

Abstract
B cell responses are required for resistance to Toxoplasma gondii; however, the events that lead to production of class-switched antibodies during T. gondii infection have not been defined. Indeed, mice challenged with the parasite exhibited an expansion of T follicular helper cells and germinal center B cells in the spleen. Unexpectedly, this was not associated with germinal center formation and was instead accompanied by profound changes in splenic organization. This phenomenon was transient and was correlated with a decrease in expression of effector proteins that contribute to splenic organization, including lymphotoxins α and β. The importance of lymphotoxin was confirmed, as the use of a lymphotoxin β receptor agonist results in partial restoration of splenic structure. Splenectomized mice were used to test the splenic contribution to the antibody response during T. gondii infection. Analysis of splenectomized mice revealed delayed kinetics in the production of parasite-specific antibody, but the mice did eventually develop normal levels of parasite-specific antibody. Together, these studies provide a better understanding of how infection with T. gondii impacts the customized structures required for the optimal humoral responses to the parasite and the role of lymphotoxin in these events.
INTRODUCTION
The intracellular protozoan parasite Toxoplasma gondii is the causative agent of toxoplasmosis, an important opportunistic infection of humans and livestock (20, 38). In mice, the acute stage of infection with T. gondii is characterized by systemic parasite dissemination and significant T and B cell activation. Control of parasite replication and transition to the chronic phase of infection are dependent on T and B cells. Although this infection has been used as a model to study cell-mediated immunity, antibodies are also critical for resistance to T. gondii; B cell-deficient mice succumb to T. gondii during the chronic phase of infection but can be rescued by passive antibody transfer (25, 39). Despite these initial studies, many questions remain about the T and B cell interactions that promote antibody responses during infection.
It has long been established that antigen-specific antibody responses develop within germinal centers (GCs), specialized regions within the lymphoid follicle that promote the T cell-B cell interactions that are crucial for isotype switching and affinity maturation. Recent work has demonstrated that the T cells involved in these events are T follicular helper (TFH) cells, a T helper cell subset defined by expression of the transcription factor Bcl6 and the chemokine receptor CXCR5, which promotes entry into the GC (5, 23, 34, 41, 51; for a review, see references 9 and 27.) This has led to the idea that the TFH cell subset is defined, at least in part, by location; although TFH cells have been found both within and outside the GC, it is unclear whether TFH cells outside GCs retain their antibody-promoting function (52).
The organization of secondary lymphoid organs is configured to facilitate the variety of cellular interactions and subsequent proliferation and maturation that occur in response to immunological challenge. Thus, the spleen is composed of red and white pulp regions; within the white pulp, lymphoid structures are further divided into B and T cell zones, the organization of which is predominantly orchestrated by chemokine signaling. In T cell zones, migration of lymphocytes is mediated by the chemokines CCL19 and CCL21, while within the B cell follicle, the chemokines CXCL13 and lymphotoxin α (LTα) and LTβ provide a feedback loop between stromal cells and B cells that is essential for the organization of this structure (31). This arrangement promotes the interaction of T and B cells at the borders of these zones, as well as within the GCs, required for the development of antibody responses (1).
In order to better understand the events that lead to antibody production during T. gondii infection, TFH cells and GC B cells were studied over the course of infection. Our studies revealed that there is an increase in TFH cells and GC B cells during acute infection; unexpectedly, this expansion is accompanied by a temporary disruption of the splenic architecture, including GCs. This disorganization coincides with a decrease in the expression of CXCL13, as well as LTα and LTβ, but can be partially reestablished through the use of an LTβ receptor (LTβR) agonist antibody. Although there is substantial parasite-specific isotype-switched antibody measured in the serum during infection, splenectomy results in only a delay in the kinetics of antibody production. Thus, as splenectomized mice do not have a severe defect in antibody production, it appears that the disorganization of the spleen may generally inhibit the production of spleen-derived parasite-specific antibody during infection.
MATERIALS AND METHODS
Animals and infections.
C57BL/6J and splenectomized C57BL/6J mice were obtained from Taconic at 6 to 8 weeks of age and kept under specific-pathogen-free conditions at the University of Pennsylvania animal facility. Tachyzoites of the Prugniaud strain of T. gondii were maintained in vitro through serial passages in human fibroblast monolayers. For infection, parasites were isolated by serial needle passage and filtration through 5-μm filters. Mice were infected intraperitoneally (i.p.) with 104 parasites in 100 μl phosphate-buffered saline (PBS). CD4+ T cell depletion was performed by treating mice i.p. with 0.5 mg anti-CD4 (GK1.5) antibody (BioXCell) every 3 days. For interleukin 12 (IL-12) treatment, mice were given 250 ng recombinant murine IL-12p70 (rmIL-12p70) (PeproTech) i.p. on days 0, 1, 2, 3, 4 and were used on day 5. LTβR agonist treatment was performed using an LTβR agonist antibody (4H8) provided by Carl Ware (Sanford Burnham Medical Research Institute). Mice were treated with 100 μg of LTβR agonist antibody i.p. every 3 days during T. gondii infection, as indicated. All experimental procedures with mice were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Cell purification and flow cytometry.
Single-cell suspensions were purified from mouse spleens and lymph nodes (LN) (cervical, axial, brachial, inguinal, or sacral, as indicated). Briefly, tissues were collected and dissociated through a 40-μm strainer, and red blood cells were lysed using 0.86% ammonium chloride (Sigma). Cells were counted, washed in flow cytometry buffer (1% bovine serum albumin [BSA] [Sigma], 2 mM EDTA [Invitrogen] in PBS), and blocked with 2.4G2 (BioXCell) and Rat IgG (Invitrogen) while stained with monoclonal antibodies (MAbs). Samples were acquired using a FACSCanto II or LSRFortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc.). The following MAbs against mouse antigens were used as fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein (PerCP)-Cy5.5, PE-Cy7, allophycocyanin (APC), APC-ef780, Pacific blue, AF450, or biotin conjugates: CD45R (B220; RA3-6B2), CD278 (ICOS; C398.4A), and IgD (11-26) (eBioscience); CD4 (RM4-5), CD8a (53-6.7), CD95 (Fas; Jo2), and CXCR5 (2G8) (BD Biosciences); and CD3 (17A2), CD19 (6D5), and CD279 (PD-1; J43) (Biolegend). Biotinylated antibodies were secondarily stained with PerCP-Cy5.5-conjugated (BD Biosciences) or APC-conjugated (eBioscience) streptavidin. FITC- or biotin-conjugated peanut agglutinin (PNA) was obtained from Vector Laboratories. The plots shown are on a Logicle scale.
Histology and immunohistochemistry.
For histological sectioning, mouse spleens and LN (cervical, axial, brachial, inguinal, and/or sacral, as indicated) were isolated and placed in 10% buffered formalin (Sigma). Tissues were paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E). All histopathology slides were reviewed by a board-certified veterinary anatomic pathologist. For immunohistochemical analysis, fresh tissues were embedded in OCT (Optimal Cutting Temperature; Sakura) and flash frozen on dry ice; 6-μm sections were cut on a Microm cryostat (Leica). The sections were fixed in either ice-cold 75% acetone with 25% ethanol or pure acetone. The sections were stained with anti-CD3 (145-2C11; eBioscience), anti-B220 (RA3-6B2) and anti-CD35 (8C12) (Pharmingen), anti-CD169 (MOMA-1; Serotec), FITC-conjugated PNA (Vector Laboratories), and DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen). Anti-rat Cy3, anti-hamster Cy3, anti-rat Dylight 488, and anti-goat Dylight 549 (Jackson Immunoresearch) were used as secondary or tertiary antibodies. Images were acquired using a Nikon Eclipse E600 microscope, and Nikon NIS Elements software was used for the capture and overlay of images. Images of the spleen were acquired using a confocal microscope (Leica 2CS SP5) on an automated XY stage and tiled together with the imaging software (Leica Microsystems LAS AF).
ELISA.
Soluble Toxoplasma antigen (STAg)-specific serum IgM and IgG levels were determined by enzyme-linked immunosorbent assay (ELISA) using an IgM-, total-IgG-, IgG2a-, or IgG2c-specific MAb (Southern Biotech); Immulon 4HBX plates (Thermo Fisher Scientific) were coated overnight at 4°C with 5 μg STAg/ml in 50 μl/well. The plates were then blocked with 10% FBS and incubated with serial dilutions of sera, followed by a peroxidase-coupled anti-IgM, IgG, IgG2a, or IgG2c conjugate and SureBlue 3,3′,5,5′-tetramethylbenzidine or ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate (KPL).
Statistical analysis.
Statistical significance was assessed using Student's t test. A P value of ≤0.05 was considered significant. Statistical analysis was performed using Prism software (GraphPad Software, Inc.).
RESULTS
Analysis of the early humoral response to T. gondii.
In order to evaluate the development of the antibody response to T. gondii, wild-type C56BL/6J mice were infected with type II Prugniaud parasites. A kinetic analysis of populations of T cells expressing TFH cell markers (CXCR5+ and ICOShi), as well as B cells expressing GC B cell markers (PNA+ IgD− Fas+), was performed. Significant populations of both TFH-like cells and GC-like B cells peaked at day 14 postinfection (Fig. 1A to D). At this time point, the TFH-like cell population reached approximately 15% of total CD4+ T cells and represented between 30 and 40% of activated (CD62Llo) CD4+ T cells (Fig. 1B). The population of phenotypically GC B cells increased in number by more than 10-fold and at 14 days postinfection represented approximately 5% of total B cells (Fig. 1D). Analysis on day 28 postinfection revealed that, although these populations contracted over time, they were maintained at or above naïve levels (Fig. 1B and D). Analysis of parasite-specific immunoglobulin in the sera of infected mice revealed that parasite-specific IgM can be detected in the serum as early as day 7 postinfection but that it reaches maximum titers by day 14 and is maintained into the chronic stage of infection (Fig. 1G), while isotype-switched antibody is detectable by day 14 but continues to rise throughout infection (Fig. 1E and F).
Fig 1.

T. gondii-infected mice develop TFH cells, GC B cells, and antibody responses in a CD4+ T cell-dependent fashion. (A to D) At several time points postinfection, single-cell suspensions were prepared from the spleen and analyzed for TFH cell (A and B) or GC B cell (C and D) markers by flow cytometry, gated on CD4+ T cells (A) or B220+ B cells (C). Gates represent TFH cells (A) and GC B cells (C), respectively. (B and D) Total cell numbers are presented as means and standard errors of the mean (SEM). (E to G) Antibody in the serum was measured at various time points and assayed by ELISA for total parasite-specific IgG (E), IgG2c (F), and IgM (G). (H and I) CD4+ T cells were depleted beginning 5 days postinfection. Spleens were analyzed at 21 days postinfection for GC B cell development shown as the gated population by flow cytometry (H) and quantified for cell numbers (I). (J to L) Antibody in the serum was measured at 21 days postinfection and assayed by ELISA for total parasite-specific IgG (J), IgG2c (K), and IgM (L). OD450, optical density at 450 nm.
The kinetics of the Toxoplasma-specific IgG response correlated with the expansion of the TFH-like cell population, suggesting that production of parasite-specific class-switched antibody is CD4+ T cell dependent. However, there are reports describing CD4+ T cell-independent antibody production (10, 18, 19, 45, 48). To examine the necessity for CD4+ T cells for the immunoglobulin response, CD4+ T cells were depleted beginning 5 days postinfection, and depletion was maintained continuously until day 20 postinfection, when GC B cells and serum antibody levels were evaluated. At that time point, CD4+ T cell depletion was greater than 99% (data not shown). Previously published work indicated that CD4+ T cell depletion does not result in an increased parasite burden (16, 21). In infected animals treated with the isotype control, GC B cell populations developed and were maintained normally, but in mice treated with a CD4+ T cell-depleting antibody, GC B cells were reduced by ∼20-fold (Fig. 1H and I). Furthermore, parasite-specific serum IgG and IgM titers were also significantly decreased (Fig. 1J to L). Similar defects in both IgG and IgM titers after CD4+ T cell depletion have been demonstrated in other infection models, indicating that some IgM production may also be CD4+ T cell dependent (6, 11, 50). These results are consistent with a known role for CD4+ T cells in the development of a GC B cell and isotype-switched antibody response.
Alterations in splenic architecture during acute toxoplasmosis.
As part of the characterization of the B cell response to T. gondii, spleens were isolated from naïve or acutely or chronically infected mice and sectioned for immunohistochemical analysis to visualize GC formation. H&E staining of splenic sections revealed that during acute infection, there is a significant loss of the distinct borders between the red and white pulp regions (Fig. 2A). A similar loss of structural organization was observed in mice infected orally with T. gondii and in the more resistant BALB/c mouse strain (data not shown). In addition, the compact and organized white pulp regions visible in naïve spleens were substantially smaller and fewer than those in the acutely infected spleen. However, by the chronic phase of infection, the compartmentalization of red and white pulp regions was largely restored (Fig. 2A).
Fig 2.
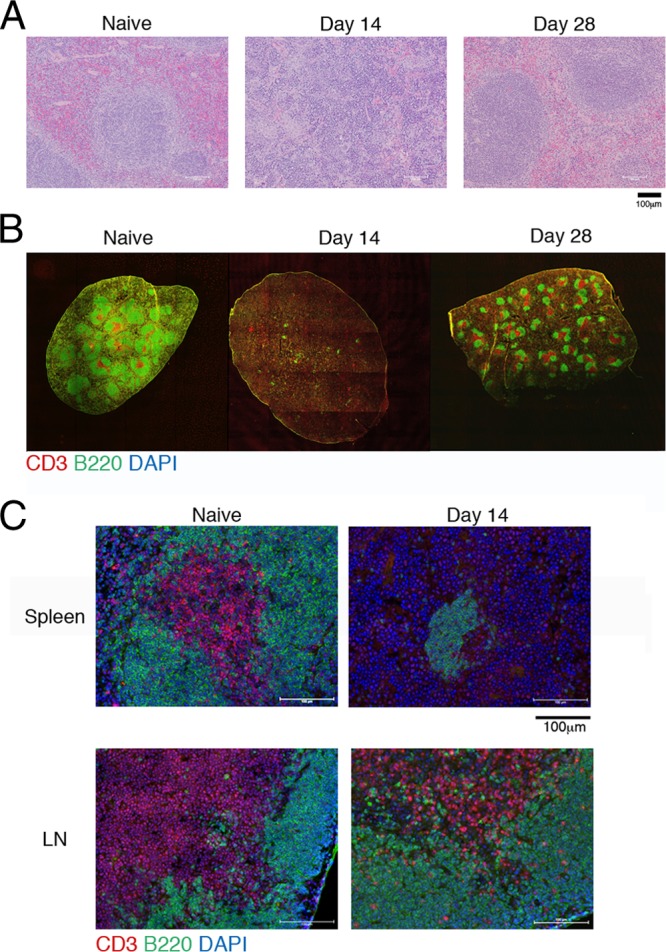
Splenic organization is transiently disrupted during acute infection. (A) Splenic sections were taken at various time points during infection and analyzed histologically using H&E staining. (B) Immunofluorescent staining of splenic sections was performed using CD3 (red), B220 (green), and DAPI (blue). The images are shown at ×10 magnification. Sections were tiled together to visualize large areas of the spleen in each image. (C) Immunofluorescent staining of sections from the spleens or LN of naïve or infected mice was performed using CD3 (red), B220 (green), and DAPI (blue); magnification, ×20.
Further analysis of the localization of T and B cell zones in the spleen was performed using immunohistochemistry. Using anti-CD3 and anti-B220 antibodies to visualize the T and B cell populations, an automated XY stage was utilized to take serial images of the full section, which were assembled to provide a comprehensive view of the organization of these lymphocyte subsets. Although activated T and B cell numbers were increased in infected mice (Fig. 1), the organization of these cells was significantly altered: B and T cell zones were lost or greatly diminished in frequency and size at the early time points (Fig. 2B). However, this disorganization is transient, and the splenic architecture returns to normal by day 28 postinfection (Fig. 2B). It should also be noted that, although the size and number of B and T cell zones in the spleen were substantially decreased 14 days postinfection, at that time point, the LN retained the follicular organization of B and T cell zones at levels comparable to those of the naïve structures (Fig. 2C).
Follicular dendritic cells (FDCs) and fibroblast reticular cells (FRCs) are stromal cells in the spleen that provide chemokine signals to B and T cells, respectively, to direct their organization into distinct zones. Immunohistochemistry staining for FDCs, identified as CD35+ cells, revealed a nearly complete loss of FDCs in spleen sections during acute infection, while FRCs, identified by ERTR-7, were somewhat reduced during acute infection, but the cells were restored in the chronic phase, indicating that changes in the stromal cell networks are correlated with the decline of B and T cell zones (Fig. 3A to C). Metallophillic macrophages, defined by MOMA-1 expression, line the outer rim of the white pulp in the marginal zone; these macrophages were absent during acute infection but were present in the spleens of chronically infected mice (Fig. 3D). Furthermore, despite the presence of phenotypically GC B cells and TFH cells by flow cytometry (Fig. 1), staining for germinal center structures, identified as PNA+ IgD− regions, revealed that in uninfected mice, there are small numbers of GCs, but they were absent during acute infection (Fig. 3E). By day 28 postinfection, GCs are again detectable, with a trend toward having slightly more and larger GCs than those found in naïve spleens (Fig. 3E). Together, these data reveal that acute infection with T. gondii leads to profound changes in splenic architecture, characterized by the loss of stromal networks and, despite the presence of TFH and GC B cells, a complete absence of GCs.
Fig 3.
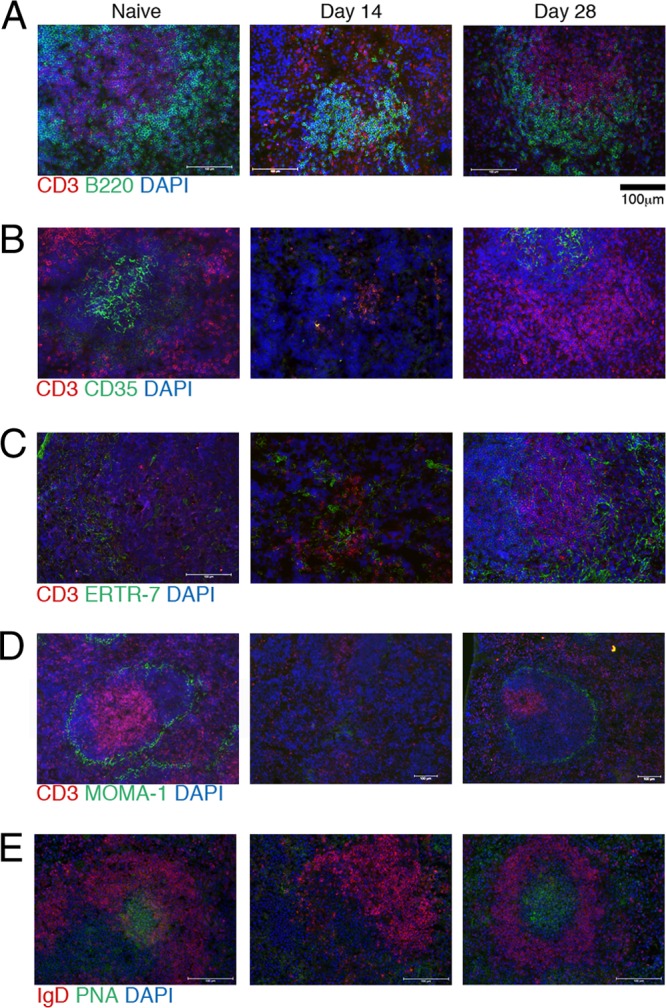
Acute infection results in the disorganization of splenic structures. (A to E) Splenic sections were taken from infected mice at the indicated time points and stained for immunofluorescent analysis. Structures were evaluated by staining for T and B cell zones with CD3 (red), B220 (green), and DAPI (blue) (A); follicular DCs with CD3 (red), CD35 (green), and DAPI (blue) (B); fibroblast reticular cells with CD3 (red), ERTR-9 (green), and DAPI (blue) (C); metallophillic macrophages using CD3 (red), MOMA-1 (green), and DAPI (blue) (D); or GCs using IgD (red), PNA (green), and DAPI (blue) (E).
EMH alone does not lead to follicular disorganization during acute toxoplasmosis.
H&E staining of the spleen in acutely infected mice revealed extramedullary hematopoiesis (EMH) beyond the baseline levels seen in naïve adult mice, characterized by the presence of megakaryocytes (asterisk) and erythroid (arrowheads) and myeloid (arrows) precursors at various stages of maturation (Fig. 4A). As EMH represents a substantial infiltration of hematopoietic precursor cells, normally located in the bone marrow (BM) and the circulation, into the spleen, it was possible that the increase in cellularity could contribute to the disruption of the splenic architecture (22). To further quantify the effects of infection on EMH, spleens and bone marrow were taken 14 or 28 days postinfection and analyzed by flow cytometry for the presence of hematopoietic precursors compared to precursor populations observed in naïve mice. Within the spleen, the numbers of bone marrow Lin− Sca1+ Kithi cells (LSKs) and multipotent progenitors (MPPs) were increased during acute infection and reduced to near naïve levels by the chronic phase of infection (Fig. 4B). Similar trends were observed with the multipotent progenitor subsets, as defined previously (2): granulocyte/monocyte progenitor (GMP), mast cell and basophil progenitor (MCP/BaP), and megakaryocyte/erythrocyte progenitor (MEP) populations, all of which were reduced at day 28 to levels below those of naïve spleens (Fig. 4B). In the bone marrow, the LSK frequency was similarly increased during acute infection and remained elevated at day 28 postinfection (Fig. 4B). Multipotent progenitor numbers remained comparable in naïve and infected mice; however, granulocyte/monocyte progenitor, mast cell and basophil progenitor, and megakaryocyte/erythrocyte progenitor populations were decreased during acute infection and restored by the chronic-infection time point (Fig. 4B). These data highlight the fact that the observed increase in EMH in the spleen during toxoplasmosis is correlated with profound changes in the bone marrow.
Fig 4.

Extramedullary hematopoiesis is not sufficient to induce significant changes in splenic organization. (A) Representative splenic section 14 days postinfection analyzed by H&E staining. Extramedullary hematopoiesis is shown by the presence of megakaryocytes (asterisk), band cells (circled), and erythroid (arrowheads) and myeloid (arrows) precursors at various stages of maturation. (B) Various hematopoietic precursor populations were analyzed by flow cytometry and quantified in the spleen and bone marrow of naïve (white bar) mice or mice infected for 14 (gray bar) or 28 (black bar) days. Total cell numbers are presented as means and SEM. (C) Naïve mice were treated with 250 ng rmIL-12p70 for 4 consecutive days. Hematopoietic precursor populations were evaluated by flow cytometry and compared to those of untreated naïve mice or mice infected with T. gondii for 14 days based on Sca1 and ckit (CD117) staining. Gated populations represent LSKs (top right gates) and MPPs (lower gates). (D) Spleens from naïve or rmIL-12p70-treated mice were evaluated by immunofluorescent staining for T and B cell zones using CD3 (red), B220 (green), and DAPI (blue) or staining for GCs using IgD (red), PNA (green), and DAPI (blue).
Previous studies have demonstrated that recombinant IL-12 treatment can promote high levels of EMH (47), and since T. gondii induces systemic levels of IL-12 (43), this likely contributes to the EMH observed in infected mice. Therefore, to determine whether the processes and cellular influx associated with EMH inherently promote disorganization of the splenic architecture, uninfected mice were treated with IL-12p70. After 4 consecutive days of IL-12p70 treatment, spleens were sectioned and evaluated for follicular structure. Although there was a significant increase in spleen cellularity and EMH, there was no observed loss of structure in the B or T cell zones (Fig. 4C and D). The EMH induced by IL-12p70 treatment was also similar to the EMH detected during T. gondii infection, in that LSKs were significantly increased, and it represented an induction of EMH even greater than that observed during infection (Fig. 4C). Moreover, despite the influx of cells, GC architecture was also preserved, as evidenced by the maintenance of PNA+ IgD− regions, which expanded in response to the IL-12p70 treatment (Fig. 4D). Thus, these data indicate that IL-12-mediated EMH alone is not sufficient to cause disruption of T and B cell zones within the spleen.
Lymphotoxin signaling can prevent the disruption of splenic architecture.
The striking loss of organized structure in the spleens of acutely infected mice suggests that the factors that maintain these distinct regions are altered during infection. This is not unique to T. gondii: multiple mechanisms have been implicated in splenic disorganization, including CD8+ T cell-mediated cytolysis of infected stromal cells (32) or FDCs and marginal-zone macrophages (36), during lymphocytic choriomeningitis virus (LCMV) infection. Unlike LCMV, however, immunohistochemistry reveals that there are few parasites in the spleen, and thus the disorganization is unlikely to be due to the killing of infected cells (data not shown). Previous reports have demonstrated the importance of chemokine and lymphotoxin signaling in lymphoid organization (4, 14, 46). Therefore, splenic tissue was processed to extract mRNA, and relative levels of CXCL13, LTα, and LTβ mRNAs were quantified by real-time PCR. This analysis revealed that acute infection resulted in a marked decrease in the expression of LTα, LTβ, and CXCL13 transcripts, but by day 28 postinfection, expression levels were partially (LTα) or fully (CXCL13 and LTβ) restored to levels at or above those in naïve mice (Fig. 5A).
Fig 5.

LT signaling can partially restore splenic architecture without altering populations of TFH or GC B cells. (A) Expression of LTα, LTβ, and CXCL13 was determined using real-time PCR from whole spleens from C57BL/6 mice on various days postinfection. (B to G) Mice were treated with an LTβR agonist antibody beginning 6 days postinfection and evaluated 14 days postinfection. (B and C) Splenic sections were analyzed using H&E staining (B), and the area of white pulp regions was quantified (C). (D) Immunofluorescent staining and tiling of splenic sections was performed using CD3 (red), B220 (green), and DAPI (blue) to visualize large representative splenic regions; magnification, ×10. (E to G) Activated CD4+ T cells (E), TFH cells (F), and GC B cells (G) were quantified by flow cytometry and are represented as mean percentages and SEM. ns, not significant.
It has been previously established that LTα-deficient animals lack organization of T and B cell zones in the spleen (14, 29, 30). To investigate whether LT signaling would antagonize the infection-induced disruption of splenic architecture, mice were treated with an LTβR agonist antibody on days 6, 9, and 12 postinfection. At day 14 postinfection, LTβR agonist-treated mice did not display the same degree of disorganization, exhibiting a higher percentage of the spleen organized in white pulp regions (Fig. 5B and C). For a more comprehensive evaluation of B and T cell zones, whole spleens were visualized using the approach described in the legend to Fig. 2. LTβR agonist-treated mice clearly demonstrate a greater ability to maintain splenic structure than untreated infected mice, with more and larger distinct B and T cell zones apparent in the spleen (Fig. 5D). In order to determine whether this increased structure resulted in an increase in TFH cell responses, the percentages of activated CD4+ T cells (Fig. 5E), TFH cells (Fig. 5F), and GC B cells (Fig. 5G) were assessed. Surprisingly, there was no difference in any of these populations between LTβR agonist-treated mice and the control animals (Fig. 5E to G).
The spleen is an early site of parasite-specific antibody production.
The disruption of the splenic compartment during acute T. gondii infection clearly alters the lymphoid environment, potentially affecting T cell-B cell interactions. As the LN structure was not disturbed, we hypothesized that the parasite-specific IgG response during acute toxoplasmosis could be primarily LN derived, with a limited contribution from the spleen. In order to test the splenic contribution to parasite-specific antibody titers, splenectomized and wild-type mice were infected and used to assay the relative parasite-specific serum antibody titers. The mice were weighed weekly, and although during infection splenectomized and WT mice initially lost weight at comparable rates, WT mice regained that weight more quickly (Fig. 6A). Surprisingly, though levels of STAg-specific IgM, IgG, and IgG2c were measured throughout acute infection and into the chronic stage of infection, the only observed differences in serum antibody titers were during the acute phase of infection (Fig. 6B). Later in infection, serum antibody levels were comparable, whether or not mice were splenectomized (Fig. 6B). These data indicate that in the initial response, the spleen contributes to the early development of a parasite-specific isotype-switched antibody response; however, in the later stages of the response, the spleen may be a less significant contributor. Alternatively, the LN could compensate for the loss of the spleen, but though there was a trend toward increased cell numbers in the LN of splenectomized mice, it was not statistically significant (Fig. 6C). Although these data must be interpreted with care, the results indicate a role for the spleen in the early antibody response to T. gondii but demonstrate that the spleen is not necessary for the long-term antibody responses associated with the chronic phase of the infection.
Fig 6.

Splenectomized mice have delayed antibody responses during T. gondii infection but ultimately reach titers comparable to those of WT mice. (A) WT or splenectomized C57BL/6 mice were infected and weighed weekly to assess disease progression. (B) Parasite-specific antibody in the serum was assessed by ELISA. Levels of total parasite-specific IgM, IgG2c, and IgG from weeks 2 and 8 postinfection are shown. (C) Cells in the LN were quantified at 8 weeks postinfection. The numbers are total cell counts from pooled cervical, axial, brachial, and lumbar LN.
DISCUSSION
Several studies have established that B cell responses are necessary for resistance to T. gondii; without B cells, mice develop severe toxoplasmic encephalitis, which can be overcome by passive transfer of high titers of Toxoplasma-specific antibodies (12, 25, 39). Furthermore, vaccination of B cell-deficient mice with a temperature-sensitive strain of T. gondii does not result in immunity, though this, too, can be rescued by passive immune serum transfer (39). In analyzing the B cell response to T. gondii, initial flow cytometry studies detailed the expected expansion of cells expressing markers associated with TFH and GC B cells, which coincided with the development of an isotype-switched, parasite-specific antibody response. Unexpectedly, at the time point when both phenotypically TFH and GC B cell populations peak in the spleen, there is a marked disruption of the splenic architecture, which is subsequently restored as inflammation is reduced and the infection progresses to the chronic phase.
Disorganization of the structures in secondary lymphoid organs has been associated with a decreased ability to mount an antibody response (36, 37). Although there are no structural GCs or B cell follicles concurrently in the spleen, T cell-dependent development of class-switched antibody production begins during acute toxoplasmosis. While this isotype switching may occur in the LN, it is also possible that during acute toxoplasmosis, early class-switched antibody is primarily derived from T cell-dependent extrafollicular responses. Previous studies have demonstrated a role for TFH cells in CD4+ T cell-dependent extrafollicular B cell responses, potentially explaining the increase in TFH cells seen during T. gondii infection, despite the lack of organized GCs, and the initial variation in antibody titers observed in splenectomized animals (28, 35). However, in current models, interaction with dendritic cells (DCs) and B cells is necessary for complete TFH cell development and maintenance (3, 8, 17, 26; for a review, see reference 49). One potential explanation for the presence of these cell populations, despite the lack of intact structures, is that TFH cells primed in the structurally intact LN subsequently traffic to the spleen. Once in the spleen, the cells may be prevented from functioning optimally, as the disorganization limits interactions between the TFH and B cells, leading to the comparable antibody responses in WT and splenectomized mice. Alternatively, priming in the spleen may begin before the GCs and follicular structures are significantly disrupted, with antibody titers not fully matured until later in infection.
In current models, the development and maintenance of B cell follicles is dependent on the ability of FDCs to produce CXCL13, which provides localization signals to B cells. This is complemented by B cell production of LTα and LTβ, which provide essential signals to FDCs that drive follicular organization and structure (13, 14, 31, 33). This pathway is disrupted during murine cytomegalovirus (MCMV) infection and, in agreement with our studies, the use of an agonistic LTβR antibody could ameliorate these changes (4). The improvements in splenic organization in response to the LTβR agonist treatment imply altered LT regulation and availability during toxoplasmosis. The loss of or changes in cell populations that produce LT would result in diminished LT signaling. Alternatively, the significant splenomegaly during T. gondii infection may simply dilute the LT signal. However, the induction of EMH by recombinant IL-12p70 treatment suggests that the influx of cells alone is not sufficient to cause the breakdown of the T and B cell zones. Regardless, the data presented here indicate that restoration of the LT axis can contribute to improved splenic structure, even at the peak of inflammation and disorganization. Despite the improved splenic architecture in LTβR agonist-treated animals, there was no change in the parasite burden compared to controls by 28 days postinfection (data not shown), indicating that the restored splenic organization does not improve the response to infection. Additionally, work by Schluter et al. showed that LTα-deficient mice challenged with T. gondii have reduced antibody responses and delayed ability to generate protective T cell responses (42), but whether this is a consequence of the altered lymphoid structures found in these mice is unclear.
Multiple studies have addressed the contribution of the spleen to the response to T. gondii, and the outcomes have varied by study: certain groups have found improved survival in splenectomized mice with lower parasite burdens and faster immune responses (24), while others demonstrate increased susceptibility to infection (44). In current studies, although splenectomized mice challenged with T. gondii demonstrated an initial defect in antibody production, they exhibited levels of parasite-specific antibody similar to those of WT mice by the later stages of infection, as well as comparable T and B cell responses in the LN. These data imply that the spleen is not essential for protective immunity to T. gondii; however, it is unclear if the cells maturing in LN are able to produce more antibody in the absence of a splenic contribution. Despite a trend toward increased cell numbers in the LN of splenectomized mice, no statistically significant difference was observed.
Although GCs and other lymphoid structures typically increase in size and number following vaccination or infection, profound disorganization in these structures is observed in many bacterial, viral, and parasitic models, although the timing and underlying causes may be very different (4, 7, 36, 37, 40, 46). For example, during LCMV infection, this has been attributed to CD8+ T cell-mediated cytolysis of infected cells and, in MCMV infection, to early Toll-like receptor (TLR)-mediated chemokine suppression (4, 36). During toxoplasmosis, infection with a virulent strain leads to destruction of the splenic architecture, which is associated with high numbers of replicating parasites (15). However, with the avirulent parasite strain used here, few parasites are detected in the spleen. Moreover, our observations that heat-killed or nonreplicating forms of T. gondii, which do not induce systemic inflammation, fail to induce alterations in the splenic structure (unpublished observations) and are more consistent with a model in which the changes in the spleen are a secondary consequence of inflammation. At this point, the signals that lead to this disorganization are not known. Our data suggest that the alterations in structure are not due to IL-12-mediated EMH; however, further studies will be required to understand these events. More broadly, this study reveals an experimental system with which to study the organization and genesis of follicular structure through the infection-induced transient disruption of lymphoid architecture and raises basic questions about whether pathways involved in initial development also contribute to the events that allow the restoration of the splenic architecture.
ACKNOWLEDGMENTS
Support for this work was provided by the state of Pennsylvania; The Lupus Foundation of America, Philadelphia Tri-State Chapter (A.G.Z.); and NIH grants T32 AI055400 (A.G.Z.), T32 AI007532 (J.S.S.), AI033068 and AI048073 (C.F.W.), and AI42334 (C.A.H.).
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Allen CD, Okada T, Cyster JG. 2007. Germinal-center organization and cellular dynamics. Immunity 27:190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arinobu Y, Iwasaki H, Akashi K. 2009. Origin of basophils and mast cells. Allergol. Int. 58:21–28 [DOI] [PubMed] [Google Scholar]
- 3. Baumjohann D, Okada T, Ansel KM. 2011. Cutting edge: distinct waves of BCL6 expression during T follicular helper cell development. J. Immunol. 187:2089–2092 [DOI] [PubMed] [Google Scholar]
- 4. Benedict CA, et al. 2006. Specific remodeling of splenic architecture by cytomegalovirus. PLoS Pathog. 2:e16 doi:10.1371/journal.ppat.0020016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Breitfeld D, et al. 2000. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192:1545–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bry L, Brenner MB. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J. Immunol. 172:433–441 [DOI] [PubMed] [Google Scholar]
- 7. Cadman ET, et al. 2008. Alterations of splenic architecture in malaria are induced independently of Toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect. Immun. 76:3924–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi YS, et al. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34:932–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crotty S. 2011. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29:621–663 [DOI] [PubMed] [Google Scholar]
- 10. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. 2010. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 28:243–273 [DOI] [PubMed] [Google Scholar]
- 11. Fossati L, Merino J, Izui S. 1990. CD4+ T cells play a major role for IgM and IgG anti-DNA production in mice infected with Plasmodium yoelii. Clin. Exp. Immunol. 79:291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frenkel JK, Taylor DW. 1982. Toxoplasmosis in immunoglobulin M-suppressed mice. Infect. Immun. 38:360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu YX, Chaplin DD. 1999. Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol. 17:399–433 [DOI] [PubMed] [Google Scholar]
- 14. Fu YX, et al. 1997. Lymphotoxin-alpha (LTalpha) supports development of splenic follicular structure that is required for IgG responses. J. Exp. Med. 185:2111–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gavrilescu LC, Denkers EY. 2001. IFN-gamma overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J. Immunol. 167:902–909 [DOI] [PubMed] [Google Scholar]
- 16. Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. 1992. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 149:175–180 [PubMed] [Google Scholar]
- 17. Goenka R, et al. 2011. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J. Immunol. 187:1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Han JH, et al. 2007. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity 27:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He B, et al. 2007. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 26:812–826 [DOI] [PubMed] [Google Scholar]
- 20. Hill DE, Chirukandoth S, Dubey JP. 2005. Biology and epidemiology of Toxoplasma gondii in man and animals. Anim. Health Res. Rev. 6:41–61 [DOI] [PubMed] [Google Scholar]
- 21. Israelski DM, et al. 1989. Treatment with anti-L3T4 (CD4) monoclonal antibody reduces the inflammatory response in toxoplasmic encephalitis. J. Immunol. 142:954–958 [PubMed] [Google Scholar]
- 22. Johns JL, Christopher MM. 2012. Extramedullary hematopoiesis: a new look at the underlying stem cell niche, theories of development, and occurrence in animals. Vet. Pathol. 49:508–523 [DOI] [PubMed] [Google Scholar]
- 23. Johnston RJ, et al. 2009. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325:1006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones TC, Alkan S, Erb P. 1987. Murine spleen and lymph node cellular composition and function during cyclophosphamide and splenectomy induced resistance to Toxoplasma gondii. Parasite Immunol. 9:117–131 [DOI] [PubMed] [Google Scholar]
- 25. Kang H, Remington JS, Suzuki Y. 2000. Decreased resistance of B cell-deficient mice to infection with Toxoplasma gondii despite unimpaired expression of IFN-gamma, TNF-alpha, and inducible nitric oxide synthase. J. Immunol. 164:2629–2634 [DOI] [PubMed] [Google Scholar]
- 26. Kerfoot SM, et al. 2011. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34:947–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. King C, Tangye SG, Mackay CR. 2008. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 26:741–766 [DOI] [PubMed] [Google Scholar]
- 28. Lee SK, et al. 2011. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 208:1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsumoto M, et al. 1996. Affinity maturation without germinal centres in lymphotoxin-alpha-deficient mice. Nature 382:462–466 [DOI] [PubMed] [Google Scholar]
- 30. Matsumoto M, et al. 1996. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science 271:1289–1291 [DOI] [PubMed] [Google Scholar]
- 31. Mueller SN, Ahmed R. 2008. Lymphoid stroma in the initiation and control of immune responses. Immunol. Rev. 224:284–294 [DOI] [PubMed] [Google Scholar]
- 32. Mueller SN, et al. 2007. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. U. S. A. 104:15430–15435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ngo VN, et al. 1999. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 189:403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nurieva RI, et al. 2009. Bcl6 mediates the development of T follicular helper cells. Science 325:1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Odegard JM, et al. 2008. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 205:2873–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Odermatt B, Eppler M, Leist TP, Hengartner H, Zinkernagel RM. 1991. Virus-triggered acquired immunodeficiency by cytotoxic T-cell-dependent destruction of antigen-presenting cells and lymph follicle structure. Proc. Natl. Acad. Sci. U. S. A. 88:8252–8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Racine R, et al. 2010. Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J. Immunol. 184:5085–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Remington JS. 1974. Toxoplasmosis in the adult. Bull. N. Y. Acad. Med. 50:211–227 [PMC free article] [PubMed] [Google Scholar]
- 39. Sayles PC, Gibson GW, Johnson LL. 2000. B cells are essential for vaccination-induced resistance to virulent Toxoplasma gondii. Infect. Immun. 68:1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scandella E, et al. 2008. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat. Immunol. 9:667–675 [DOI] [PubMed] [Google Scholar]
- 41. Schaerli P, et al. 2000. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192:1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schluter D, et al. 2003. Both lymphotoxin-alpha and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J. Immunol. 170:6172–6182 [DOI] [PubMed] [Google Scholar]
- 43. Sher A, et al. 2003. Induction and regulation of IL-12-dependent host resistance to Toxoplasma gondii. Immunol. Res. 27:521–528 [DOI] [PubMed] [Google Scholar]
- 44. Stahl W, Matsubayashi H, Akao S. 1966. Modification of subclinical toxoplasmosis in mice by cortisone, 6-mercaptopurine and splenectomy. Am. J. Trop. Med. Hyg. 15:869–874 [DOI] [PubMed] [Google Scholar]
- 45. Stavnezer J, Guikema JE, Schrader CE. 2008. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 26:261–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. St John AL, Abraham SN. 2009. Salmonella disrupts lymph node architecture by TLR4-mediated suppression of homeostatic chemokines. Nat. Med. 15:1259–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tare NS, et al. 1995. Administration of recombinant interleukin-12 to mice suppresses hematopoiesis in the bone marrow but enhances hematopoiesis in the spleen. J. Interferon Cytokine Res. 15:377–383 [DOI] [PubMed] [Google Scholar]
- 48. Ueda Y, Liao D, Yang K, Patel A, Kelsoe G. 2007. T-independent activation-induced cytidine deaminase expression, class-switch recombination, and antibody production by immature/transitional 1 B cells. J. Immunol. 178:3593–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vinuesa CG, Cyster JG. 2011. How T cells earn the follicular rite of passage. Immunity 35:671–680 [DOI] [PubMed] [Google Scholar]
- 50. Xu R, Johnson AJ, Liggitt D, Bevan MJ. 2004. Cellular and humoral immunity against vaccinia virus infection of mice. J. Immunol. 172:6265–6271 [DOI] [PubMed] [Google Scholar]
- 51. Yu D, et al. 2009. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31:457–468 [DOI] [PubMed] [Google Scholar]
- 52. Yusuf I, et al. 2010. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J. Immunol. 185:190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]