K124R) were left untreated or treated with IL-17 (50 ng/ml) for 15 or 30 min. Lysates were analyzed by immunoblotting with antibodies against Act1, M2, and actin. Arrowheads indicate the shifted (phosphorylated) Act1 band. L.E., long exposure; S.E., short exposure.
TRAF6 is essential for IL-17-induced TBK1 activation and its association with Act1.
As TRAF6 is required for Act1 phosphorylation, we determined whether TRAF6 mediates the activation of the IKK-related kinases. We found that siRNA-mediated TRAF6 knockdown also blocked IL-17-induced TBK1 phosphorylation (Fig. 11A), suggesting that TRAF6 is required for IL-17-induced TBK1 activation. We next asked whether TRAF6 is required for IKKi activation. As there is no p-IKKi antibody, we did an in vitro kinase assay and found that TRAF6 appeared not to be essential for IKKi activation, although the assay did not show strong kinase activity (Fig. 11B). It is reported that Act1 is required for IKKi activation (1). We also found that Act1 was essential for TBK1 activation (Fig. 11C). These data suggest that the IKK-related kinases are differentially activated. The detailed activation mechanisms remain to be further explored.
Fig 11.

TRAF6 is essential for IL-17-induced TBK1 activation and its association with Act1. (A) TRAF6 is required for TBK1 activation. HeLa cells were transfected with either scrambled (si-NC) or Traf6 (si-Traf6) siRNA oligonucleotides. After 72 h, the cells were left untreated or treated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with the indicated antibodies. (B) TRAF6 is not essential for IKKi activation. WT and Traf6−/− MEFs infected with retrovirus for Flag-IKKi were stimulated with IL-17 (50 ng/ml) for the indicated times. Lysates were immunoprecipitated with anti-M2 and subjected to in vitro kinase assay. Whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (C) Act1 is required for TBK1 activation. HeLa cells were transfected with either scrambled (si-NC) or Act1 siRNA (si-Act1) oligonucleotides. After 72 h, the cells were left untreated or treated with IL-17 (50 ng/ml) for the indicated times. Lysates were analyzed by immunoblotting with the indicated antibodies. (D) TRAF6 is critical for IL-17-induced association of Act1 with the IKK-related kinases. WT or Traf6−/− MEFs infected with retrovirus encoding HA-TBK1 were treated with IL-17 (50 ng/ml) for 0, 15, or 30 min, and then lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis with the indicated antibodies. WCL, whole-cell lysates. (E) TRAF6 E3 ligase activity is critical for IL-17-induced Act1 association with the IKK-related kinases. Traf6−/− MEFs infected with control virus (EV) or retrovirus encoding Flag-TRAF6 (WT) or its E3 mutants (Flag-Traf6-ΔRing and Flag-Traf6-C70A) were left untreated or treated with IL-17 (50 ng/ml) for the indicated times. Lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis with the indicated antibodies. (F) The dominant negative form of TRAF6 severely reduces IL-17-induced Act1 association with the IKK-related kinases in human cells. Human U87-MG cells infected with retroviral control or retrovirus for Flag-TRAF6-ΔRing (the dominant negative form) were treated with IL-17 (50 ng/ml) for 0, 15, or 30 min. Lysates were immunoprecipitated with anti-Act1, followed by immunoblot analysis with the indicated antibodies.
We then determined whether TRAF6 is also required for the association of Act1 with the IKK-related kinases. We found that TRAF6 deficiency completely blocked IL-17-induced association of Act1 with TBK1 (Fig. 11D). TRAF6 deficiency also reduced but did not completely block IL-17-induced binding of Act1 to IKKi (Fig. 11D), indicating that IKKi has TRAF6-dependent and -independent roles, while TBK1 has only TRAF6-dependent function in IL-17R signaling. Consistent with the dual roles of IKKi, we also found that mutation (the 3SA mutant) of Act1 phosphorylation by the IKK-related kinases had no effect on IL-17-induced mRNA stability of KC and did not affect IL-17-induced association of Act1 with IKKi (data not shown). We then checked whether TRAF6 E3 ligase activity was important for the association of Act1 with the IKK-related kinases. We found that the TRAF6 E3 deletion mutant or point mutant failed to restore IL-17-induced association of Act1 with the IKK-related kinases in the TRAF6-deficient MEFs, while wild-type TRAF6 did restore this association (Fig. 11E), suggesting that TRAF6 E3 activity was critical for the association. We then asked if the human system has the same phenomenon as the mouse system. The ring deletion mutant of human TRAF6 was used as a dominant negative form to test its effect on the association of Act1 with the IKK-related kinases in human U87-MG cells. The dominant negative form severely reduced IL-17-induced binding of Act1 to IKKi or phosphorylated TBK1 as well as NF-κB activation (Fig. 11F), indicating that TRAF6 E3 ligase activity was also critical for IL-17-induced association of Act1 with the IKK-related kinases in the human system. We next sought to determine whether TRAF6 directly associated with the IKK-related kinases for their subsequent interaction with Act1. We did not detect IL-17-induced association of TRAF6 with the IKK-related kinases (data not shown), indicating that TRAF6 may indirectly mediate the association of Act1 with the IKK-related kinases.
DISCUSSION
IL-17 is a key proinflammatory cytokine critically involved in the pathogenesis of variety of inflammatory and autoimmune disorders. Act1 and TRAF6 are essential signaling components for IL-17-mediated activation of NF-κB. In this study, we discovered a new role for the IKK-related kinases (TBK1 and IKKi) in suppressing IL-17-induced NF-κB activation through TRAF6-dependent Act1 phosphorylation. Both kinases directly phosphorylated Act1 on three serine sites for their suppressive effects. TRAF6 was critical for IL-17-induced TBK1 activation and the association of Act1 with the IKK-related kinases, and the consequent Act1 phosphorylation. Our study demonstrates a negative-feedback loop (Act1-TRAF6-TBK1/IKKi-Act1) in regulation of IL-17R-mediated NF-κB activation (Fig. 12).
Fig 12.

Proposed model for the IKK-related kinase-mediated negative-feedback loop in the regulation of IL-17-induced NF-κB activation.
Very recently, Bulek et al. reported that IKKi phosphorylates Act1 on Ser 311 to mediate TRAF6-independent mRNA stability (1). We show here that TBK1 is another direct kinase for Act1 phosphorylation. We found that TBK1 was activated and associated with Act1 upon IL-17 stimulation. Instead of regulating TRAF6-independent mRNA stability, TBK1 phosphorylated Act1 on three other serine sites and suppressed IL-17-mediated TRAF6-dependent NF-κB activation in both human and mouse systems. Here we show that in addition to the reported TRAF6-independent role, IKKi also has a TRAF6-dependent role. We found that TBK1 and IKKi did not depend on each other to phosphorylate Act1, they phosphorylated Act1 on the same three Ser sites, and Act1 phosphorylation on these sites interfered with its association with TRAF6 for continuous positive signaling, suggesting that IKKi plays a redundant role with TBK1 in Act1 phosphorylation to repress TRAF6-dependent NF-κB activation in IL-17R signaling. In Bulek's study, IL-17-induced NF-κB activation was not shown to be affected in IKKi-deficient airway epithelial cells, consistent with the redundant role of the IKK-related kinases in our study. However, we noticed in that report that IKKi has some positive effects on the gene production induced by IL-17 alone, although the effects are minor compared to the essential role of IKKi for IL-17-mediated mRNA stability of the genes induced by TNF in airway epithelial cells. In this study, we found that IKKi and TBK1 redundantly mediated negative effects on IL-17-induced gene production. The difference may be due to the fact that the negative effects mediated by the two kinases together exceed the minor positive effect by IKKi alone. It may also be due to cell-type-specific effects.
TBK1 and IKKi are structurally related to IKK kinases (IKKα and IKKβ) and share some substrates, such as IκBα and P65, for NF-κB activation (9, 30). Instead of activating NF-κB, the IKK-related kinases repressed IL-17-mediated NF-κB activation. This could be due to their substrate specificity, because we observed that TBK1 and IKKi but not IKKα and IKKβ phosphorylated Act1, although Act1 has been reported to associate with the IKK complex (20, 21). TAK1 was also found not to be involved in Act1 phosphorylation. We noticed that IL-17-induced Act1 phosphorylation was not totally blocked, although it was severely impaired, by the double deficiency of TBK1 and IKKi, suggesting that there may be other kinases for Act1. Although the IKK-related kinases have differential expression patterns in that TBK1 is constitutively expressed while IKKi is an inducible kinase (9, 30), the two kinases redundantly phosphorylate mouse Act1 on the three Ser sites (Ser 147, Ser 209, and Ser 222). It is reported that Act1 is also phosphorylated on Ser 311 by IKKi in IL-17R signaling (1). Consistent with the extra site, our data showed that IL-17-induced Act1 phosphorylation was not totally blocked in the mutant (3SA) with changes of the three Act1 phosphorylation sites, although it was severely reduced. We further found that mutation of the three sites (3SA) did not affect Act1 association with IKKi or IL-17-induced mRNA stability of KC, indicating that the phosphorylation of these three Ser sites may not affect Ser 311 phosphorylation, which is required for IL-17-induced mRNA stability of KC. On the other hand, Ser311 phosphorylation did not affect the TRAF6-dependent NF-κB pathway, and Ser 311 mutation only reduced and did not block IL-17-induced Act1 phosphorylation in Bulek's study, suggesting that Ser 311 phosphorylation may not affect the phosphorylation of the three Ser sites in Act1 described in our study. It is worth noting that the shifted band detected by the Act1 antibody is not sensitive enough to detect all Act1 phosphorylation. It remains to be determined whether there are other potential phosphorylation sites in Act1 by the sensitive mass spectrometry approach in addition to the four determined sites.
The IKK-related kinases have been well studied for their roles in antiviral signaling. TRAF3, but not TRAF6, is the upstream adaptor for activation of the IKK-related kinases to phosphorylate the transcription factor IRF3 for antiviral responses (10, 24). However, we found that TRAF6, but not TRAF3, was important for IL-17-induced Act1 phosphorylation, indicating the functional specificity for the IKK-related kinases in IL-17R signaling. We have previously reported that Act1 is an upstream adaptor to recruit TRAF6 to IL-17R (27). Considering that TRAF6-dependent Act1 phosphorylation plays a negative role in IL-17-induced NF-κB activation and that IL-17-induced binding kinetics of Act1 and the IKK-related kinases is slower than that of Act1 and TRAF6, it is likely that after TRAF6 activation, Act1 is phosphorylated to immediately turn off the NF-κB pathway. Consistent with that, the double deficiency of the IKK-related kinases or mutation of the Act1 phosphorylation sites (3SA) enhanced IL-17-induced interaction of Act1 and TRAF6, suggesting that Act1 phosphorylation may lead to its conformation change which interferes with its association with TRAF6. Although TBK1 and IKKi redundantly phosphorylate Act1 on the same three Ser sites, the activation of the kinases and their association with Act1 are differentially regulated. TRAF6 is essential only for TBK1 activation, while Act1 is required for activation of both kinases. The regulation of Act1 association with TBK1 or IKKi is also different. IL-17-induced association of TBK1 with Act1 is totally TRAF6 dependent, while the association of Act1 with IKKi is both TRAF6 dependent and independent. Although the detailed mechanisms for the differential regulation remain to be further identified, these data are consistent with the differential roles of the IKK-related kinases in IL-17R signaling; that is, TBK1 only regulates TRAF6 dependent signaling, while IKKi plays dual roles in both TRAF6-dependent NF-κB activation and TRAF6-independent mRNA stability. The redundant and differential roles of TBK1 and IKKi have also been observed in other signaling involved in antiviral and antibacterial immunity and in cancer (5, 9, 30).
The E3 ubiquitin ligase activity and autoubiquitination of TRAF6 are essential for IL-17-induced NF-κB activity, similar to other TRAF6-mediated signaling pathways (4, 22). Here we found TRAF6 E3 ligase activity was also critical for IL-17-induced association of Act1 with the IKK-related kinases and consequent Act1 phosphorylation. However, TRAF6 autoubiquitination was found to not be important for IL-17-induced TRAF6-dependent Act1 phosphorylation, suggesting that TRAF6 likely ubiquitinates its downstream signaling molecules for Act1 phosphorylation. We did not detect direct association of TRAF6 with the IKK-related kinases in IL-17R signaling. Considering that TRAF6 mediates Lys 63-type ubiquitination and we did not detect IL-17-induced Lys 63 ubiquitination of Act1 (data not shown), it is not likely for TRAF6 to ubiquitinate Act1 for its association with the IKK-related kinases. Therefore, TRAF6 may ubiquitinate unknown molecules to mediate the association of Act1 with the IKK-related kinases for subsequent Act1 phosphorylation.
In conclusion, we demonstrate that TBK1 is another direct kinase for Act1 phosphorylation in IL-17R signaling. TBK1 and IKKi redundantly phosphorylate Act1 to suppress IL-17-induced NF-κB activation. TRAF6 is required for IL-17-induced association of Act1 with the IKK-related kinases and consequent phosphorylation of Act1. Our findings identify a new role for the IKK-related kinases in suppressing IL-17-induced TRAF6-dependent NF-κB activation and demonstrate a negative-feedback loop mechanism in the regulation of IL-17R signaling.
ACKNOWLEDGMENTS
This work was supported by grants from National Natural Science Foundation of China (no. 30930084, no. 91029708, and no. 30871298) and Science and Technology Commission of Shanghai Municipality (no. 10JC1416600).
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Bulek K, et al. 2011. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat. Immunol. 12: 844–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chang SH, Dong C. 2009. IL-17F: regulation, signaling and function in inflammation. Cytokine 46: 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang SH, Park H, Dong C. 2006. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J. Biol. Chem. 281: 35603–35607 [DOI] [PubMed] [Google Scholar]
- 4. Chen ZJ. 2005. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7: 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clément JF, Meloche S, Servant MJ. 2008. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 18: 889–899 [DOI] [PubMed] [Google Scholar]
- 6. Cua DJ, Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10: 479–489 [DOI] [PubMed] [Google Scholar]
- 7. Gaffen SL. 2011. Recent advances in the IL-17 cytokine family. Curr. Opin. Immunol. 23: 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaffen SL. 2009. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9: 556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Häcker H, Karin M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006: re13. [DOI] [PubMed] [Google Scholar]
- 10. Hacker H, et al. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439: 204–207 [DOI] [PubMed] [Google Scholar]
- 11. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. 2007. IL-17 enhances chemokine gene expression through mRNA stabilization. J. Immunol. 179: 4135–4141 [DOI] [PubMed] [Google Scholar]
- 12. Hartupee J, et al. 2009. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J. Immunol. 182: 1660–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hemmi H, et al. 2004. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199: 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho AW, et al. 2010. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J. Immunol. 185: 1063–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishii KJ, et al. 2008. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451: 725–729 [DOI] [PubMed] [Google Scholar]
- 16. Iwakura Y, Ishigame H, Saijo S, Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity 34: 149–162 [DOI] [PubMed] [Google Scholar]
- 17. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11: 373–384 [DOI] [PubMed] [Google Scholar]
- 18. Kishore N, et al. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277: 13840–13847 [DOI] [PubMed] [Google Scholar]
- 19. Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 Cells. Annu. Rev. Immunol. 27: 485–517 [DOI] [PubMed] [Google Scholar]
- 20. Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. 2000. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 97: 10494–10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li X, et al. 2000. Act1, an NF-kappa B-activating protein. Proc. Natl. Acad. Sci. U. S. A. 97: 10489–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu C, et al. 2009. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci. Signal. 2: ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McAleer JP, Kolls JK. 2011. Mechanisms controlling Th17 cytokine expression and host defense. J. Leukoc. Biol. 90: 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oganesyan G, et al. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439: 208–211 [DOI] [PubMed] [Google Scholar]
- 25. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to Toll-like receptor activation and viral infection. J. Exp. Med. 199: 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qian Y, Kang Z, Liu C, Li X. 2010. IL-17 signaling in host defense and inflammatory diseases. Cell. Mol. Immunol. 7: 328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qian Y, et al. 2007. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8: 247–256 [DOI] [PubMed] [Google Scholar]
- 28. Schwandner R, Yamaguchi K, Cao Z. 2000. Requirement of tumor necrosis factor receptor-associated factor (Traf)6 in interleukin 17 signal transduction. J. Exp. Med. 191: 1233–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen F, et al. 2009. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci. Signal. 2: ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen RR, Hahn WC. 2011. Emerging roles for the non-canonical IKKs in cancer. Oncogene 30: 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi P, et al. 2011. Persistent stimulation with interleukin-17 desensitizes cells through SCFbeta-TrCP-mediated degradation of Act1. Sci. Signal. 4: ra73. [DOI] [PubMed] [Google Scholar]
- 32. Sønder SU, et al. 2011. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J. Biol. Chem. 286: 12881–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song X, et al. 2011. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat. Immunol. 12: 1151–1158 [DOI] [PubMed] [Google Scholar]
- 34. Sun D, et al. 2011. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 12: 853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tenoever BR, et al. 2007. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 315: 1274–1278 [DOI] [PubMed] [Google Scholar]
- 36. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. 2009. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10: 1215–1221 [DOI] [PubMed] [Google Scholar]
- 37. Zepp JA, et al. 2012. Cutting edge: TNF receptor-associated factor 4 restricts IL-17-mediated pathology and signaling processes. J. Immunol. 189: 33–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu S, et al. 2010. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J. Exp. Med. 207: 2647–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu S, et al. 2012. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nat. Med. 18: 1077–1086 [DOI] [PubMed] [Google Scholar]
- 40. Zhu S, Qian Y. 2012. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin. Sci. (Lond.) 122: 487–511 [DOI] [PubMed] [Google Scholar]