

Abstract
Toll-like receptors (TLRs) play a major role in host mucosal and systemic defense mechanisms by recognizing a diverse array of conserved pathogen-associated molecular patterns (PAMPs). TLR2, with TLR1 and TLR6, recognizes structurally diverse bacterial products such as lipidated factors (lipoproteins and peptidoglycans) and nonlipidated proteins, i.e., bacterial porins. PorB is a pan-neisserial porin expressed regardless of organisms' pathogenicity. However, commensal Neisseria lactamica organisms and purified N. lactamica PorB (published elsewhere as Nlac PorB) induce TLR2-dependent proinflammatory responses of lower magnitude than N. meningitidis organisms and N. meningitidis PorB (published elsewhere as Nme PorB). Both PorB types bind to TLR2 in vitro but with different apparent specificities. The structural and molecular details of PorB-TLR2 interaction are only beginning to be unraveled and may be due to electrostatic attraction. PorB molecules have significant strain-specific sequence variability within surface-exposed regions (loops) putatively involved in TLR2 interaction. By constructing chimeric recombinant PorB loop mutants in which surface-exposed loop residues have been switched between N. lactamica PorB and N. meningitidis PorB, we identified residues in loop 5 and loop 7 that influence TLR2-dependent cell activation using HEK cells and BEAS-2B cells. These loops are not uniquely responsible for PorB interaction with TLR2, but NF-κB and MAP kinases signaling downstream of TLR2 recognition are likely influenced by a hypothetical “TLR2-binding signature” within the sequence of PorB surface-exposed loops. Consistent with the effect of purified PorB in vitro, a chimeric N. meningitidis strain expressing N. lactamica PorB induces lower levels of interleukin 8 (IL-8) secretion than wild-type N. meningitidis, suggesting a role for PorB in induction of host cell activation by whole bacteria.
INTRODUCTION
Neisseriae are Gram-negative bacteria comprising both pathogenic and nonpathogenic microorganisms. The pathogenic species include Neisseria gonorrhoeae, which colonizes the human reproductive system, and Neisseria meningitidis, which colonizes the human respiratory system. N. meningitidis, carried by approximately 15% of the adult population, occasionally invades the host and causes meningococcal disease and septicemia (2, 59). The commensal Neisseria lactamica is also carried in the human upper respiratory tract (14, 39), but reports of systemic infections are very rare (10, 57).
All Neisseria species express porins, major outer membrane proteins that belong to the Gram-negative porin superfamily (5, 20). N. meningitidis expresses two porins, PorA and PorB, while N. lactamica expresses only PorB (13). Porins are trimeric proteins composed of ∼35-kDa monomers with a 16-strand β-barrel fold and eight surface-exposed, variable, hydrophilic loops (11, 48). These proteins share sequence homology in the transmembrane domains, but the sequences of extracellular loops 1 through 8 have a high degree of variability among strains (11, 52).
The known effects of neisserial porins on eukaryotic cells include induction of cell activation and immune stimulation (immune adjuvant effect) (56), contribution to serum resistance to Neisseria infections (19, 46), modulation of host cell survival (31), and involvement in bacterial invasion of host cells (36). Both N. meningitidis PorB and N. lactamica PorB have been identified as nonlipidated TLR2 ligands that require TLR2-TLR1 heterodimerization for inducing cell activation via a MyD88-dependent pathway (30, 33).
Toll-like receptors (TLRs) are cellular pattern recognition receptors (PRRs) that recognize microbial products (pathogen-associated molecular patterns [PAMPs]) (35). Cell activation via TLR engagement triggers intracellular signaling pathways, such as NF-κB nuclear translocation and mitogen-activated protein kinase (MAPK) phosphorylation and activation, that regulate acute inflammatory responses, host innate and adaptive immune responses, and site-specific defense mechanisms (1).
N. lactamica PorB and N. meningitidis PorB (published elsewhere as Nlac PorB and Nme PorB, respectively) have been shown to elicit TLR2-dependent cell activation of different magnitudes, likely due to their different binding affinities for TLR2 (26, 33). Similarly, N. lactamica whole organisms induce lower TLR2-dependent inflammatory responses than N. meningitidis whole organisms in human airway epithelial cells and meningeal cells (12). Regulation of TLR-dependent cell activation is a common mechanism employed by several microorganisms to actively prevent or downregulate host cell responses that control local inflammation. For example, Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pneumoniae, and Pseudomonas aeruginosa induce different amounts of the inflammatory mediators interleukin 8 (IL-8) and RANTES (55). An inverse correlation between serum levels of IL-8 and RANTES has also been shown in patients with meningococcal infections, where high levels of IL-8 and low levels of RANTES correlate with severe disease and poor prognosis (i.e., acute bacterial meningitis and meningococcal septic shock) while low IL-8 and high RANTES levels correlate with mild systemic meningitis and are associated with survival (15). It is possible that the interaction of PorB with TLR2 helps to shape the local host inflammatory response following initial airway epithelial cell colonization by Neisseria strains. The TLR2-PorB binding specificity may then influence the quality and the magnitude of cell response.
In the past decade, much progress has been made in defining how TLR signaling modulates host immune responses, but less is known about the molecular mechanisms of TLR-ligand interactions. The mechanism of PorB-TLR2 interaction is not known; a recent study suggested that it may occur via electrostatic interaction of a ring of positively charged residues on the porin surface-exposed loops and negatively charged residues on the TLR2 ectodomain (49). Thus, differences in the sequence of the PorB surface-exposed loops putatively involved in TLR2 recognition could be crucial for such interaction and for the subsequent induction of intracellular signaling pathways leading to cell activation.
Our work attempted to determine whether a TLR2-binding signature is expressed within the sequence of the putative TLR2 binding site(s) of PorB and whether sequence variability within such regions may explain the different intensities of the cell responses induced by distinct PorB molecules. First, by using a loop mutagenesis approach, surface-exposed loop residues in N. meningitidis PorB in a recombinant background were mutated based on the sequence of N. lactamica PorB. Second, a chimeric N. meningitidis organism expressing N. lactamica PorB was constructed. TLR2-dependent cell activation and PorB-TLR2 interaction in vitro were examined using HEK cells overexpressing TLR2 and BEAS-2B cells, which are naturally TLR2-competent human airway epithelial cells.
MATERIALS AND METHODS
Cell lines and culture conditions.
Transfected HEK cells stably overexpressing TLR2 and TLR4 or transfected with an empty vector (pcDNA-HEK cells) (9) were grown in Dulbecco's modified Eagle medium (DMEM) with 5% fetal bovine serum (FBS), 2 mM l-glutamine, and 10 μg/ml ciprofloxacin. Adenovirus-12 simian virus 40 (SV40) hybrid virus-transformed, nontumorigenic human bronchial epithelial cells (BEAS-2B, ATTC CRL-9609) were grown at 37°C with 5% CO2 in DMEM F-12 supplemented with 5% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in flasks coated with 0.01 mg/ml BSA, 0.03 mg/ml bovine collagen type I, and 0.01 mg/ml fibronectin. Prior to stimulation, BEAS-2B cells were serum starved overnight and then stimulated in FBS-free medium, unless otherwise specified. BEAS-2B cell cultures were passaged no more than 30 times from frozen stocks.
Construction of PorB loop mutants.
All site-specific sequence changes in porB were generated with a QuikChange Lightning site-directed mutagenesis kit (Stratagene) according to the manufacturer's recommendations. Briefly, plasmid pNVK15 carrying N. meningitidis porB (originally cloned from N. meningitidis strain 8765 [48]; the recombinant N. meningitidis PorB protein is referred to here as wtPorB) was amplified by PCR with Pfu Ultra HF DNA polymerase and with the primers indicated in Table S1 in the supplemental material. The PCR products were digested with DpnI to remove the original methylated pNVK15 DNA and transformed into competent Escherichia coli DH5α (New England BioLabs). Positive colonies were selected on LB agar plates containing 50 μg/ml of kanamycin, purified by streaking, and inoculated into large-volume cultures for overnight growth and DNA extraction (Qiagen). The presence of the porB gene was confirmed by digestion with NdeI and BamHI restriction enzymes. The presence of the desired nucleotide change was confirmed by DNA sequence analysis (Tufts DNA Core Facility, Boston, MA). pNVK15 (encoding the protein designated wtPorB) and the plasmids encoding each PorB loop mutant protein (designated PorBGEG, PorBDDE255-262AKR, and PorBL143P) were used to transfect E. coli BL21(DE3) for subsequent protein expression.
Porin purification.
Expression and purification of recombinant wtPorB and loop mutants PorBGEG, PorBDDE25-262AKR, and PorBL143P were performed as previously described (41). Native PorB from N. lactamica strain Y92-1002 was purified as previously described (25). Conventional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and modified SDS-PAGE under nondenaturing conditions, followed by Coomassie staining or silver staining, were performed as previously described (32).
Bacterial strains, growth conditions, and PorB mutagenesis.
N. meningitidis serogroup B, strain H44/76 (18), its derivative lacking PorA and Rmp (N. meningitidis H44/76 Δ1Δ4) (51), and N. lactamica strain Y92-1009 (38, 25) were used. The entire porB gene in N. meningitidis H44/76 Δ1Δ4 was replaced with the porB gene from N. lactamica Y92-1009. Briefly, this was achieved using a plasmid that contained (5′ to 3′) N. lactamica porB, a kanamycin resistance determinant, and DNA homologous to the 1.1-kb region immediately downstream of N. meningitidis porB. The aphA-3 kanamycin resistance cassette (amplified by PCR with he primers kan-ndeI-f1 and kan-ndeI-r1) was cloned into pGem T-Easy (Promega Corporation), and then the region immediately downstream of N. meningitidis porB (amplified with the primers PorMC58Down F SpeI and PorMC58Down R SacI) was cloned directionally into SpeI and SacI sites downstream of aphA-3; lastly, N. lactamica porB (amplified with the primers nl_porF ApaI and nl_porR SacII) was cloned directionally into ApaI and SacII sites located upstream of aphA-3. The resultant plasmid was linearized with ScaI and used to transform N. meningitidis H44/76 Δ1Δ4 as previously described (23). Transformants were selected using 100 μg/ml kanamycin and screened for incorporation of N. lactamica porB by PCR using the primers NLporloop1_F and NLporloop7_R, which are specific to loops 1 and 7 of N. lactamica porB and do not amplify N. meningitidis porB. Positive transformants were confirmed by amplification and sequencing of the entire N. lactamica porB locus. All the primers used are listed in Table S1 in the supplemental material. Bacteria were plated on chocolate agar plates in a 5% CO2 incubator at 37°C. The next day, colonies were inoculated into gonococcal (GC) liquid medium containing 1% IsoVitaleX and grown to exponential phase. Culture density was determined spectrophotometrically (an optical density at 600 nm [OD600] of 0.1 = 108 CFU/ml), and bacterial growth rates and viability were compared over time. Expression levels of PorB were examined by SDS-PAGE and Western blotting of bacterial lysates from approximately 8.25 × 105-CFU/ml liquid cultures using anti-wtPorB rabbit polyclonal sera (25, 31) (1:1,000 dilution).
Cell stimulation.
HEK cells and BEAS-2B cells (105 cells/ml) were incubated for 18 h with purified N. lactamica PorB, wtPorB, PorB loop mutants (10 μg/ml), Pam3CSK4 (100 ng/ml) (EMC Microcollections, Tubingen, Germany), phenol-purified E. coli lipopolysaccharide (LPS) (100 ng/ml), and recombinant human tumor necrosis factor alpha (TNF-α) (20 ng/ml) (Sigma). BEAS-2B cells were incubated in medium with or without 5% FBS as specified in Results. BEAS-2B cells were also incubated with whole Neisseria organisms at a multiplicity of infection (MOI) of 10 bacteria/cell in antibiotic-free medium for 18 h. For studies on NF-κB and MAPK signaling pathways, the following inhibitors were used at a concentration of 25 μM (in 10 mM dimethyl sulfoxide [DMSO]) for 1 h prior to cell stimulation: U0126 (MEK1/2 inhibitor, upstream kinase for ERK1/2 phosphorylation), SB203580 (inhibitor of p38 phosphorylation), and SP600125 (inhibitor of JNK phosphorylation) (Sigma). NF-κB inhibitory ligand (25 μg/ml; Millipore) was also used. For coincubation experiments, 10 μg/ml of PorB loop mutants was added to cells stimulated with wtPorB (10 μg/ml), N. lactamica PorB (10 μg/ml), or Pam3CSK4 (100 ng/ml) and incubated for 18 h.
Cytokine ELISA.
Production of IL-8 was quantified in supernatants of HEK cells and BEAS-2B cells incubated as described above using an OptEIA enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences). RANTES was detected using a DuoSet ELISA kit (R&D).
NF-κB luciferase reporter assay.
HEK cells (105 cells/ml) were transfected with an NF-κB luciferase reporter vector as previously described (9). After overnight adherence, the transfected cells were incubated with wtPorB, N. lactamica PorB, PorB loop mutants, Pam3CSK4, E. coli LPS, and TNF-α as described above for 18 h, followed by measurement of luciferase activity using commercial reagents (Promega) per manufacturer's protocol. The luminescence was assessed using a Wallac Victor2 luminometer.
RT-PCR.
BEAS-2B cells (5 × 105/ml) were incubated as described above, and total mRNA was extracted using an RNeasy minikit and a reverse transcriptase kit (Qiagen). The cDNA was amplified by PCR with specific primers for IL-8, RANTES, and GAPDH (see Table S1 in the supplemental material). The PCRs were carried out as follows: for IL-8 and GAPDH, 50°C for 30 min, 94°C for 1 min, 57°C for 1 min, and 72°C 1 min for 30 cycles; for RANTES, 50°C for 30 min, 94°C for 15 s, 53°C for 20 s, and 72°C for 30 s for 38 cycles and 72°C for 10 min. The PCR products were analyzed on 1.5% agarose gels.
TLR2 cell-binding assays.
PorB binding to TLR2 on the cell surface was measured by flow cytometry as previously described (33). Briefly, N. lactamica PorB and wtPorB were labeled with the fluorescent dye Alexa Fluor-594 (Molecular Probes), and increasing concentrations of fluorescent porin (1.5 μg/ml up to 100 μg/ml) were incubated with TLR2-HEK cells or BEAS-2B cells (106/ml) for 1 h at 0°C. Cells were washed with 2% FBS–phosphate-buffered saline (PBS) and fixed with 1% paraformaldehyde. Binding inhibition studies were carried out by adding excess unlabeled competitors (wtPorB, N. lactamica PorB, PorBGEG, and PorBDDE25-262AKR) (100 μg/ml). Binding was measured as cell-associated fluorescence using a FACScan flow cytometer with gating to exclude cell debris associated with necrosis and expressed as mean fluorescence intensity (MFI) (means ± standard errors) normalized to that of fluorescent PorB molecules in the absence of competitors.
Immunoprecipitation.
PorB-TLR2 interaction was measured by Western blotting as previously described (33). Briefly, biotinylated PorB molecules (10 μg/ml) were incubated with TLR2-HEK cells or pcDNA-HEK cells (∼8 × 106 cells) for 1 h in 5 ml of medium, washed, and lysed with ice-cold lysis buffer (20 mM Tris [pH 8], 137 mM NaCl, 1% Triton X-100, 2 mM EDTA, 10% glycerol, 1 nM phenylmethylsulfonyl fluoride [PMSF], and protease inhibitor cocktail). Clarified lysate supernatants were incubated with 20 μl of packed streptavidin-agarose beads (Sigma) for 1 h at 4°C. The pellets were washed extensively in lysis buffer lacking the protease inhibitors and resuspended in 1× SDS-PAGE loading buffer. Equal volumes of protein samples were examined by 10% SDS-PAGE and Western blotting using an anti-TLR2 rabbit polyclonal antibody (Imgenex) as specified by the manufacturer, followed by horseradish peroxidase (HRP)-conjugated secondary antibody. Immunoreactive bands were detected by enhanced chemiluminescence (ECL).
Soluble-TLR2 binding assay.
TLR2 binding in vitro was performed using a soluble TLR2:Fc chimera in a modified ELISA as previously described (54). Briefly, high-binding-capacity ELISA plates were coated with increasing concentrations of PorB molecules (0.002 μM to 0.47 μM), followed by incubation with TLR2:Fc (2 μg/ml) and an HRP-conjugated anti-mouse IgG secondary antibody. Binding was detected as OD450 and is expressed as a percentage of the values obtained for the highest PorB concentration used for each curve (mean ± standard error).
Statistical analysis.
Where applicable, experiments were performed in triplicate and repeated a minimum of three times, as indicated in Results. Statistical analyses were calculated using GraphPad PRISM software. P values were calculated by unpaired t test and by Wilcoxon signed rank test as indicated in Results.
RESULTS
PorB sequence analysis.
The sequences of PorB from commensal N. lactamica strains Y92-1009 (38) and ATCC 23970 (17) were compared to those of PorB from pathogenic N. meningitidis serogroup B strains H44/76 (40), 8765 (60) (identical to strain H44/76), and MC58 (50) and from serogroup A strain A115 (58) using the ClustalW2 electronic platform (8) based on the sequences described in http://pubmlst.org/neisseria/porB/. Large regions of sequence homology were identified in the PorB transmembrane domains among all the strains, while regions of sequence variability were identified in the surface-exposed loops. In Fig. 1A, loops 1, 4, 5, 6, 7, and 8 are shaded, and loops 2 and 3, involved in porin function, are boxed (3, 11). The alignment revealed single substitutions or, in some cases, multiple residue substitutions in the sequence of PorB from commensal and pathogenic strains. Based on the sequence of N. lactamica PorB, residues in loops 4, 5, and 7 (bold in Fig. 1A) were chosen for mutagenesis of wtPorB (recombinant PorB from N. meningitidis 8765 [42]): in loop 4, leucine 143 was exchanged for proline (PorBL143P) to introduce a bend in the loop conformation; in loop 5, a glycine, a glutamic acid, and a glycine were inserted between histidines 177 and 178 (PorBGEG) to elongate the loop and to interrupt an area of positive charge potentially involved in TLR2 interaction; in loop 7, negatively charged aspartic acids 255 and 256 and glutamic acid 262 were mutated to alanine (nonpolar), lysine, and arginine (positive) (PorBDDE25-262AKR) to modify the local residue charges (Fig. 1A and B; also, see Table S2 in the supplemental material).
Fig 1.

PorB sequence comparison and analysis of PorB mutants. (A) Alignment of PorB sequences from N. meningitidis serogroup B strains 8765 (*) (homologous to H44/76), MC58, and H44/76 and serogroup A strain A115 and N. lactamica strains Y92-1009 (*) and ATCC 23970, performed with the ClustalW2 alignment tool. Loop 1, 4, 5, 6, 7, and 8 are shaded. Loops 2 and 3 are boxed. Residues in loops 4, 5, and 7 chosen for mutagenesis are in bold. (B) Theoretical model of PorB monomer (N. meningitidis strain 8765) based on the N. meningitidis PorB template (PBID 3A2R) (49) created with EasyPre3D server (21a) using RasMol (43a) and Jalview (9a, 55a). The position of mutated residues in loops 4, 5, and 7 is indicated by the arrows, and the effect on the corresponding loop regions is indicated in the insets. (C) Coomassie staining of wtPorB, representative of purified recombinant PorB molecules. (Left) Conventional SDS-PAGE; (right) modified PAGE (SDS free). Prior to electrophoresis, PorB aliquots were heated in sample loading buffer in the presence or absence of SDS, as indicated. Bands with molecular weights corresponding to monomers, dimers, trimers, and oligomers were observed.
Construction of PorB loop mutants.
Using a site-directed mutagenesis approach, PorB loop mutants of wtPorB were constructed (41). Recombinant PorB molecules were expressed in E. coli strain BL21(DE3) in inclusion bodies (41) and chromatographically purified so as to be free of E. coli LPS contamination, as previously described (32). The PorB-containing fractions were identified by SDS-PAGE and Coomassie staining, pooled, and included in detergent-free proteosome micelles. The PorB proteosomes obtained by this method are correctly folded and suitable for use in vitro and in vivo without toxic effects.
To monitor PorB folding and trimeric state, gel electrophoresis under nondenaturing conditions was used (32). Aliquots of wtPorB and PorB loop mutants were subjected to conventional electrophoresis in the presence of SDS or to modified electrophoresis in the absence of SDS. The samples were dissolved in SDS-containing gel loading buffer or SDS-free gel loading buffer and heated at 100°C prior to electrophoresis on 12% acrylamide gel and visualization of the bands by Coomassie blue staining. In Fig. 1C, wtPorB is shown as a representative recombinant PorB molecule. Using conventional SDS-PAGE (Fig. 1C, left) and SDS-free PAGE (Fig. 1C, right), PorB monomers were visible as a ∼34-kDa band when the sample was dissolved in denaturing sample buffer containing 2% SDS. When SDS-free loading buffer was used, traces of PorB monomers were visible in SDS-PAGE (Fig. 1C, left) and higher-molecular-weight forms, corresponding to dimers, trimers, and oligomers, were observed in SDS-free PAGE (Fig. 1C, right). Similar results were obtained for each PorB mutant (see Fig. S1 in the supplemental material), confirming that the purified proteins retained a trimeric form (32). No E. coli LPS contamination was detected by SDS-PAGE and silver staining of the recombinant PorB molecules (data not shown), consistent with what was previously shown for the purification of endotoxin-free neisserial porins (32).
TLR2-dependent activity of PorB loop mutants.
N. meningitidis PorB and N. lactamica PorB are established TLR2/TLR1 agonists. However, they induce cell activation levels of different magnitudes, with N. lactamica PorB being less active than N. meningitidis PorB (26, 30, 33). The effect of loop mutations on the activity of PorB was examined by measuring induction of IL-8 and RANTES using the TLR2 overexpression cell line, HEK cells. TLR2-HEK cells (105/ml) were stimulated for 18 h with medium alone, with PorB loop mutants, or with wtPorB and N. lactamica PorB at 10 μg/ml, a concentration that has been optimized in both in vitro and in vivo studies (25, 26, 30, 33). The TLR2 agonist Pam3CSK4 (100 ng/ml) and TNF-α (20 ng/ml) (TLR-independent control) were also used. pcDNA-HEK cells were used as a negative control. As shown in Fig. 2A, wtPorB induced higher levels of IL-8 secretion than native N. lactamica PorB (P = 0.0024 by unpaired t test, n > 12), measured by ELISA of cell supernatants. A similar difference was previously shown for native N. meningitidis PorB and N. lactamica PorB (26). When the activity of PorB loop mutants was examined, PorBGEG and PorBDDE25-262AKR, in particular, induced significantly less IL-8 secretion than wtPorB. The effect of PorBL143P was less dramatic and not statistically significant (Fig. 2A). PorBDDE25-262AKR also induced less IL-8 than N. lactamica PorB, while IL-8 levels induced by PorBGEG and PorBL143P were comparable to those induced by N. lactamica PorB (Fig. 2A). Pam3CSK4 induced significantly more IL-8 secretion than PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB (Fig. 2A) and was comparable to wtPorB and PorBL143P. Stimulation with TNF-α as a non-TLR2-dependent control induced significantly higher IL-8 levels than PorBGEG, PorBDDE25-262AKR, PorBL143P, and N. lactamica PorB.
Fig 2.

TLR2-dependent HEK cell activation. (A) TLR2-HEK cells and pcDNA-HEK cells were incubated with wtPorB, PorB loop mutants, or N. lactamica PorB (Nlac PorB; all at 10 μg/ml), Pam3CSK4 (100 ng/ml), and TNF-α (20 ng/ml) for 18 h, and IL-8 secretion was measured by ELISA of cell supernatants. IL-8 is expressed in pg/ml (mean ± standard error). P values relative to wtPorB: **, P = 0.0024, ***, P = 0.0002, and **, P = 0.023 by unpaired t test; n > 12. P values relative to N. lactamica PorB: *, P = 0.017; **, P = 0.0024. P values relative to Pam3CSK4: **, P = 0.0013; ***, P = 0.0002; **, P = 0.0011. P values relative to TNF-α: **, P = 0.004; ****, P < 0.0001. (B) RANTES secretion induced as described for panel A and expressed as pg/ml fold change (mean ± standard error). P values relative to N. lactamica PorB: **, P = 0.003, **, P = 0.005 and *, P = 0.02 by unpaired t test; n = 6. (C) NF-κB-dependent luciferase activity induced as described for panel A. Luciferase is expressed in arbitrary units (mean ± standard error) normalized to values for nonstimulated cells. P values relative to wtPorB: *, P < 0.04 by unpaired t test; n = 6. P values relative to N. lactamica PorB: *, P < 0.04. P values relative to Pam3CSK4: *, P < 0.04. P values relative to TNF-α: *, P = 0.01; **, P = 0.009.
In contrast, N. lactamica PorB induced significantly higher TLR2-dependent secretion of RANTES than wtPorB and PorB loop mutants (Fig. 2B). No statistically significant differences in RANTES levels were induced by Pam3CSK4 or TNF-α (Fig. 2B).
As a control for potential contamination by E. coli LPS in the recombinant PorB preparations, HEK cells overexpressing TLR4 (9) were also tested. In this cell system, high levels of IL-8 were induced only by the TLR4 agonist E. coli LPS (100 ng/ml) (see Fig. S2A in the supplemental material) (P < 0.005 by unpaired t test; n = 6). Representative purified recombinant PorB molecules (wtPorB and PorBGEG) and N. lactamica PorB are also shown in Fig. S2A.
As an additional indicator of TLR2-dependent HEK cell activation, NF-κB-driven luciferase activity was measured (33). Similar to IL-8, wtPorB induced higher TLR2-dependent luciferase activity than PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB (Fig. 2C) (P < 0.04 by unpaired t test; n = 6), while it was comparable to PorBL143P (Fig. 2C). Similar to what was observed for IL-8 secretion, N. lactamica PorB induced lower NF-κB-driven luciferase activity than wtPorB and PorBL143P (Fig. 2C) (P < 0.04). As a TLR2-positive control, Pam3CSK4 induced higher NF-κB-driven luciferase activity than PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB (Fig. 2C) (P < 0.04). TNF-α induced higher luciferase activity than PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB (Fig. 2C). None of the PorB molecules induced significant luciferase activity above baseline level via TLR4 signaling (see Fig. S2B in the supplemental material).
Intracellular signaling pathways that regulate IL-8 and RANTES production by N. lactamica PorB and PorB loop mutants.
Different signaling pathways regulate IL-8 and RANTES production: maximal IL-8 expression is achieved by activation of the NF-κB pathway and the AP-1 signaling pathway (via ERK1/ERK2, JNK, and p38 MAP kinases) (21, 24), while RANTES production is regulated only by the NF-κB pathway (4, 16). To examine the contribution of these pathways to the observed difference in IL-8 and RANTES induction by N. lactamica PorB and wtPorB, specific inhibitors were used in TLR2-HEK cells. Cells were stimulated with each of the PorB molecules in the presence and in the absence of specific inhibitors [SB03580 (p38 inhibitor), U0126 (ERK1/2 inhibitor), SP600125 (JNK inhibitor), and NF-κB inhibitory ligand] and secretion of IL-8 was quantified by ELISA. For each stimulation set, the amount of secreted IL-8 was normalized to that induced in the absence of inhibitor and reported as a percentage.
IL-8 induced by wtPorB was inhibited by blocking p38, ERK1/2, JNK, and NF-κB (Fig. 3A). Similar results were obtained for PorBGEG (Fig. 3B), PorBDDE25-262AKR (Fig. 3C), and PorBL143P (Fig. 3D), as well as Pam3CSK4 (Fig. 3F). In contrast, inhibition of p38, ERK1/2, and JNK pathways did not reduce IL-8 levels induced by N. lactamica PorB, while this was affected by inhibition of NF-κB (Fig. 3E). TLR-independent IL-8 secretion induced by TNF-α was also susceptible to inhibition via both AP-1 and NF-κB signaling pathways (Fig. 3G). Predictably, inhibition of NF-κB signaling prevented TLR2-dependent and -independent luciferase activity in TLR2-HEK cells (see Fig. S3A in the supplemental material).
Fig 3.

Intracellular cell signaling pathway inhibition. TLR2-HEK cells were pretreated for 1 h with 25 μM p38 inhibitor SB03580, the ERK1/2 inhibitor U0126, the JNK inhibitor SP600125, and 25 μg/ml of the NF-κB inhibitory ligand prior to stimulation with 10 μg/ml of the indicated PorB molecules, 100 ng/ml of Pam3CSK4 or 20 ng/ml of TNF-α. IL-8 was measured in the cell supernatant by ELISA and is expressed as a percentage (mean ± standard error) of that induced by each stimulation in the absence of inhibitors. *, P < 0.05, and **, P < 0.005, by Wilcoxon signed rank test; n = 6.
TLR2 engagement by PorB loop mutants on the cell surface.
TLR2-mediated cell activation is dependent on ligand binding to TLR2. Our previous work has characterized N. meningitidis PorB and N. lactamica PorB as TLR2 ligands (26, 33). Using a flow cytometry-based binding assay, it was established that wtPorB also binds to TLR2 on the cell surface in a dose-dependent manner (data not shown). To determine whether insertion or exchange of wtPorB loop residues with N. lactamica PorB loop residues affected binding to TLR2 on the cell surface, a binding competition assay was performed (33). TLR2-HEK cells were coincubated with fluorescently labeled wtPorB (1.5 μg/ml to 25 μg/ml) and with an excess amount (100 μg/ml) of unlabeled wtPorB or PorB loop mutants as competitors. Binding was measured by fluorescence-activated cell sorting (FACS) as cell-associated fluorescence, and inhibition was expressed as mean fluorescence intensity (MFI) normalized to that of fluorescent wtPorB in the absence of competitors. When unlabeled wtPorB was used as the “homologous” inhibitor, binding of fluorescent wtPorB to the surface of TLR2-HEK cells was significantly decreased (Fig. 4A). Similarly, binding was also inhibited by unlabeled PorBGEG, PorBDDE25-262AKR, and PorBL143P as competitors (Fig. 4A).
Fig 4.
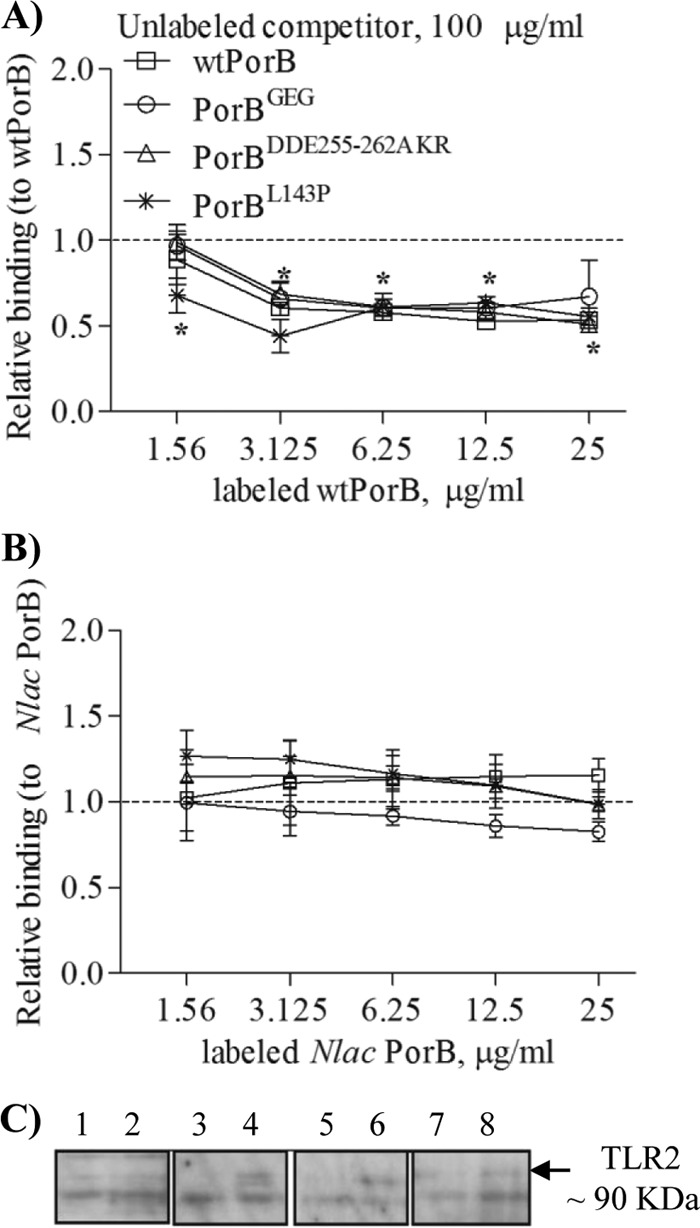
Cell surface TLR2 binding. (A) Inhibition of dose-dependent binding of fluorescently labeled wtPorB to TLR2-HEK cells measured by flow cytometry using an excess (100 μg/ml) of unlabeled wtPorB, PorBGEG, PorBDDE25-262AKR, and PorBL143P. (B) Inhibition of fluorescently labeled N. lactamica PorB binding as described for panel A. Binding is expressed as mean fluorescence intensity (MFI) normalized to that of fluorescent PorB ± standard deviations. *, P = 0.05 by Wilcoxon signed rank test; n = 6. (C) TLR2 pulldown assay of HEK cell lysates examined by Western blotting with anti-TLR2 antibody. Lanes 1, 3, 5, and 7, pcDNA HEK cells. Lanes 2, 4, 6, and 8, TLR2-HEK cells. Lanes 1 and 2, medium alone; lanes 3 and 4, biotinylated PorBGEG; lanes 5 and 6, biotinylated PorBDDE25-262AKR; lanes 7 and 8, biotinylated PorBL143P. A band of approximately 90 kDa, corresponding to TLR2, was detected in lanes 4, 6, and 8.
In contrast, binding of fluorescent N. lactamica PorB was not inhibited by unlabeled wtPorB (Fig. 4B) or PorB loop mutants as competitors (Fig. 4B) in a “heterologous” binding competition assay.
Porin binding to TLR2 on the cell surface was also tested in cells of the human airway epithelial line BEAS-2B, which constitutively express low surface amounts of TLR2 (26) and have been used to characterize the effect of N. lactamica PorB. Also in this cell model, binding of wtPorB to surface TLR2 was subject to homologous competition by wtPorB and PorB loop mutants, while binding of N. lactamica PorB was not inhibited (data not shown).
Previously, evidence of direct binding of N. meningitidis PorB and N. lactamica PorB to TLR2 in HEK cells was shown by immunoprecipitation of a TLR2–biotin-PorB complex (33); using this approach, binding of biotin-labeled PorB loop mutants to TLR2 was examined. pcDNA-HEK cells and TLR2-HEK cells were incubated with 10 μg/ml of biotin-labeled PorB mutants for 1 h followed by cell lysis and incubation with streptavidin-agarose beads to pull down the biotin-PorB–TLR2 complexes. The proteins associated with streptavidin beads were then separated by SDS-PAGE, and Western blotting was performed using an anti-TLR2 antibody to detect TLR2 coimmunoprecipitation. In the pulldown assay, a band of approximately 90 kDa, corresponding to TLR2, was detected in lysates from TLR2-HEK cells incubated with biotinylated PorBGEG, PorBDDE25-262AKR, and PorBL143P (Fig. 4C, lanes 4, 6, and 8, respectively), while it was absent in lysates from pcDNA-HEK (Fig. 4C, lanes 3, 5, and 7), as well as from lysates from cells incubated with medium alone (Fig. 4C, lanes 1 and 2).
Activation of human airway epithelial cells by PorB loop mutants.
The different magnitudes of the TLR2-dependent cell responses induced by N. lactamica PorB and N. meningitidis PorB are amplified in cell systems that naturally express TLR2, such as human airway epithelial cells (26). The effect of PorB loop residue mutations was examined in human airway epithelial BEAS-2B cells. Production of IL-8 and RANTES was measured in cell culture supernatants following stimulation as described for TLR2-HEK cells. Since these cells do not express MD-2, activation via TLR4 signaling can be achieved only by addition of FBS as a source of soluble exogenous MD-2 (26, 34). FBS-free culture medium was thus used for measuring cell stimulation exclusively via TLR2, unless otherwise specified. High IL-8 levels were induced by wtPorB and PorBL143P, while N. lactamica PorB, PorBGEG, and PorBDDE25-262AKR induced significantly lower IL-8 levels than wtPorB (Fig. 5A). PorBDDE25-262AKR also induced significantly less IL-8 than N. lactamica PorB (Fig. 5A) (P = 0.002). As a TLR2-positive control, Pam3CSK4 was used and was found to induce IL-8 levels that were significantly higher than those induced by PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB (Fig. 5A) but similar to those induced by wtPorB and PorBL143P. When BEAS-2B cells were stimulated in the presence of 5% FBS, only E. coli LPS induced high levels of IL-8 (see Fig. S2B in the supplemental material), while the porin molecules failed to induce IL-8 above baseline level, consistent with a lack of TLR4 contribution to the effect of PorB and with the absence of LPS in the purified PorB molecules. Data for representative PorB molecules are shown in Fig. S2B in the supplemental material. Also in BEAS-2B cells, N. lactamica PorB induced higher levels of RANTES than wtPorB, PorB loop mutants, and Pam3CSK4 (Fig. 5B).
Fig 5.
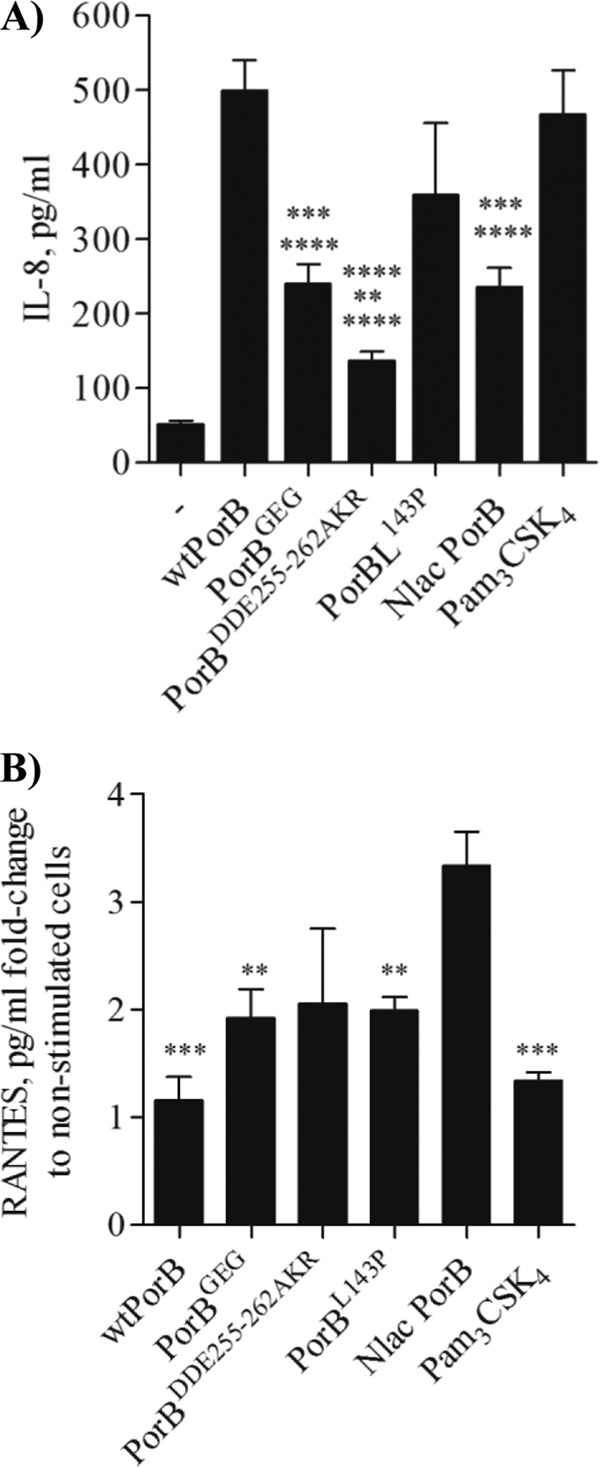
BEAS-2B cell activation. BEAS-2B cells were incubated with wtPorB, PorB loop mutants, or N. lactamica PorB (all at 10 μg/ml), Pam3CSK4 (100 ng/ml), TNF-α (20 ng/ml), and LPS (100 ng/ml) in FBS-free medium for 18 h. (A) Secretion of IL-8 measured by ELISA and expressed in pg/ml (mean ± standard error). P values relative to wtPorB: ****, P < 0.0001 by unpaired t test; n > 15. P values relative to N. lactamica PorB: **, P = 0.002. P values relative to Pam3CSK4: ***, P = 0.0002; ****, P < 0.0001. (B) Secretion of RANTES measured as described for panel A and expressed as fold change (in pg/ml) relative to nonstimulated cells (mean ± standard error). P values relative to Nlac PorB: ***, P < 0.0005, and **, P < 0.009, by unpaired t test; n = 6.
To determine whether the observed differences in levels of secreted IL-8 and RANTES correlated with transcription levels, RT-PCR was used to measure IL-8 and RANTES mRNA expression in BEAS-2B cells. Consistent with previous results (26) and with IL-8 protein secretion levels, N. lactamica PorB induced lower levels of IL-8 mRNA than wtPorB (see Fig. S4A in the supplemental material). Although both PorBGEG and PorBDDE25-262AKR induced smaller amounts of secreted IL-8 than wtPorB, the corresponding mRNA levels were not dramatically decreased, suggesting that both transcriptional and posttranscriptional regulation processes may be involved. In contrast, N. lactamica PorB and PorBDDE25-262AKR induced higher RANTES mRNA levels than wtPorB and, to a certain extent, than PorBGEG, similar to RANTES protein secretion. A similar discrepancy between secreted IL-8 and RANTES and mRNA levels was observed in response to Pam3CSK4 (Fig. 5; also, see Fig. S4 in the supplemental material), similar to findings reported in previous studies using airway epithelial cells. This has been attributed to differential regulation of these inflammatory mediators by Pam3CSK4 (4, 29). The effect of LPS on IL-8 and RANTES mRNA expression was appreciable only in the presence of 5% FBS (see Fig. S4A in the supplemental material). mRNA levels relative to a representative gel were quantified by densitometric analysis of PCR products normalized to the levels of a reference gene, GAPDH, relative to control cells stimulated with medium alone (see Fig. S4B in the supplemental material).
PorB loop mutants binding to TLR2 in vitro.
A difference in the specific interactions of TLR2 with N. lactamica PorB and with N. meningitidis PorB was shown previously (26, 33). To determine whether insertion or exchange of loop residues from N. lactamica PorB into wtPorB had an effect on TLR2 binding in vitro, a modified ELISA binding assay was used. Binding of soluble hTLR2 (TLR2:Fc) (54) to wtPorB and PorB loop mutants was compared using increasing concentrations of plate-bound PorB molecules (up to 0.47 μM) and 2 μg/ml of soluble TLR2:Fc, followed by detection with HRP-labeled anti-mouse IgG secondary antibody, as previously described (33). In Fig. 6, binding is expressed as a percentage (mean ± standard error) of the value obtained for the highest concentration of ligand used for each curve (n = 10). Molar concentrations of PorB molecules are based on the molecular mass of the trimer (∼105 kDa). No significant difference was observed between binding of wtPorB, PorBGEG, and PorBDDE25-262AKR to TLR2 in vitro (Fig. 6), despite their different ability to induce cell activation. Consistent with previous results, N. lactamica PorB binding to TLR2 in vitro was lower than that of wtPorB or PorB loop mutants (Fig. 6) (26).
Fig 6.
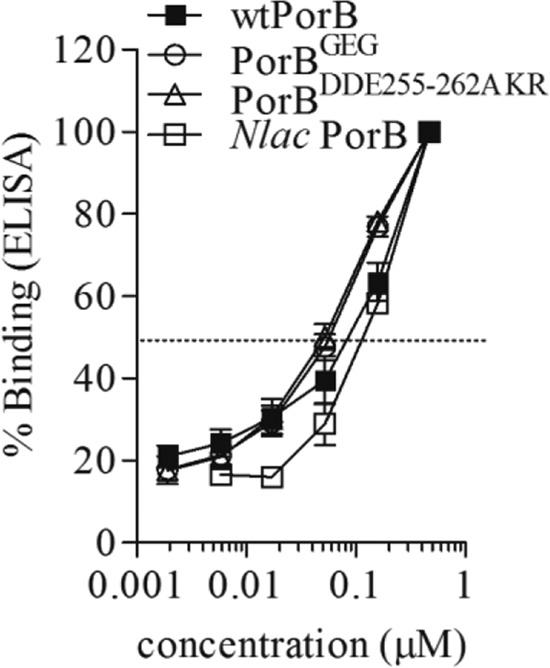
In vitro TLR2/PorB binding assay. Increasing concentrations (up to 0.47 μM) of wtPorB, PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB were used to coat high-binding-capacity ELISA plates and incubated with 2 μg/ml of soluble TLR2 (TLR2:Fc). Binding was detected using an HRP-labeled anti-mouse IgG secondary antibody and expressed as a percentage (mean ± standard error) of the value obtained for the highest concentration of ligand used for each curve (n = 9). Molar concentrations for PorB molecules are based on the molecular mass of the trimer (∼105 kDa).
Effect of PorB loop mutants as competitive inhibitors of TLR2-induced IL-8 secretion.
Our results so far show that PorB loop mutants engage TLR2, but the activities of PorBGEG and, particularly, PorBDDE25-262AKR were considerably lower than those of N. lactamica PorB and wtPorB. To determine whether TLR2 occupancy by PorB loop mutants influenced the ability of other TLR2 ligands to induce cell activation, N. lactamica PorB and wtPorB (10 μg/ml) and Pam3CSK4 (100 ng/ml) were incubated individually with BEAS-2B cells or in coincubation assays with PorB loop mutants (10 μg/ml) for 18 h. IL-8 secretion was measured in the cell supernatant and quantified by ELISA.
First, to determine the effect of PorB loop mutants on TLR2-dependent cell activation induced by wtPorB, the amount of secreted IL-8 was normalized to that induced by wtPorB alone (Fig. 7A) and expressed as fold change. While coincubation with PorBGEG or N. lactamica PorB did not affect IL-8 induction by wtPorB, coincubation with PorBDDE25-262AKR significantly inhibited IL-8 production (Fig. 7A) (P < 0.0001 by Wilcoxon signed rank test; n = 15). PorBGEG, PorBDDE25-262AKR, and N. lactamica PorB alone induced lower levels of IL-8 than wtPorB alone (Fig. 7A) (P = 0.0001).
Fig 7.

Inhibition of IL-8 secretion. BEAS-2B cells were incubated with wtPorB, N. lactamica PorB, PorBGEG, PorBDDE25-262AKR, or PorBL143P (all at 10 μg/ml) and with Pam3CSK4 (100 ng/ml) individually or in coincubations for 24 h. IL-8 secretion was quantified by ELISA and expressed as fold-change (mean ± standard error) normalized to wtPorB alone (*, P < 0.0001 by Wilcoxon signed rank test; n = 15) (A), N. lactamica PorB alone (*, P = 0.02) (B), or Pam3CSK4 alone) (*, P < 0.0001) (C).
Next, IL-8 secretion induced by coincubation of PorB loop mutants with N. lactamica PorB was measured and normalized to that induced by N. lactamica PorB alone (Fig. 7B). While PorBDDE25-262AKR also decreased N. lactamica PorB-induced IL-8, coincubation with both wtPorB and PorBGEG enhanced IL-8 secretion (Fig. 7B), possibly due to an additive effect. PorBGEG and PorBDDE25-262AKR alone, but not wtPorB alone, induced less IL-8 than N. lactamica PorB alone (Fig. 7B).
Lastly, the effect of PorB loop mutants on IL-8 induced by Pam3CSK4 was examined. While PorBDDE25-262AKR moderately inhibited IL-8 production induced by Pam3CSK4 alone, neither PorBGEG, N. lactamica PorB, nor wtPorB had a similar effect (Fig. 7C). PorB loop mutants alone induced significantly less IL-8 than Pam3CSK4 alone, while the level induced by the latter was comparable to that induced by wtPorB and N. lactamica PorB (Fig. 7C).
Influence of PorB on induction of cell activation by whole bacteria.
Lastly, to verify whether the observed effects of purified PorB in vitro correlated with differences in induction of airway epithelial cell activation by whole Neisseria, a chimeric N. meningitidis organism was constructed in which N. meningitidis PorB was exchanged with N. lactamica PorB. For this purpose, a mutant of N. meningitidis H44/76 lacking PorA expression (N. meningitidis H44/76 Δ1Δ4) was used, to avoid confounding effects due to expression of PorA (PorA is not expressed in N. lactamica). No effects of PorB exchange were observed on bacterial viability or on the organism's growth rates, which were comparable to that of N. meningitidis H44/76 Δ1Δ4 as well as the N. meningitidis H44/76 parent strain and N. lactamica Y92-1009 (see Fig. S5A in the supplemental material). To compare and quantify PorB expression among strains, the bacterial cultures were diluted to 8.25 × 105 CFU/ml, and aliquots of approximately 1.6 × 104 CFU total were lysed, separated by SDS-PAGE, and examined by Western blotting using an anti-PorB rabbit polyclonal serum (25, 31) (1:1,000 dilution). As shown in Fig. S5B in the supplemental material, a band of approximately 34 to 37 kDa was identified by the anti-PorB antibody, and it appeared to be expressed at comparable levels in all the strains.
Next, IL-8 secretion induced in BEAS-2B cells in response to incubation with these different organisms was examined by ELISA. Cells were incubated as previously described with each strain at an MOI of 10 bacteria/cell in antibiotic-free medium, and culture supernatants were assayed for cytokine production. As previously demonstrated, levels of IL-8 induced by N. lactamica Y92-1009 were lower than those induced by N. meningitidis H44/76 Δ1Δ4 (Fig. 8) (26). Similarly, IL-8 levels induced by N. lactamica Y92-1009 were significantly lower than those induced by the N. meningitidis H44/76 parent strain (Fig. 8). No statistically significant difference was measured in IL-8 production induced by the N. meningitidis H44/76 parent strain compared to the PorA/Rmp mutant (Fig. 8), although the latter was slightly reduced, possibly due to lack of PorA contribution. However, levels of IL-8 induced by the mutant N. meningitidis H44/76 Δ1Δ4 expressing N. lactamica PorB were considerably reduced compared to those induced by both the N. meningitidis H44/76 parent strain and the N. meningitidis H44/76 Δ1Δ4 mutant strain (Fig. 8), to levels that were close to those induced by N. lactamica Y92-1009. Similar results were observed when induction of IL-6 secretion was measured (data not shown).
Fig 8.
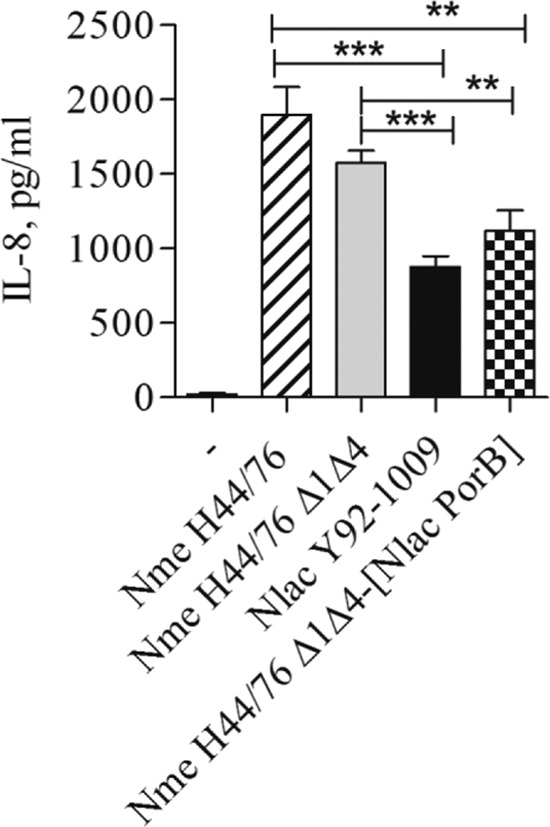
BEAS-2B cell activation by whole bacteria. BEAS-2B cells were incubated with whole Neisseria organisms (MOI of 10 CFU/cell) for 18 h in antibiotic-free medium, and IL-8 secretion was measured by ELISA of culture supernatants. Strains used include the N. meningitidis H44/76 parent strain, N. meningitidis H44/76 Δ1Δ4, N. meningitidis H44/76 Δ1Δ4 expressing N. lactamica PorB (Nme H44/76 Δ1Δ4-[Nlac PorB]), and N. lactamica Y92-1009. IL-8 is expressed in pg/ml (mean ± standard error). ***, P < 0.0001, and **, P < 0.0058, by unpaired t test; n = 9.
DISCUSSION
In previous studies, our group and others have reported significant differences in the outcomes of cell stimulation with purified porins from pathogenic and commensal Neisseria and with whole bacteria (12) via TLR2 signaling. PorB is a well-established TLR2 ligand (30, 32, 33, 47). Although the molecular mechanism of PorB interaction with TLR2 has not been identified yet, recent work from Tanabe et al. (49) proposed that positively charged amino acids in the surface exposed loops of PorB might mediate binding to TLR2 via electrostatic interactions with negatively charged residues on the TLR2 ecto-domain.
Differences in the amino acid sequences of the surface exposed loops of PorB (loop variable regions) have been identified in the majority of neisserial strains. Our PorB sequence analysis of commensal N. lactamica strains and pathogenic N. meningitidis strains revealed that certain loop residues appear predominantly expressed in strain subsets. Thus, to explore the contribution of the surface-exposed loops to the molecular and functional interaction of PorB with TLR2, hybrid mutant porins were engineered in a meningococcal PorB recombinant background (originally cloned from the pathogenic N. meningitidis strain 8765 [41]). This particular PorB, referred to as wtPorB, shares 100% sequence homology with PorB from N. meningitidis strain H44/76, used as a model molecule for both in vitro and in vivo studies (27, 47, 56). Our mutagenesis approach focused on amino acid sequences that were different between N. lactamica PorB and N. meningitidis PorB and that could possibly influence local loop charges and/or conformation, based on molecular modeling prediction tools and the published structural analysis of PorB (48). Since loops 2 and 3 are mostly involved in PorB monomer interaction, pore formation, and gating function, they are less likely to participate in protein-protein interactions (11) and were not mutagenized. The mutation of leucine 143 in loop 4 to proline (PorBL143P), according to the N. lactamica PorB loop 4 sequence, was predicted to affect the loop tertiary structure. The insertion of a glycine-glutamic acid-glycine tripeptide (GEG) between histidines 177 and 178 in loop 5 (PorBGEG) was predicted to elongate this loop and likely shift the position of residues potentially exposed to interaction with TLR2. Finally, a switch from the negatively charged aspartic acids 255 and 256 and glutamic acid 262 in loop 7 to nonpolar, hydrophobic alanine and positively charged lysine and arginine (PorBDDE25-262AKR) was expected to modify the charge of loop 7.
Analysis of the biological activity of such PorB mutants revealed that loop 5 and loop 7 mutants induced lower levels of cell activation compared to wtPorB and N. lactamica PorB. No indication of cell toxicity was observed for any of the PorB molecules used. To rule out the possibility that such inferior activity was due to incorrect protein folding or lack of trimeric structure (32), purified PorB mutants were examined by nondenaturing SDS-PAGE. All PorB molecules used in this study were found to retain proper folding and trimeric conformation, as demonstrated by an abundance of high-molecular-weight forms corresponding to trimers. Furthermore, the method employed for PorB purification ensures the absence of contaminating bacterial endotoxin (25, 32), which was confirmed both biochemically, by silver staining of SDS-PAGE gels, and functionally, by the lack of TLR4-dependent activity in a TLR4-HEK cell overexpression system (9). Our results were thus consistent with functional, LPS-free, purified PorB trimers.
The use of HEK cells overexpressing TLRs is a well-established tool for characterization of the activity of various TLR agonists. However, the overabundance of TLR expression may, in part, supersede variations of activity of different agonists. For this reason, naturally TLR2-competent cells were also used to test the activity of PorB loop mutants. Differences in TLR2-dependent activity between N. lactamica PorB and meningococcal PorB have been previously shown using the human airway epithelial cell lines BEAS-2B and Detroit 562 (26). These are very relevant cell models for studying the outcomes of cell stimulation with neisserial products, since both N. meningitidis and N. lactamica are colonizers of the human nasopharyngeal epithelium. An additional advantage of these cells is their unresponsiveness to TLR4 agonists, due to lack of expression of MD-2 (34), which is required for LPS signaling.
In both TLR2-HEK cells and BEAS-2B cells, PorBGEG and PorBDDE25-262AKR induced lower levels of IL-8 secretion and NF-κB-dependent luciferase activity (in TLR2-HEK cells) than wtPorB, indicating that mutation of residues in loops 5 and 7 attenuated the porin activity. While PorBGEG induced cell activation levels similar to those induced by N. lactamica PorB, PorBDDE25-262AKR induced dramatically lower cell activation levels, suggesting that residue charge variations in loop 7 had a profound effect on PorB activity. The activity of PorBL143P was comparable to that of wtPorB, suggesting a negligible effect of a proline insertion in loop 4. Interestingly, in both TLR2-HEK cells and BEAS-2B cells, secretion of IL-8 and RANTES was induced by N. lactamica PorB and wtPorB to different levels. In particular, N. lactamica PorB induced lower IL-8 secretion than wtPorB and PorBL143P, while it induced higher RANTES secretion than wtPorB or PorB loop mutants. In the case of IL-8, however, mRNA expression levels were less dramatically different among PorB molecules, suggesting that both transcriptional and posttranscriptional regulation processes may be involved. Similar evidence of secreted IL-8 and RANTES and mRNA levels discrepancy and differential gene regulation was observed in response to Pam3CSK4, consistent with what was previously reported for airway epithelial cells (4, 29).
It is well established that, following TLR stimulation, maximal expression of IL-8 is achieved by combined activation of NF-κB and AP-1 signaling pathways (via ERK1/ERK2, JNK, and p38 MAP kinases) in a stimulus-specific fashion, particularly in nonhematopoietic cells (21, 24, 43, 45). In contrast, RANTES expression is only regulated by the NF-κB pathway (4, 44). Thus, high IL-8 and low RANTES levels induced by wtPorB and, conversely, low IL-8 and high RANTES levels induced by N. lactamica PorB could result from activation of different signaling pathways. The cartoon in Fig. 9 summarizes this observation and provides a preliminary model for the different effects of N. lactamica PorB and wtPorB. Supporting this concept, TLR2-dependent B-cell proliferation and CD86 surface expression are induced by N. meningitidis PorB via NF-κB, protein tyrosine phosphorylation, and ERK1/2 activation but are not susceptible to ERK1/2 inhibition (28). Furthermore, induction of anti-apoptotic genes by PorB is TLR2- and NF-κB-independent and possibly due to activation of alternative signaling pathways (29).
Fig 9.
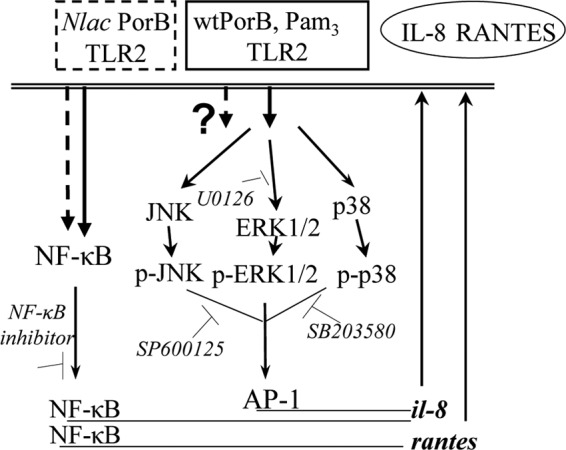
Schematic cartoon of the signaling pathways hypothetically induced by N. lactamica PorB and wtPorB following differential TLR2 interaction. While both wtPorB and N. lactamica PorB induce NF-κB-dependent IL-8 and RANTES production downstream of TLR2 activation, only wtPorB also induces IL-8 production via activation of MAP kinases, resulting in enhanced IL-8 levels.
The role of MAP kinases p38 and JNK in the activity of PorB has not been explored so far. By using specific inhibitors of NF-κB and MAP kinase phosphorylation and activation, an essential role for NF-κB was identified for IL-8 induction by both N. lactamica PorB and wtPorB via TLR2 signaling (similar to Pam3CSK4) and by TNF-α via non-TLR2-dependent signaling. Inhibition of p38, ERK1/2, and JNK signaling did not affect IL-8 induced by N. lactamica PorB, while it prevented TLR2-dependent cell activation by wtPorB, PorB loop mutants, and Pam3CSK4, as well as TLR2-independent cell activation by TNF-α. These results are in agreement with the multiple signaling pathways involved in TLR2-dependent (and independent) induction of IL-8 and suggest that only the NF-κB pathway appears to be induced by N. lactamica PorB.
Since TLR2-mediated cell activation by PorB is dependent on binding to TLR2 (32, 33), it is possible that mutation of PorB surface-exposed loops may alter the hypothetical TLR2 binding site(s). If this site(s) is the same across PorB types, and if PorB-TLR2 binding affinity is the same across PorB types, then any PorB type would inhibit TLR2 binding when used in a competitive binding assay (referred to as homologous competition). Our previous work established that N. lactamica PorB and meningococcal PorB have different apparent specificities for TLR2 in vitro and on the cell surface and that they are not subject to reciprocal binding inhibition, thus behaving as heterologous competitors (26, 33). Upon the introduction of N. lactamica PorB residues into wtPorB, different outcomes on TLR2 interaction could have been predicted, such as (i) disruption of the TLR2 binding site(s) and complete loss of activity (this was not observed), (ii) no effect of mutations on binding or activity (this was observed for PorBL143P), and (iii) no evident effect of mutations on TLR2 binding combined with alteration of activity (this was observed for PorBGEG and PorBDDE25-262AKR, possibly due to changes in binding specificity or in the ability to recruit TLR1, the TLR2 coreceptor required for PorB activity) (33). Interestingly, when the ability of an anti-wtPorB rabbit polyclonal serum to recognize wtPorB and PorB loop mutants was examined by ELISA, wtPorB, PorBL143P, and PorBGEG were efficiently recognized, and similar IgG titers were measured, while lower IgG titers were observed when N. lactamica PorB and particularly PorBDDE25-262AKR were used (data not shown). This observation confirms that selected amino acid sequence differences between N. lactamica PorB and wtPorB are important for loop conformation and/or for crucial residues exposure and indirectly support the possibility that mutation of these residues may affect the quality of PorB-TLR2 interaction.
Our results suggested that the effect of residue mutations in loops 4, 5, and 7 on PorB-TLR2 binding was negligible, maybe because not enough sequence variation was introduced to modify the TLR2 binding site(s). However, particularly for PorBDDE25-262AKR, competitive occupancy of TLR2 resulted in an inhibitory effect on cellular activation by wtPorB. A significant reduction of IL-8 secretion was observed when PorBDDE25-262AKR was coincubated with wtPorB, N. lactamica PorB, or even Pam3CSK4. The hypothesis that engagement of TLR2 by PorBDDE25-262AKR partly blocks access to TLR2 by other agonists is tempting; however, the possibility that TLR1 recruitment and TLR2-TLR1 heterodimerization are also affected cannot be excluded. Further studies are necessary to clarify these possibilities.
While PorBGEG did not affect IL-8 induced by wtPorB, it enhanced IL-8 induced by N. lactamica PorB and Pam3CSK4, possibly via an additive effect, similar to that of wtPorB. If IL-8 induced by N. lactamica PorB is regulated only by NF-κB, while a MAP kinase signaling component is also induced by wtPorB (and possibly PorB loop mutants), the observed enhancement of IL-8 production would likely be due to activation of such signaling pathways.
We conclude that signaling events downstream of TLR2 recognition of neisserial PorB are influenced by a hypothetical TLR2-binding signature within the sequence of PorB surface-exposed loops among different Neisseria strains. Thus, the amino acid sequence of PorB surface-exposed loops is a key component of TLR2 recognition of neisserial PorB. Based on our results, loop 5 and loop 7 appear to play a role in the porin activity and likely contribute to interaction with TLR2 but are not uniquely responsible for this interaction. The effect of loop mutations of PorB on the TLR2-TLR1 signaling complex is under investigation. Characterization of additional loop mutants will provide further structural and functional details about the activity of PorB. Studies using additional loop mutants, including mutants in which positively charged amino acids in each loop have been replaced with alanine to specifically address the electrostatic interaction model proposed by Tanabe et al. (49), are ongoing.
Our observations might have implications beyond simple in vitro incubation of cell lines with different purified PorB types. It is possible that expression of distinct PorB variants by N. meningitidis and N. lactamica organisms influences TLR2-dependent host cell inflammatory responses following bacterial colonization, consistent with previous findings in vitro (12). In vivo, high levels of IL-8 and low levels of RANTES are found in sera from meningitis patients and correlate with severe infections, acute bacterial meningitis, and meningococcal septic shock, while a reversed inflammatory pattern characterizes mild systemic meningitis (6, 7, 37). Similar findings have also been reported for other microorganisms, such as pathogenic and commensal Streptococcus strains (S. aureus and S. epidermis) (55) and P. aeruginosa (22). Although in the case of neisserial infections, this effect has been mostly ascribed to lipooligosaccharide (LOS) (15, 53), a role for other bacterial cell wall components, including PorB, is plausible (37). To expand our findings on the role of PorB on induction of cell responses to whole neisseriae, a chimeric N. meningitidis organism expressing N. lactamica PorB was constructed. Upon stimulation of BEAS-2B cells with the chimeric bacteria, levels of IL-8 secretion lower than those in the original organisms were observed, suggesting that substitution of N. lactamica PorB in N. meningitidis contributes to differential induction of cell activation. Thus, the molecular recognition of PorB by TLR2 likely influences cellular inflammatory responses to Neisseria organisms and may be particularly relevant in the context of PorB sequence variants found in pathogenic N. meningitidis strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniela Vecchio and Sanjay Ram for critical reading of the manuscript.
This work was supported by the BUSM I.D. Basic Research Transitional Program Award and by the BU Pilot Grant to P.M.
Footnotes
Published ahead of print 23 July 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Akira S. 2003. Toll-like receptor signaling. J. Biol. Chem. 278:38105–38108 [DOI] [PubMed] [Google Scholar]
- 2. Ala'Aldeen DA, et al. 2000. Dynamics of meningococcal long-term carriage among university students and their implications for mass vaccination. J. Clin. Microbiol. 38:2311–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett JS, et al. 2010. Independent evolution of the core and accessory gene sets in the genus Neisseria: insights gained from the genome of Neisseria lactamica isolate 020-06. BMC Genomics 11:652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berube J, Bourdon C, Yao Y, Rousseau S. 2009. Distinct intracellular signaling pathways control the synthesis of IL-8 and RANTES in TLR1/TLR2, TLR3 or NOD1 activated human airway epithelial cells. Cell Signal. 21:448–456 [DOI] [PubMed] [Google Scholar]
- 5. Blake MS, Gotschlich EC. 1986. Functional and immunological properties of pathogenic neisserial surface proteins, p 377–400 In Inouye M. (ed), Bacterial outer membranes as model systems. John Wiley, New York, NY [Google Scholar]
- 6. Carrol ED, Thomson AP, Jones AP, Jeffers G, Hart CA. 2005. A predominantly anti-inflammatory cytokine profile is associated with disease severity in meningococcal sepsis. Intensive Care Med. 31:1415–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carrol ED, Thomson AP, Mobbs KJ, Hart CA. 2000. The role of RANTES in meningococcal disease. J. Infect. Dis. 182:363–366 [DOI] [PubMed] [Google Scholar]
- 8. Chenna R, et al. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31:3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274:10689–10692 [DOI] [PubMed] [Google Scholar]
- 9a. Clamp M, Cuff J, Searle SM, Barton GJ. 2004. The Jalview Java alignment editor. Bioinformatics 20:426–427 [DOI] [PubMed] [Google Scholar]
- 10. Denning DW, Gill SS. 1991. Neisseria lactamica meningitis following skull trauma. Rev. Infect. Dis. 13:216–218 [DOI] [PubMed] [Google Scholar]
- 11. Derrick JP, Urwin R, Suker J, Feavers IM, Maiden MC. 1999. Structural and evolutionary inference from molecular variation in neisseria porins. Infect. Immun. 67:2406–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fowler MI, Yin KY, Humphries HE, Heckels JE, Christodoulides M. 2006. Comparison of the inflammatory responses of human meningeal cells following challenge with Neisseria lactamica and with Neisseria meningitidis. Infect. Immun. 74:6467–6478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frasch CE, Zollinger WD, Poolman JT. 1985. Serotype antigens of Neisseria meningitidis and a proposed scheme for designation of serotypes. Rev. Infect. Dis. 7:504–510 [DOI] [PubMed] [Google Scholar]
- 14. Gold R, Goldschneider I, Lepow ML, Draper TF, Randolph M. 1978. Carriage of Neisseria meningitidis and Neisseria lactamica in infants and children. J. Infect. Dis. 137:112–121 [DOI] [PubMed] [Google Scholar]
- 15. Halstensen A, et al. 1993. Interleukin-8 in serum and cerebrospinal fluid from patients with meningococcal disease. J. Infect. Dis. 167:471–475 [DOI] [PubMed] [Google Scholar]
- 16. Henriquet C, Gougat C, Combes A, Lazennec G, Mathieu M. 2007. Differential regulation of RANTES and IL-8 expression in lung adenocarcinoma cells. Lung Cancer 56:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hollis DG, Wiggins GL, Weaver RE. 1969. Neisseria lactamicus sp. n., a lactose-fermenting species resembling Neisseria meningitidis. Appl. Microbiol. 17:71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holten E. 1979. Serotypes of Neisseria meningitidis isolated from patients in Norway during the first six months of 1978. J. Clin. Microbiol. 9:186–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jarva H, et al. 2007. Molecular characterization of the interaction between porins of Neisseria gonorrhoeae and C4b-binding protein. J. Immunol. 179:540–547 [DOI] [PubMed] [Google Scholar]
- 20. Jeanteur D, Lakey JH, Pattus F. 1991. The bacterial porin superfamily: sequence alignment and structure prediction. Mol. Microbiol. 5:2153–2164 [DOI] [PubMed] [Google Scholar]
- 21. Lakshminarayanan V, Drab-Weiss EA, Roebuck KA. 1998. H2O2 and tumor necrosis factor-alpha induce differential binding of the redox-responsive transcription factors AP-1 and NF-kappaB to the interleukin-8 promoter in endothelial and epithelial cells. J. Biol. Chem. 273:32670–32678 [DOI] [PubMed] [Google Scholar]
- 21a. Lambert C, Leonard N, De Bolle X, Depiereux E. 2002. ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18:1250–1256 [DOI] [PubMed] [Google Scholar]
- 22. Leidal KG, Munson KL, Denning GM. 2001. Small molecular weight secretory factors from Pseudomonas aeruginosa have opposite effects on IL-8 and RANTES expression by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 25:186–195 [DOI] [PubMed] [Google Scholar]
- 23. Lewis LA, et al. 1999. Phase variation of HpuAB and HmbR, two distinct haemoglobin receptors of neisseria meningitidis DNM2. Mol. Microbiol. 32:977–989 [DOI] [PubMed] [Google Scholar]
- 24. Li J, et al. 2002. Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L690–L699 [DOI] [PubMed] [Google Scholar]
- 25. Liu X, Wetzler LM, Massari P. 2008. The PorB porin from commensal Neisseria lactamica induces Th1 and Th2 immune responses to ovalbumin in mice and is a potential immune adjuvant. Vaccine 26:786–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X, Wetzler LM, Nascimento LO, Massari P. 2010. Human airway epithelial cell responses to Neisseria lactamica and purified porin via Toll-like receptor 2-dependent signaling. Infect. Immun. 78:5314–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mackinnon FG, et al. 1999. The role of B/T costimulatory signals in the immunopotentiating activity of neisserial porin. J. Infect. Dis. 180:755–761 [DOI] [PubMed] [Google Scholar]
- 28. MacLeod H, Bhasin N, Wetzler LM. 2008. Role of protein tyrosine kinase and Erk1/2 activities in the Toll-like receptor 2-induced cellular activation of murine B cells by neisserial porin. Clin. Vaccine Immunol. 15:630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Massari P, Gunawardana J, Liu X, Wetzler LM. 2010. Meningococcal porin PorB prevents cellular apoptosis in a toll-like receptor 2- and NF-kappaB-independent manner. Infect. Immun. 78:994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Massari P, et al. 2002. Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 168:1533–1537 [DOI] [PubMed] [Google Scholar]
- 31. Massari P, Ho Y, Wetzler LM. 2000. Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc. Natl. Acad. Sci. U. S. A. 97:9070–9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Massari P, King CA, Macleod H, Wetzler LM. 2005. Improved purification of native meningococcal porin PorB and studies on its structure/function. Protein Expr. Purif. 44:136–146 [DOI] [PubMed] [Google Scholar]
- 33. Massari P, et al. 2006. Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J. Immunol. 176:2373–2380 [DOI] [PubMed] [Google Scholar]
- 34. Mayer AK, Dalpke AH. 2007. Regulation of local immunity by airway epithelial cells. Arch. Immunol. Ther. Exp. (Warsz.) 55:353–362 [DOI] [PubMed] [Google Scholar]
- 35. Medzhitov R, Janeway C., Jr 2000. The Toll receptor family and microbial recognition. Trends Microbiol. 8:452–456 [DOI] [PubMed] [Google Scholar]
- 36. Merz AJ, So M. 2000. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16:423–457 [DOI] [PubMed] [Google Scholar]
- 37. Moller AS, et al. 2005. Chemokine patterns in meningococcal disease. J. Infect. Dis. 191:768–775 [DOI] [PubMed] [Google Scholar]
- 38. Oliver KJ, et al. 2002. Neisseria lactamica protects against experimental meningococcal infection. Infect. Immun. 70:3621–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen SF, et al. 1991. Pharyngeal carriage of Neisseria meningitidis and Neisseria lactamica in households with infants within areas with high and low incidences of meningococcal disease. Epidemiol. Infect. 106:445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piet JR, et al. 2011. Genome sequence of Neisseria meningitidis serogroup B strain H44/76. J. Bacteriol. 193:2371–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qi HL, Tai JY, Blake MS. 1994. Expression of large amounts of neisserial porin proteins in Escherichia coli and refolding of the proteins into native trimers. Infect. Immun. 62:2432–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qin H, et al. 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399:549–557 [DOI] [PubMed] [Google Scholar]
- 43. Roebuck KA, et al. 1999. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J. Leukoc. Biol. 65:291–298 [DOI] [PubMed] [Google Scholar]
- 43a. Sayle RA, Milner-White EJ. 1995. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 20:374. [DOI] [PubMed] [Google Scholar]
- 44. Schall TJ. 1991. Biology of the RANTES/SIS cytokine family. Cytokine 3:165–183 [DOI] [PubMed] [Google Scholar]
- 45. Schmeck B, et al. 2006. Streptococcus pneumoniae induced c-Jun-N-terminal kinase- and AP-1-dependent IL-8 release by lung epithelial BEAS-2B cells. Respir. Res. 7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shaughnessy J, Lewis LA, Jarva H, Ram S. 2009. Functional comparison of the binding of factor H short consensus repeat 6 (SCR 6) to factor H binding protein from Neisseria meningitidis and the binding of factor H SCR 18 to 20 to Neisseria gonorrhoeae porin. Infect. Immun. 77:2094–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singleton TE, Massari P, Wetzler LM. 2005. Neisserial porin-induced dendritic cell activation is MyD88 and TLR2 dependent. J. Immunol. 174:3545–3550 [DOI] [PubMed] [Google Scholar]
- 48. Tanabe M, Iverson TM. 2009. Expression, purification and preliminary X-ray analysis of the Neisseria meningitidis outer membrane protein PorB. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65:996–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanabe M, Nimigean CM, Iverson TM. 2010. Structural basis for solute transport, nucleotide regulation, and immunological recognition of Neisseria meningitidis PorB. Proc. Natl. Acad. Sci. U. S. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tettelin H, et al. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809–1815 [DOI] [PubMed] [Google Scholar]
- 51. Tommassen J, Vermeij P, Struyvé M, Benz R, Poolman JT. 1990. Isolation of Neisseria meningitidis mutants deficient in class 1 (Por A) and class 3 (Por B) outer membrane proteins. Infect. Immun. 58:1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Urwin R, Holmes EC, Fox AJ, Derrick JP, Maiden MC. 2002. Phylogenetic evidence for frequent positive selection and recombination in the meningococcal surface antigen PorB. Mol. Biol. Evol. 19:1686–1694 [DOI] [PubMed] [Google Scholar]
- 53. van Deuren M, et al. 1995. Correlation between proinflammatory cytokines and antiinflammatory mediators and the severity of disease in meningococcal infections. J. Infect. Dis. 172:433–439 [DOI] [PubMed] [Google Scholar]
- 54. Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. 2005. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J. Immunol. 175:6465–6472 [DOI] [PubMed] [Google Scholar]
- 55. Wanke I, et al. 2011. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J. Investig. Dermatol. 131:382–390 [DOI] [PubMed] [Google Scholar]
- 55a. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wetzler LM, Ho Y, Reiser H. 1996. Neisserial porins induce B lymphocytes to express costimulatory B7-2 molecules and to proliferate. J. Exp. Med. 183:1151–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilson HD, Overman TL. 1976. Septicemia due to Neisseria lactamica. J. Clin. Microbiol. 4:214–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang L, et al. 2009. Distribution of surface-protein variants of hyperinvasive meningococci in China. J. Infect. 58:358–367 [DOI] [PubMed] [Google Scholar]
- 59. Yazdankhah SP, Caugant DA. 2004. Neisseria meningitidis: an overview of the carriage state. J. Med. Microbiol. 53:821–832 [DOI] [PubMed] [Google Scholar]
- 60. Zollinger WD, Moran EE, Devi SJ, Frasch CE. 1997. Bactericidal antibody responses of juvenile rhesus monkeys immunized with group B Neisseria meningitidis capsular polysaccharide-protein conjugate vaccines. Infect. Immun. 65:1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.