

Abstract
Tuberculosis (TB) is a leading cause worldwide of human mortality attributable to a single infectious agent. Recent studies targeting candidate genes and “case-control” association have revealed numerous polymorphisms implicated in host susceptibility to TB. Here, we review current progress in the understanding of causative polymorphisms in host innate immune genes associated with TB pathogenesis. We discuss genes encoding several types of proteins: macrophage receptors, such as the mannose receptor (MR, CD206), dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN, CD209), Dectin-1, Toll-like receptors (TLRs), complement receptor 3 (CR3, CD11b/CD18), nucleotide oligomerization domain 1 (NOD1) and NOD2, CD14, P2X7, and the vitamin D nuclear receptor (VDR); soluble C-type lectins, such as surfactant protein-A (SP-A), SP-D, and mannose-binding lectin (MBL); phagocyte cytokines, such as tumor necrosis factor (TNF), interleukin-1β (IL-1β), IL-6, IL-10, IL-12, and IL-18; chemokines, such as IL-8, monocyte chemoattractant protein 1 (MCP-1), RANTES, and CXCL10; and other important innate immune molecules, such as inducible nitric oxide synthase (iNOS) and solute carrier protein 11A1 (SLC11A1). Polymorphisms in these genes have been variably associated with susceptibility to TB among different populations. This apparent variability is probably accounted for by evolutionary selection pressure as a result of long-term host-pathogen interactions in certain regions or populations and, in part, by lack of proper study design and limited knowledge of molecular and functional effects of the implicated genetic variants. Finally, we discuss genomic technologies that hold promise for resolving questions regarding the evolutionary paths of the human genome, functional effects of polymorphisms, and corollary impacts of adaptation on human health, ultimately leading to novel approaches to controlling TB.
INTRODUCTION
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis, kills approximately 2 million people annually. TB is exacerbated by the emergence of multidrug- and extensively drug-resistant (MDR and XDR) bacterial strains (258) and represents a major public health problem on a global scale. Humans are the natural reservoir for M. tuberculosis, a highly host-adapted intracellular bacterial pathogen of macrophages. TB occurs predominantly in parts of the world such as Africa and South Asia (80, 87). The occurrence of TB at different rates among particular races, ethnicities, and families indicates a genetic predisposition to TB susceptibility. Complex interactions of M. tuberculosis with environmental and host genetic factors play a critical role in TB development. Host genetic factors explain, at least in part, why some people are more or less susceptible to infection. Several lines of evidence, including twin studies (48, 148, 242), genome-wide linkage studies (21, 49, 136, 144, 148, 217), and recently published genome-wide association studies (GWAS) (135, 172, 229), demonstrate that host genetics strongly influences susceptibility to TB. However, assessing the contributions and functional consequences of specific genetic variations (polymorphisms) in the human genome to host susceptibility or resistance to TB remains a longstanding challenge of population genetics research, with many questions unanswered. Also, in the vast majority of implicated genes, the molecular functions of candidate polymorphisms have remained unknown (Table 1).
Table 1.
Association studies on host innate immune genes related to TB pathogenesis
| Gene | Polymorphism(s) (genetic location) | Population(s)a | Association with TBb | Molecular mechanism known? | Reference(s) |
|---|---|---|---|---|---|
| MR | 1186G/A (exon) | China | Yes | No | 272 |
| DC-SIGN | −336G/A (promoter) | South Africa | Yes | No | 17 |
| Colombia, Tunisia | No | 25,88 | |||
| SSA, Gambia | Yes | No | 247 | ||
| India, China | No | 202,275 | |||
| −871G/A (promoter) | South Africa | Yes | No | 17 | |
| China | No | 275 | |||
| Dectin-1 | Not identified | ||||
| TLR1 | N248S, S602I (exon) | USA (African-American) | Yes | No | 131 |
| Europe (Caucasian) | Yes | No | 239 | ||
| TLR2 | R753Q (exon) | Turkey | Yes | No | 162 |
| R677W (exon) | Tunisia | Yes | No | 24 | |
| Insertion/deletion (promoter) | Guinea-Bissau, USA (Caucasian) | Yes | No | 251 | |
| TLR4 | D299G (exon) | Gambia, Guinea-Bissau | No | 157,166 | |
| TLR8 | rs3764880 (exon) | Indonesia, Russia | Yes | No | 56 |
| TLR9 | rs352143, rs574386 (exon) | USA (Caucasian), USA (African-American) | Yes | No | 251 |
| TIRAP | S180L (exon) | West Africa | Yes | Yes | 110 |
| Indonesia, Russia, Ghana | No | 156 | |||
| Meta-analysis (China) | No | 142 | |||
| CR1 | Q1022H (exon) | Malawi | Yes | Yes | 76 |
| CR3 | Not identified | ||||
| NOD1 | Not identified | ||||
| NOD2 | P268S, R702W, A725G (exon) | USA (African-American) | Yes | No | 10 |
| CD14 | −159C/T (promoter) | Mexico | Yes | No | 184 |
| Korea | Yes | Yes | 108 | ||
| P2X7 | 1513A/C (exon) | Gambia, China | No | 119,262 | |
| Mexico, Russia | Yes | No | 145,161 | ||
| India | Yes | No | 205,209 | ||
| Meta-analysis (China) | Yes | No | 263 | ||
| Meta-analysis (China) | No | 257 | |||
| −762T/C (promoter) | Gambia, India | Yes | No | 119,209 | |
| Mexico, Russia, China | No | 145,161,262 | |||
| Meta-analysis (China) | No | 263 | |||
| VDR | ApaI (exon) | Guinea-Bissau | Yes | No | 166 |
| Tanzania | No | 211 | |||
| BsmI (exon) | Meta-analysis (Asia) | Yes | No | 85 | |
| Meta-analysis (Africa) | No | 85 | |||
| FolkI (exon) | China | Yes | No | 125 | |
| Tanzania | No | 211 | |||
| TaqI (exon) | Gambia | Yes | No | 22 | |
| Cambodia, Tanzania | No | 57,211 | |||
| FolkI-BsmI-ApaI-TaqI | West Africa | Yes | No | 33 | |
| haplotype | South Africa | Yes | No | 127 | |
| SP-A1 | 1416C/T (intron) | India | Yes | No | 134 |
| 307G/A,776C/T (exon) | Ethiopia | Yes | No | 138 | |
| SP-A2 | 1382C/G (intron) | India | Yes | No | 134 |
| 355C/G, 751A/C (exon) | Ethiopia | Yes | No | 138 | |
| MBL | O allele | India | Yes | No | 203 |
| O, X alleles | Denmark | Mixed | 212 | ||
| B allele | USA (African-American) | Yes | No | 68 | |
| O allele | USA (Caucasian) | No | 68 | ||
| O,H,L,X,Y,P,Q alleles | China | No | 126 | ||
| AO genotype | Tanzania | No | 211 | ||
| HYA/HYA, LYB/LYD haplotypes | Italy | Yes | No | 39 | |
| AA,AO,OO genotypes | Meta-analysis (Australia) | No | 59 | ||
| B allele | India | Yes | No | 210 | |
| G57E (exon) | Ghana | Yes | No | 228 | |
| TNF | −238G/A (promoter) | Colombia | Yes | No | 53 |
| Iran | No | 5 | |||
| −238GG genotype | Iran | Yes | No | 5 | |
| −308G/A (promoter) | Colombia, Iran | Yes | No | 53,141 | |
| Korea | No | 163 | |||
| Meta-analysis (Brazil) | No | 169 | |||
| Meta-analysis (China) | No | 256 | |||
| IL-1β | −511T/C (promoter) | Gambia | Yes | No | 11 |
| Colombia | No | 90 | |||
| 3953T/C (exon) | Gambia | No | 11 | ||
| 3953 T allele | Colombia | Yes | No | 90 | |
| IL-6 | −174G/C (promoter) | Colombia, India | No | 97,201 | |
| −174GG genotype | Iran | Yes | Yes | 5 | |
| −174G allele | Canada (aboriginals) | Yes | Yes | 115 | |
| IL-8 | −251T/A (promoter) | Gambia | No | 50 | |
| −251A allele | USA (African-American) | Yes | No | 132 | |
| 781T/C (exon) | Gambia | No | 50 | ||
| IL-10 | −1082G/A (promoter) | Cambodia | Yes | No | 57 |
| Brazil, Spain, Pakistan | No | 7,128,169 | |||
| Meta-analysis (China) | Yes | No | 270 | ||
| −1082G allele | Turkey | Yes | No | 9,168 | |
| −1082 allele/genotype | Egypt | No | 154 | ||
| −592A/C (promoter) | Korea | Yes | No | 207 | |
| IL-18 | −607C/A, −137G/A | India | No | 94 | |
| −667G/T, −618A/C | Korea | No | 117 | ||
| AGA, CCA haplotypes | China | Yes | No | 93 | |
| MCP-1 (CCL2) | −2518A/G (promoter) | Zambia, Tunisia | Yes | No | 28,37 |
| Hong Kong | No | 45 | |||
| −2518 GG genotype | Mexico, South Korea | Yes | Yes | 78 | |
| −2518AA genotype | Zambia, Mexico, South Korea | No | 37,78 | ||
| −362G/C (promoter) | Ghana | Yes | No | 227 | |
| RANTES (CCL5) | −403G/A,−28C/G,ln1T/C | Hong Kong | No | 45 | |
| ACT, GCC haplotypes | Hong Kong | Yes | No | 45 | |
| −403G/A, −28C/G or GG, AC haplotypes | Spain, Tunisia | Yes | No | 26,192 | |
| CXCL10 | −135G/A (promoter) | China | Yes | No | 225 |
| −872G/A, −1447A/G | China | No | 225 | ||
| iNOS (NOS2) | −1026G allele | Brazil | Yes | No | 102 |
| −954G/C (promoter) | Mexico | No | 78 | ||
| CCTTT (microsatellite) | Colombia | Yes | No | 89 | |
| TAAA (microsatellite) | Colombia | No | 89 | ||
| rs2274894 (intron), rs7215373 (3′-UTR) | USA (African-American) | Yes | No | 250 | |
| rs9282799, rs8078340 (promoter) haplotypes | South Africa | Yes | No | 150 | |
| SLC11A1 | 274C allele (exon) | USA (Children) | Yes | No | 137 |
| (NRAMP1) | 3′-UTR | Gambia, South Korea | Yes | No | 23,187 |
| China | Yes | No | 103,118 | ||
| D543N (exon) | Gambia, China | Yes | No | 23,118 | |
| 5′(GT)n (promoter) | Gambia, South Africa | Yes | No | 12,23,99 | |
| USA, Tanzania | Yes | No | 130,211 | ||
| USA, Morocco | No | 67,249 | |||
| INT4 (intron) | Gambia, China | Yes | No | 23,271 | |
| China | No | 103 | |||
| 3′-UTR, D543N | Morocco, Thailand | No | 67,248 | ||
| Cambodia | Yes | No | 57 | ||
| 3′-UTR, D543N, 5′(GT)n | Meta-analysis (China) | Yes | No | 121 | |
| 3′-UTR, D543N, 5′(GT)n, INT4 | Turkey | No | 8 | ||
| Meta-analysis (China) | Yes | No | 122 |
SSA, sub-Saharan Africa.
Refers to mainly pulmonary TB.
Widespread M. tuberculosis infection of the human race over prolonged time periods suggests powerful evolutionary pressures in the interactions between host and pathogen genomes (15, 38, 170). Because of ready access to the microbial genome, the majority of studies have thus far focused on the evolution of Mycobacterium species (46, 254, 268), while the adaptive pressures on the human genome have been such that only incremental insights into candidate genes have been yielded. Nevertheless, clear signatures of evolutionary selection pressure can be found in multiple human gene families involved in TB pathogenesis, as well as associated processes, such as autophagy (60, 61). Strong evidence for selection of genetic variants modulating infectivity and resistance exists for genes encoding major histocompatibility complex/human leukocyte antigen (MHC/HLA) (31, 235), tumor necrosis factors (TNFs) and their receptors (259), immune-related GTP-ases (IRGs) (20), NRAMP1 (solute carrier protein 11A1 [SLC11A1]) (18, 38), Toll-like receptors (TLRs) (16, 265), vitamin D nuclear receptor (VDR) (38, 260), and cell surface proteins, such as lectins (159). As yet, however, an integrative understanding is still lacking of how these factors act in concert to mitigate the effects of TB or render resistance, versus the ability of M. tuberculosis to evade such mechanisms.
It is also critical to understand that genetic adaptation leading to resistance of the host does not occur without cost. For example, MHC-associated diseases may be the price paid for an effective immune response (235). Similarly, the same polymorphisms conveying partial resistance to M. tuberculosis infection—for example, promoter variants in TNF and NRAMP (199) that increase resistance—conversely enhance autoimmune reactions (1). A link between the spread of TB, as a result of human urbanization and high population density leading to enhanced infection rates, and rheumatoid arthritis was suggested in one study, paralleling the observation that therapy of rheumatoid arthritis with TNF antibodies results in increased TB rates (1). The evolutionary history of the IRGM locus highlights the important role of the IRGM gene in autophagy and Crohn's disease in response to pathogenesis (20). Other consequences can occur in unexpected ways, even involving psychiatric disorders, such as schizophrenia, suggested to occur via a kynurenine pathway and NAD+ biosynthesis (143). Thus, cumulative information from the literature suggests a fundamental evolutionary concept, that environmental factors have exerted selection pressure on the human host genome, leading to the accumulation of genetic variants relevant to TB susceptibility.
Here, we review polymorphisms present in host innate immune genes having either a significant or a possible association with TB. For completeness, in each section, we first summarize the biological attributes of selected proteins, focusing on macrophage pattern recognition receptors (PRRs), soluble C-type lectins (including the collectins), cytokines, and chemokines as major subsets of innate immune determinants (Table 1). In view of the evolutionary history of M. tuberculosis-human interactions, we expect many of the genetic variants to occur at relatively high frequencies, but negative consequences to the host could limit selective sweeps, as homozygous carriers can suffer corollary damage that itself will be under negative selection pressure. We highlight cases where evidence of selection pressure is established as a sign that the genetic variant indeed has a strong phenotypic influence on TB. We also review current challenges in genomic approaches to TB and provide examples of opportunities for the road forward in this critical area of study.
PATTERN RECOGNITION RECEPTORS
Macrophage PRRs and phagocytic receptors contributing to mycobacterial diseases, especially TB, have been discussed in several recent reviews (104, 193, 231, 266). These include cell membrane-bound receptors, such as the mannose receptor (MR, CD206), dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN) (CD209), Dectin-1, TLRs, and complement receptor 3 (CR3, CD11b/CD18), and intracellular cytosolic receptors, such as nucleotide oligomerization domain 1 (NOD1) and NOD2 (Fig. 1).
Fig 1.
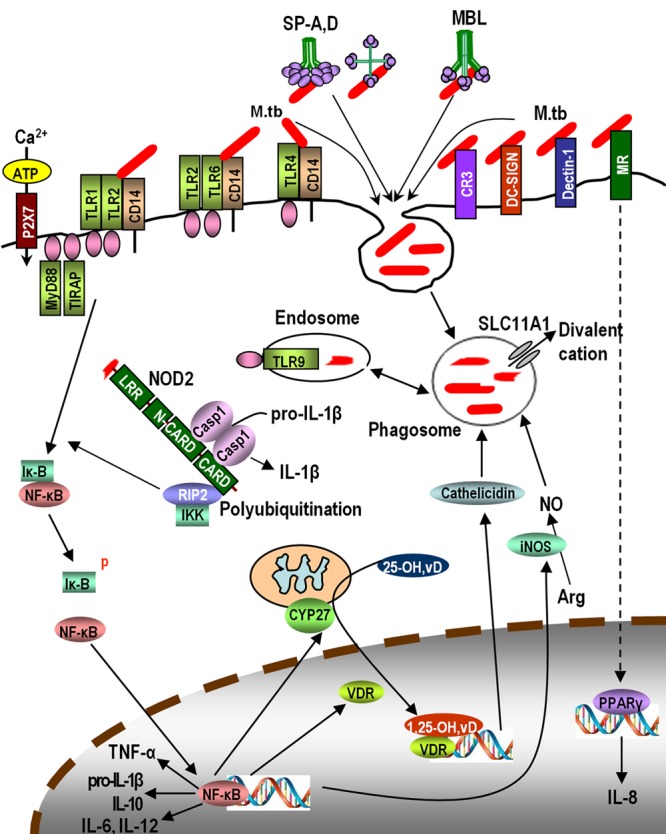
A representative scheme of the macrophage innate immune network in response to M. tuberculosis (M.tb) infection discussed in this review. LRR, leucine-rich repeat; IKK, IκB kinase; 25-OH,vD: 25-hydroxyvitamin D; 1,25-OH,vD: 1,25-dihydroxyvitamin D.
MR.
The MR or MRC1, also known as CD206, is a type I transmembrane protein expressed abundantly on macrophages and DCs (139, 213, 267). Phagocytosis of M. tuberculosis by macrophages is mediated in part by the MR (107, 197), and its engagement by bacterial surface mannosylated lipoglycans results in limited phagosome-lysosome fusion within macrophages (106, 230). MR ligation by the M. tuberculosis surface lipoglycan, mannose-capped lipoarabinomannan (ManLAM), creates an immunosuppressive environment in the host (reviewed in references 70 and 231). Our recent studies on an MR-specific signaling pathway (179) demonstrate that the MR-mediated uptake of live M. tuberculosis or ManLAM by macrophages leads to upregulation of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPAR-γ) (Fig. 1), which is linked to macrophage anti-inflammatory pathways. The MR also serves as a molecular link between innate and adaptive immune responses (174, 213).
Genetic studies.
In a recent case-control study with a Chinese population, a nonsynonymous single-nucleotide polymorphism (SNP), 1186G/A (rs34039386; Gly→Ser, G396S in exon 7), was found to be associated with pulmonary TB (272). The Ser allele was previously shown to be significantly associated with leprosy (caused by the closely related species Mycobacterium leprae) in Vietnamese, whereas the Gly allele was associated in Brazilian cases (4). Such discrepancies can arise if the candidate SNP serves only as a surrogate for a functional variant with different haplotype structures present in different populations or if these associations are false positives. While a genetic association of the MR with TB or TB-related diseases is plausible, the causative polymorphisms and underlying molecular mechanisms remain uncertain.
DC-SIGN.
DC-SIGN (CD209) is a type II transmembrane protein predominantly expressed on DCs (245), whereas its presence on macrophages depends on the tissue type and state of activation (175, 204). DC-SIGN is induced in alveolar macrophages from M. tuberculosis-infected patients (220). It directly mediates phagocytosis of M. tuberculosis by DCs (221) and interferes with DC maturation (86). The cytoplasmic tail of DC-SIGN has three conserved motifs that are involved in ligand binding and receptor signaling, phagocytosis, and intracellular trafficking of ligand particles (13, 75).
Genetic studies.
Two polymorphisms in the promoter region of DC-SIGN (−871A/G and −336A/G) have shown significant associations with TB (Table 1). In a separate study with both Caucasian Canadian and indigenous African populations, SNPs within the 5′- and 3′-untranslated regions (UTRs) of both DC-SIGN and DC-SIGNR revealed possible associations but also showed significantly different frequencies among the two populations, which emphasizes the need to consider geographical and ethnic diversity in assessing susceptibility to TB (32). These regulatory polymorphisms were assumed to affect gene transcription and translation, but further studies are needed to establish a causative chain of events.
Dectin-1.
Dectin-1, a β-glucan PRR, is a type II transmembrane protein expressed on macrophages, DCs, and neutrophils, regulating proinflammatory responses to microbial pathogens (35, 36). Although no specific mycobacterial ligands for Dectin-1 have been identified so far, this receptor interacts with mycobacteria in concert with TLR2 to produce cytokines such as TNF and interleukin-12p40 (IL-12p40) (186, 264). Recent studies have proposed additional roles of Dectin-1 in M. tuberculosis infection (243, 269).
Genetic studies.
There are no reports to date of Dectin-1 polymorphisms that can be linked directly to TB. Because of the potential cross talk of Dectin-1 with other PRRs on the cell surface (240), polymorphisms in this C-type lectin should be tested for their impact on TB pathogenesis. Because of the interaction with other susceptibility factors, studies should focus on gene-gene interactions (epistasis) whereby a genetic variant in Dectin-1 may show significance only in the context of variants in interacting genes.
TLRs.
TLRs are expressed on many cell types, including host immune cells, serving as critical mediators of the immune response to a variety of pathogens, including M. tuberculosis (104, 109). TLRs are either expressed on the cell surface (e.g., TLR2 and 4) or intracellularly (e.g., TLR8 and 9) (185). M. tuberculosis and its cell wall components are recognized by several TLRs, including TLR1, TLR2, TLR4, TLR6, and TLR9 (96, 105, 140, 177, 224). Among them, evidence for genetic variants associated with TB is most abundant for TLR2, which functions alone or as a heterodimer with TLR1 or TLR6 (Fig. 1). The strong proinflammatory response to M. tuberculosis infection observed via the TLR2 signaling pathway is mediated through its adaptor proteins MyD88 and TIR domain-containing adaptor protein (TIRAP) (178, 185) (Fig. 1).
Genetic studies.
Several common polymorphisms associated with TB have been reported for different TLRs, including TLR1, -2, -4, -8, and -9 (Table 1). The TLR2 variant R753Q appears to influence the progression of infection to TB disease in children (55). Another exonic SNP of TLR2 (597T/C) was found to be strongly associated with TB meningitis and miliary TB in Vietnam (226). Although the TLR4 polymorphism D299G did not show any association with pulmonary TB (Table 1), the same variant has been found to be a risk factor for TB in HIV-infected patients in Tanzania and Spain (74, 176). HIV is thus a factor interacting with certain host genetic variants to influence susceptibility to TB. In a diversified racial study assessing 71 reference SNPs in five TLRs (TLR1, -2, -4, -6, and -9) within populations of U.S. Caucasian, African-American, and West African TB cases, significant associations of TB were observed with two variants each from TLR2 (an insertion/deletion at −196 to −174) and TLR9 across certain, but not all, study populations (251) (Table 1). Adaptor protein TIRAP, central to signaling from both TLR2 and TLR4 to NF-κB activation, is also implicated in association with TB. A reported role of the nonsynonymous SNP S180L (975C/T) in TIRAP (Table 1) in protection against TB results from attenuation of TLR2 signal transduction (110). Another polymorphic variant (558C/T) in TIRAP, discovered in the Vietnamese population, showed an association with TB meningitis but not pulmonary TB (95). These results support TLRs and their signaling partners as true susceptibility genes; however, the evidence is varied for individual genes and alleles, making it difficult to assess the clinical relevance of specific genetic factors related to TLRs at this point.
CR3 (CD11b/CD18).
CR3 is one member of the CR family, which also includes CR1 (CD35) and CR4 (CD11c/CD18). The role of C3 opsonization and the contributions of CR1, CR3, and CR4 in the phagocytosis of M. tuberculosis have been described (reviewed in references 70 and 196). CR3 expressed on human monocytes and macrophages (Fig. 1) plays an important role in both opsonic and nonopsonic uptake of M. tuberculosis (196). The role of CR3 during M. tuberculosis infection in vivo remains elusive.
Genetic studies.
To date, little is known about the role of genetic polymorphisms in CR family members and TB. Among five polymorphisms in CR1, including Q1022H, identified in a Malawian study, homozygous 1022H carriage was reported to increase the risk of TB (76). This nonsynonymous SNP impairs ligand binding and, thus, may alter M. tuberculosis uptake. Such functional analysis strengthens the case for a role of CR polymorphisms in M. tuberculosis infectivity.
NOD1 and NOD2.
NOD1 and NOD2 (also known as CARD4 and CARD15, respectively), prototype members of the NOD-like receptor family, are cytoplasmic sensor proteins. M. tuberculosis contains a unique N-glycolyl muramyl dipeptide (MDP) in its cell wall peptidoglycan which is a particularly potent NOD2 ligand (54). NOD proteins are implicated in a variety of inflammatory diseases, including Crohn's disease (40, 100), while a contribution to mycobacterial diseases is also emerging (111). We have recently found that NOD2 regulates proinflammatory responses and the intramacrophage survival of M. tuberculosis in human macrophages (34).
Genetic studies.
To date, any role of polymorphisms in NOD1 in TB pathogenesis remains uncertain. Two independent studies in an African population failed to reveal associations with known NOD2 polymorphisms (2936insC, R675W, G1881R, R702W, G908R, and 1007fs) that had previously been associated with Crohn's disease (149, 218). Amino acid substitutions resulting from three NOD2 polymorphisms (Table 1) are proposed to change the protein's hydrophobicity and increase its stability, thereby enhancing ligand sensing (10). Taken together, genetic studies should include NOD2 to test its potential role as a TB modulator.
Other receptors.
Macrophage receptors, such as the membrane-bound CD14, human purinergic receptor P2X7, and vitamin D nuclear receptor (VDR), also play a role in the host innate immune response to TB (Fig. 1) (for reviews, see references 29, 148, 200, and 266). Association of a promoter region CD14 polymorphism (−159C/T) with TB (Table 1) was attributed to higher promoter activity of the variant allele, increased soluble CD14 production, and decreased secretion of gamma interferon (IFN-γ) (108). The P2X7 receptor mediates ATP-induced killing of mycobacteria through induction of phospholipase D and phagolysosomal fusion (69, 112). Two polymorphisms in P2X7 (1513A/C and −762T/C) have been tested for TB associations, with various results (Table 1). In addition to its potential role in pulmonary TB, the association of P2X7 1513A/C with increased susceptibility to extrapulmonary TB was attributed to perturbation of ATP-mediated killing of M. tuberculosis by macrophages (73). The promoter polymorphism −762T/C was suggested to alter P2X7 gene expression, but experimental evidence is lacking. Vitamin D has been linked to TB pathogenesis through the action of VDR (29, 190, 200), suppressing M. tuberculosis growth in macrophages (29) through induction of the antimicrobial peptide cathelicidin, potentially also involving TLR signaling (Fig. 1) (124, 190). Several VDR polymorphisms with nomenclature derived from their restriction enzyme cleavage sites, such as FokI, TaqI, BsmI, and ApaI, and their combination as a haplotype, have been tested in population studies for TB associations (Table 1), but a meta-analysis yielded inconsistent results (85).
SOLUBLE C-TYPE LECTINS (COLLECTINS)
Collectins are collagen like-containing calcium-dependent (C-type) lectins, including lung surfactant proteins A and D (SP-A and SP-D) and mannose-binding lectin (MBL) (Fig. 1). Interactions of M. tuberculosis with SP-A, SP-D, and MBL have been described previously (231).
SP-A and SP-D.
Present in the lung alveoli, SP-A and SP-D are predominantly expressed by alveolar type II epithelial cells (261). SP-A consists of two highly similar isoforms encoded by separate genes, SP-A1 and SP-A2. SP-A and SP-D participate in the lung innate immune system for a spectrum of lung pathogens, including M. tuberculosis. We have demonstrated that SP-A and SP-D modulate the phagocytosis of M. tuberculosis by human macrophages in opposite directions (19, 72). SP-D treatment of M. tuberculosis causes increased phagosome-lysosome fusion and reduced intracellular growth (71). SP-A's interaction with macrophages regulates the expression and function of TLR2 and -4 (98).
Genetic studies.
Polymorphisms in SP-A and SP-D that occur with considerable frequency are considered genetic determinants for a number of pulmonary infectious diseases, including TB (92). Allelic variations in both SP-A and SP-D have been shown to influence host susceptibility to TB in a Mexican population (79). Several intronic and exonic SP-A1 and SP-A2 SNPs associated with TB are listed in Table 1. Malik et al. (138) suggested that these polymorphisms may affect splicing and/or mRNA maturation, but a rigorous analysis of the underlying mechanism remains to be done.
MBL.
Secreted by the liver, MBL is an acute-phase protein which recognizes microbial surface carbohydrates, especially mannose- and N-acetylglucosamine-terminated glycoproteins. It is a multimeric protein with a structure similar to those of SP-A and SP-D (Fig. 1). MBL plays an important role in host defense against pathogens, including M. tuberculosis. By binding to M. tuberculosis, MBL acts as an opsonin, enhances both complement-dependent and -independent phagocytosis, and promotes inflammation with the release of cytokines (65, 222).
Genetic studies.
Interindividual variations in human serum MBL levels have been associated with three common SNPs in exon 1 of the MBL2 gene, at codons 52, 54, and 57 (reviewed in reference 266). Mutations at these codons result in low or nearly absent serum MBL levels in hetero- and homozygote individuals, respectively. However, several studies with MBL alleles, genotypes, or haplotypes, mostly based on these polymorphisms, have yielded conflicting results (Table 1). Clearly, more work is needed to establish a causative link between MBL variants and TB.
CYTOKINES AND CHEMOKINES
Cytokine networks established and maintained by macrophages in the innate immune system play a critical role in controlling M. tuberculosis infection. Upon infection, macrophages are activated to produce proinflammatory cytokines, including TNF, IL-1β, IL-6, IL-12, and IL-18, and the regulatory IL-10 (Fig. 1). Chemokines relevant to M. tuberculosis infection include IL-8 (CXCL8), monocyte chemoattractant protein 1 (MCP-1, CCL2), RANTES (CCL5), and CXCL10 (IP-10). The role of these cytokines/chemokines and their genetic variations in TB pathogenesis has been reviewed (29, 193, 201, 266). Many of the implicated polymorphisms reside in promoter regions, with the potential to affect transcription and, hence, cytokine/chemokine activity.
TNF.
Nineteen isoforms of cytokines have been identified for the TNF family, and TNF (formerly known as TNF-α) is a prototypic proinflammatory cytokine produced by monocytes, macrophages, and DCs when stimulated with mycobacteria or mycobacterial products. TNF is involved in strong protective host responses against M. tuberculosis infection by a variety of mechanisms (77, 81, 183).
Genetic studies.
Two common SNPs in the promoter region of the TNF gene (−238G/A and −308G/A) have been extensively studied, with various outcomes (Table 1), while other studies have also focused on TNF receptor (TNFR). An association between TB and TNFR1 was reported in a Ugandan population (216). A single TNFR1 SNP (rs3397) and a 3′-UTR haplotype (GTT) were associated with TB resistance in Ghana and South Africa (147). Possible evolutionary selection pressure appears to affect TNF gene expression rather than protein structure (1, 259), a common finding among the TB susceptibility genes, as regulation of gene expression may be a prevalent selection mechanism in evolution.
IL-1β.
Expressed in a variety of immune cells in an inactive precursor form (pro-IL-1β) and then cleaved by caspase 1 to an active form via the inflammasome (Fig. 1), IL-1β upregulates essential mediators necessary for the control of M. tuberculosis infection (63, 82).
Genetic studies.
Two IL-1β polymorphisms, one in the promoter region (−511) and the other exonic (+3953), have been associated with pulmonary TB (Table 1). Extrapulmonary TB was also associated with the +3953CT genotype of the IL-1β gene (155). Another IL-1β polymorphic locus was identified at +3962 in a Macedonian population; the +3962TT genotype was significantly associated with susceptibility, while the +3962CT genotype appeared to be protective (232), suggesting a dominant effect for the T allele. However, the underlying mechanisms have not been investigated.
IL-6.
With both pro- and anti-inflammatory properties, IL-6 is produced early during mycobacterial infection and is involved in macrophage and cytotoxic T-cell differentiation (114, 195, 208).
Genetic studies.
A genome-wide scan of Ugandans with TB revealed a linkage to a locus on chromosome 7 which contains the IL-6 gene (217); however, direct association of IL-6 variants with TB could not be confirmed by fine mapping in a subsequent study (14). Increased frequency of the G allele or GG genotype in a common variant in the promoter region of IL-6 (−174) in TB patients (Table 1) was associated with high IL-6 production (5, 115), which may have promoted M. tuberculosis infection by inhibiting the production of other potent cytokines, such as TNF and IL-1β.
IL-8.
The chemokine IL-8 (CXCL8) is produced by phagocytic cells upon stimulation with M. tuberculosis or its products (182, 273), leading to recruitment of inflammatory cells to the site of infection. Increased levels of IL-8 in the bronchoalveolar lavage fluid of M. tuberculosis-infected humans have been associated with increased mortality of TB patients (189). We found an increased production of IL-8 by M. tuberculosis-infected human macrophages through MR-directed PPAR-γ activation (179) (Fig. 1). Only a few studies have addressed the association of IL-8 polymorphisms with TB (Table 1). A causative link between IL-8 and TB, however, remains elusive.
IL-10.
Expressed by activated monocytes/macrophages, DCs, B cells, and regulatory T cell subsets (188), IL-10 is considered a macrophage-deactivating cytokine since it suppresses the proinflammatory response by downregulating the production of several cytokines. M. tuberculosis phagocytosis induces IL-10 production in human monocytic cells (206). During latent M. tuberculosis infection, increased IL-10 production promotes reactivation of disease in mice (238). IL-10 potentially helps M. tuberculosis persistence in humans by blocking phagosome maturation in macrophages (165).
Genetic studies.
IL-10 SNPs (−1082G/A and −592A/C) have been associated with increased protein expression (237). Despite extensive studies, especially with −1082G/A, associations with TB were inconsistent (Table 1). In a candidate gene linkage study using microsatellite markers, the IL-10 gene was implicated in clinical TB (216). This study found a strong linkage between the IL-10 polymorphisms and TNF levels and confirmed the previous association of TB with IL-10, especially for the SNP at −592.
IL-12.
IL-12 is mainly secreted by phagocytic cells (monocytes, macrophages, neutrophils, and DCs), induces IFN-γ production by T cells by signaling through IL-12 receptors (IL-12BR1 and IL-12BR2), and promotes T-cell differentiation into Th1 cells (233, 234). IL-12 is one of the key players in host defense against M. tuberculosis infection (51, 84).
Genetic studies.
Discrepant results have been reported among various populations regarding the link between TB and IL-12 polymorphisms (especially for IL-12B, the gene encoding the IL-12p40 subunit). Several IL-12B polymorphisms in the introns, promoter, or 3′-UTR were found to be associated with TB in some studies (83, 151, 236) but not others (132, 173, 201). An IL-12B 3′-UTR SNP (rs3212227) was identified as a risk factor for pulmonary TB in populations of African origin in a confirmation study (152). Polymorphisms in the IL-12 receptor gene, IL-12RB1, have also been implicated in the development of TB in Moroccan (181) and Japanese (113) populations but not in Koreans (116).
IL-18.
Sharing many features with IL-1, IL-18 is another proinflammatory cytokine involved in the induction of IFN-γ, synergistically with IL-12 (64, 164). IL-18 production by M. tuberculosis-infected macrophages is increased by contact with activated CD4+ T cells (244). There is evidence for a protective role of IL-18 during mycobacterial infections; IL-18 knockout mice were found to be highly susceptible to Mycobacterium bovis BCG and M. tuberculosis (219).
Genetic studies.
Several promoter polymorphisms for IL-18 have been reported, but only haplotypes were significantly associated with TB (Table 1). This result suggests a role for SNP phasing for determining functionality or that any causative variants are insufficiently represented by single SNP markers.
MCP-1 (CCL2).
Monocyte chemoattractant protein-1, or MCP-1, is a β-chemokine produced by and acting on monocytes and macrophages. M. tuberculosis preferentially induces MCP-1 production by human monocytes (123). MCP-1 deficiency in mice impaired monocyte recruitment and granuloma formation, and yet it conferred decreased susceptibility to M. tuberculosis (129).
Genetic studies.
Discrepant results obtained from genotype- and allele-based TB association studies with MCP-1 variants (especially at −2518) (Table 1) failed to yield solid evidence for genetic effects of MCP-1. Carriers of the −2518 GG genotype were reported to produce high levels of MCP-1, which inhibits IL-12 production in response to M. tuberculosis and promotes active pulmonary TB (78), whereas Thye et al. (227) initially found association of the −2518G allele with TB resistance in Ghana, and further genotyping led to the identification of the −362C allele as the putatively “true” protective variant of MCP-1, reflecting linkage disequilibrium between −2518 and −362. This study is a cautionary note for the need to carry out a thorough genetic investigation to search for the actual causative polymorphism.
RANTES (CCL5).
RANTES (regulated upon activation, normal T-cell expressed, and secreted), a member of the C-C chemokine subfamily, acts as a chemokine for T cells, monocytes/macrophages, eosinophils, and basophils. RANTES promotes granuloma formation in M. tuberculosis-infected lungs in a mouse model (44, 252). M. tuberculosis infection of human alveolar macrophages induces the production of RANTES, which reduces intracellular bacterial growth (194). The role of RANTES in protective immunity to M. tuberculosis infection has also been reported in mouse macrophages (191) and in knockout mice (252).
Genetic studies.
A few studies have focused on −403G/A and −28C/G promoter variants of the RANTES gene (Table 1), providing evidence that genotypes or haplotypes derived from a combination of SNPs could confer resistance or susceptibility to TB (26, 45, 192). However, the actual causative variants need to be established before definitive conclusions can be made.
CXCL10(IP-10).
A member of the CXC chemokine subfamily, CXCL10 (C-X-C motif chemokine 10), is also known as gamma interferon-induced protein-10 (IP-10) because it is secreted by several cell types in response to IFN-γ. In addition to its chemotactic properties for immune cells, including monocytes/macrophages, CXCL10 is also involved in stimulation of NK cells and migration of T cells following M. tuberculosis infection (276).
Genetic studies.
One (−135G/A) of three CXCL10 promoter polymorphisms examined was moderately associated with TB in a Chinese population (Table 1). The same variant had previously been associated with hepatitis B virus infection (58) and was suggested to contribute to CXCL10 expression by NF-κB transactivation (225). Taken together, these observations justify further studies on the role of CXCL10 variants.
OTHER INNATE IMMUNE MOLECULES
iNOS (NOS2).
Inducible nitric oxide synthase, or iNOS (also known as NOS2 and encoded by the human NOS2A), expressed by mycobacterium-infected macrophages and epithelial cells, produces NO, an effector molecule with bactericidal activity against M. tuberculosis (42, 43) (Fig. 1). Although iNOS (human NOS2A) expression has been reported in lung macrophages from patients infected with M. tuberculosis (160), iNOS activity and NO production are more robust in mouse macrophages than in human macrophages (198).
Genetic studies.
An iNOS locus has been found to be protective against TB in a mouse study (133), and variants in the human NOS2A locus have been tested in TB association studies (Table 1). Together with functional evidence described in these studies, the genetic associations found between NOS2A and TB support the importance of NO in TB and infectious diseases in general.
SLC11A1 (NRAMP1).
Solute carrier family 11A, member 1, or SLC11A1 (also known as natural resistance-associated macrophage protein 1, or NRAMP1), acts as a divalent cation transporter or antiporter across phagosomal membranes (91, 101) (Fig. 1). It was originally identified in mice as a regulator of resistance to intracellular pathogens, including M. bovis BCG (253). By inducing microbicidal activities in infected macrophages, SLC11A1 is a critical mediator in the innate immune response to mycobacterial infection, but the precise function of this protein remains unclear.
Genetic studies.
Four SLC11A1 polymorphisms [3′-UTR, D543N, 5′(GT)n, and INT4] and their associations with TB have been studied globally (Table 1). Association of most of the variants has been confirmed in a meta-analysis in China (121), whereas a small case study in Turkey on all four variants did not reveal any association with TB (8). Further results of a recent meta-analysis in China support SLC11A1's role in host defense against TB (122). SLC11A1 polymorphisms at the D543N and INT4 loci have been suggested to contribute to TB progression from infection rather than to M. tuberculosis infection (271). Another SLC11A1 polymorphism (rs3731865) in African-Americans was found to be significant when considering an interaction with TLR2 (249), which emphasizes the importance of gene-gene interaction in TB-host genetic studies (see below). It is clear from these studies that there is a great deal of allelic heterogeneity within SLC11A1. It is possible that the reported associations with SNPs in this gene are due to other adjacent or remote SNPs which are in some linkage disequilibrium with these reported variants, and discovering the actual regulatory variant is a point of emphasis of this current review. However, the 5′(GT) repeat in the promoter region of SLC11A1, consisting of 4 alleles, was found to affect promoter activity in a reporter gene assay (199), with the frequent allele 3 showing higher basal and lipopolysaccharide (LPS)-stimulated activity, possibly mediated by an adjacent LPS-related response element. While reporter gene activity alone is insufficient to establish clear evidence for in vivo activity, high activity was strongly associated with resistance and low activity with susceptibility to TB (130).
ROLE OF INNATE IMMUNE GENE POLYMORPHISMS IN SUSCEPTIBILITY TO INFECTION AND DISEASE PROGRESSION
In the immunocompetent human host, the outcome of a primary infection is typically asymptomatic, i.e., the primary infection (during which time innate immune genes are critical) is silent (subclinical), and therefore, it is difficult to ascertain the time of primary infection. Following primary infection (considered a 2- to 12-week process), M. tuberculosis can either remain latent or, on occasion, reactivate to cause clinical disease, when adaptive immune genes play a major role. As a result, most studies have tested associations of innate immune genes with clinical TB, not with primary infection. In addition, there are few studies that address the question of how innate immune genes participate in the progression of primary infection to the clinical disease. Stein et al. (216) showed in a linkage and association study that IL-10 and TNFR (along with IFNGR1) are implicated in the progression to active TB disease. In other instances, polymorphisms in innate immune genes, such as INT4 and D543N of SLC11A1 (271) and R753Q of TLR2 (55), were found to be involved in altering the progression of M. tuberculosis infection to active TB. Further evidence for the potential contribution of innate immune genes in clinical TB stems from reported interactions of the innate immune genes NOS2A and TLR4 with the adaptive immunity-related gene IFNGR1 (250) or NRAMP1 with IFNGR1 (62) in influencing susceptibility to TB. Taken together, the results overwhelmingly support a strong genetic component in infectivity, disease progression, and the clinical manifestation of TB, while the relative contribution of each gene in these processes needs to be clarified. Moreover, the evidence suggests gene-gene interactions (epistasis) as an important component of genetic influence in TB, with many of the candidate genes interacting in pathways or physically at the protein level. Future progress will depend on resolving such interactions.
CAUSES OF VARIABILITY IN GENETIC ASSOCIATION WITH TB
Table 1 clearly illustrates a high degree of variability and discrepancy between the association of innate immune genes and TB from study to study, region to region, and population to population. Some polymorphisms have been associated with susceptibility or resistance to TB with inconsistent or even opposing findings, depending on the study. We can propose several hypotheses for these inconsistencies. First, a portion of the reported associations is spurious; this is likely to occur even if the significance level was adjusted for testing of multiple hypotheses, in view of the many association studies performed with a few select polymorphisms detected some time ago. Many more large-scale studies are needed to sort out false positives and negatives. Second, multiple variants may have emerged independently in the same gene during evolution to enhance TB susceptibility or resistance, a likely scenario in the presence of positive selection pressure. Third, differences in ethnic and geographic characteristics can account for multiple genetic variants in these genes associated with TB, owing to the prevalence of genetically distinct M. tuberculosis strains and different allele frequencies in human populations. Fourth, the likely influence of epistasis emphasizes the genetic background against which selectable genes acquire favorable mutations. Today, all of these concepts are readily amenable to experimental scrutiny, but scant knowledge is available as yet. Here, we will focus on two major reasons, evolutionary selection pressure and study design, that can account for inconsistent results from linkage and association studies.
Evolutionary selection pressure.
The accumulation of frequent genetic variants detected in TB association studies is probably a result of strong evolutionary selection favoring resistance to TB. For example, the SLC11A1 locus appears to have been subjected to evolutionary selection pressure over a long time span (30, 199). The observation that Europeans have greater resistance to TB than populations of sub-Saharan African descent has been attributed to the longer time period that Europeans have been exposed to M. tuberculosis compared to Africans (66). Similarly, a study of a nursing home in the United States (214) showed that individuals of African descent were infected with M. tuberculosis twice as often as individuals of European descent in the same environment. The inverse relationship between autoimmune disorders and TB infectivity mentioned earlier provides an additional cue and points to balancing selection whereby heterozygous carriers might be protected while homozygous carriers could experience adverse effects unrelated to TB. Taken together, the broad spectrum of evidence demonstrates the pervasive effect of evolutionary selection pressure on TB susceptibility and disease resistance. In this review, we have summarized the many innate immune genes playing a critical role in TB, some of which already have documented signatures endorsing positive selection pressure. With large-scale human sequence data now available, it is imperative that each of these genes be tested for its evolutionary history, either neutral (random genetic drift or bottleneck selection) or under selection pressure. The latter is revealed by statistical methods, estimating the likelihood that a given variant and its associated haplotype have accumulated at a faster pace than expected from neutral processes. Where such evidence is detected, those genes should become the focus of future study. Moreover, the same genes need to be tested for epistatic interactions that can reveal hidden penetrance as a result of different ethnic backgrounds.
Study design.
The quality of the study design has a great impact on the final results of association and linkage studies (215). Important factors include population sample size, diagnostic criteria, and the phenotype definition of TB. The availability of large populations will improve the power to yield statistically significant data. In many cases, nonreproducibility of genetic association results may have been due to false-positive or false-negative associations caused by low sample numbers, a particular concern when multiple investigators study the same few SNPs but adjust their significance values only to their own study design. Population stratification poses a particular challenge in mixed populations where any given SNP can travel fortuitously with an ethnic subgroup more susceptible to TB—all will show significant associations. Use of Ancestry Informative Markers (AIMs) should be mandatory to minimize such problems while alternative solutions are being developed. Genome-wide association studies (GWAS) compound the problem of multiple-hypothesis testing and therefore require a much larger cohort, and yet, GWAS is similarly sensitive to stratification while providing data elements that can be used to minimize the problem. None of these study designs have systematically addressed the issue of epistasis and the selection of evolutionary target genes, a concept that could resolve some of the intractable problems encountered thus far. Other factors influencing study design are population substructures with various degrees of linkage disequilibrium, a main criterion for selecting marker SNPs, and differences in M. tuberculosis strains within a population. Significant advances are further impeded by a common absence of definitive information on what the causative variants are and their underlying biological consequences—the selection of surrogate SNP markers introduces much variability that can be avoided. Clarifying the underlying mechanism should be of high priority; we think that epistatic interaction in particular cannot be readily studied without selecting the causative variants, a topic discussed in more detail below.
Diagnostic criteria or differences in methods used to diagnose TB lead to various phenotype definitions used to classify disease severity, which results in potential misclassification of TB disease status (whether latent or active). For example, in case-control association studies, “controls” could be latently infected with M. tuberculosis if not properly diagnosed, generating significant errors in the evaluation of disease association with candidate genes. As pointed out by Moller et al. (146), the evaluation of an association study of TB is “exquisitely sensitive to phenotype definition.” Therefore, the phenotype definition of TB as a disease should be unambiguous to achieve reliable data.
FACTORS THAT INFLUENCE THE FUNCTIONS OF INNATE IMMUNE GENE POLYMORPHISMS
Gene-gene interaction.
TB is polygenic with respect to host genetics (146). Gene-gene interactions play an important role in an individual's susceptibility to develop a disease, but gene-gene interactions in infectious diseases such as TB have been poorly studied until recently. Multilocus analyses have identified significant interactions between SNPs in NOS2A and those in TLR4 and IFNGR1 in promoting TB susceptibility (250). These findings are particularly interesting because the SNPs in TLR4 and IFNGR1 alone did not show significant effects and would not have been identified as associated with TB without the interaction with NOS2A. In a similar way, gene-gene interactions between variants of SLCA11A and TLR2 in African-Americans (249), SLCA11A1 and IFNGR1 in South Africans (62), and TNF and IL-10 in Tunisians (27) yielded significant associations with TB. Those multilocus and multigene approaches can identify critical variants that strongly influence the incidence of TB when combined with other gene variants, while alone they would not meet the criteria for statistical significance. However, even among only the innate immune genes, there are a large number of possible combinations, exacerbating the potential for attaining false-positive results. Rigorous statistical correction for multiple-hypothesis testing is essential (but may invalidate many findings by forcing too much stringency), while the use of validated causative variants in genes undergoing positive selection would greatly facilitate the search for the critical gene-gene interactions.
M. tuberculosis genotype and/or phenotype variation.
Interactions between genotypes of the human host and those of a particular bacterial strain have been proposed to determine the onset and the progression of disease (146). A study of Vietnamese subjects detected an association between the C allele of TLR2 597T/C and TB caused by East Asian/Beijing genotype M. tuberculosis strains (41). Genetic variations in the 3′-UTR and the D543N SNP of SLC11A1 (NRAMP1) were significantly associated with susceptibility to infection by M. tuberculosis Beijing strains in Indonesian TB patients (241) and with the incidence of MDR-TB cases in Japan (223), which suggests an influence of bacterial genotype/phenotype on host innate immunity. To what extent these same variants also interact with other M. tuberculosis strains remains to be tested. Because of ambiguities in such clinical association studies, the question of strain-specific human resistance variants has yet to be resolved, but functional genomics of both the host and the infectious agent are now feasible and need to be performed.
HIV coinfection and other nongenetic factors.
The relationship between TB incidence and HIV coinfection is well established (47). A candidate gene association study in Uganda has reported an interaction between the TNFR gene and HIV in TB patients (216), which represents an example of gene-gene (epistasis) or gene-environment interaction. A homozygous genotype (YY) at the −221 codon of the MBL gene, associated with high MBL levels, conveyed susceptibility to TB in HIV-infected patients in South India (3).The same authors identified an association of the VDR regulatory haplotype b-A-T with protection against HIV infection and of the haplotype B-A-t with susceptibility to the development of TB in HIV patients (2), emphasizing HIV coinfection as a factor for polymorphism-mediated TB susceptibility. Since many studies have excluded HIV-positive cases, this area of study remains relatively unexplored. Other nongenetic factors influencing the functions of gene polymorphisms include diabetes, malnutrition, aging, socioeconomic conditions, environmental factors, etc. In an ambitious study by van der Eijk et al. (242), environmental factors were shown to contribute more to the development of TB than hereditary factors. However, such assertions are based on the assumption that environment and genetics are additive to account for 100% of the trait, while one needs to consider dynamic interactions where each factor is a main player, each potentially contributing more than 50%.
Epigenetic factors.
Recent progress in human genome research indicates that epigenetic factors modulate disease risk and influence the impact of genetic variation in disease association. Epigenetic factors include DNA methylation, histone acetylation, and mediators such as microRNAs (miRNAs), etc. DNA methylation and histone acetylation regulate the transcription rate and/or tissue-specific expression of genes without altering the DNA sequence. MicroRNAs are short-nucleotide RNAs which play a role in posttranscriptional regulation of gene expression. The first report of a polymorphism in miRNA in relation to TB pathogenesis was recently published (120), where an SNP in miR-146a (rs2910194G/C), which regulates the TLR signaling pathway, was shown to be associated with an increased risk of pulmonary TB in a Chinese population. This SNP has been proposed to act by altering miRNA target selection and/or its expression, resulting in functional and/or phenotypic changes. More studies are needed to demonstrate epigenetic regulation of innate immune genes via miRNAs that can be linked to TB. For example, miR-125b is highly expressed in response to M. tuberculosis infection and results in reduced TNF production by human macrophages (180); therefore, the genetics of miR-125b should be studied for the presence of any polymorphisms in miR-125b target sites. A recent report on VDR polymorphisms (6) has revealed the interaction between epigenetic processes and genetic variants, linking CpG island methylation at the 3′ end of the VDR gene to its expression, an effect considered to be modulated by a TaqI allele. These epigenetic changes appeared to be ethnicity specific, suggesting an interesting case of gene-environment-epigenetic interactions.
CURRENT CHALLENGES AND POTENTIAL SOLUTIONS
Strong evidence supports a critical role for genetic factors in susceptibility and resistance to TB, involving multiple genes of the innate immune system. For a number of these genes, evolutionary selection of variants can be demonstrated, presumably a result of pervasive pathogen-host interactions. Relatively unbiased approaches, such as GWAS (158), should continue to reveal additional candidate genes. One recent TB GWAS report (229) did not support previous candidate gene-based findings, although it identified a novel association locus (rs4331426) at chromosome 18q11.2 and was suggested to represent proof-of-principle for the utility of GWAS in understanding underlying molecular mechanisms of the disease. However, an integrated understanding of the concerted genetic factors underlying an individual's susceptibility to TB remains elusive. We can look forward to significant progress with the combined use of large-scale technologies that enable full sequencing of genomes and transcriptomes and functional genomics approaches (153, 158, 171, 246), leading to a systems biology analysis of host-pathogen interaction dynamics (47). In particular, we believe that further progress is needed with new approaches in the following areas.
(i) With increasing availability of human genomic sequences, it has become feasible to construct phylogenetic trees and overlay these onto TB traits such as infectivity and resistance, taking into account population dynamics and geographic distributions of host populations and M. tuberculosis strains (15, 38, 46, 170, 254, 268). Such an approach might resolve the question of which pathways are under evolutionary constraint, beyond an analysis of single genes, and allow for the integration of human and M. tuberculosis evolutionary paths.
(ii) A majority of the clinical association results have not been validated by molecular studies (Table 1) to reveal an underlying mechanism that could buttress the clinical results. The whole field is built on surrogate markers, with few exceptions. It is unlikely that the field will progress meaningfully unless the major relevant variants with both associated clinical evidence and underlying biological mechanism have been identified. The determination of functional SNPs and biochemical phenotypes (e.g., differential expression and function of innate immune molecules like receptors, cytokines, etc., by the host) is necessary. A novel molecular technique called “allelic expression imbalance (AEI),” utilized with great success in one of our laboratories, can be used to detect functional SNPs in heterozygous carriers (255, 274). AEI is used as a host phenotype to scan the gene locus surrounding the selected reference SNP to detect functional variant(s). Deep-sequencing technology can be used to determine both AEI and functional SNPs on a large scale.
(iii) Genetic variants under evolutionary selection often have accumulated to relatively high frequencies, e.g., in NRAMP1 and TLR2. High frequencies of functionally relevant variants could represent a path toward a complete allele sweep, or it might show a signature of balancing evolution, wherein a polymorphism conveys protection against TB but homozygous carriers have reduced reproductive fitness or are at high risk of other diseases (1, 143). In the latter case, allele frequencies are limited to a maximum level balancing negative and positive fitness attributes. A better understanding of these relationships is germane to the management of TB disease.
(iv) It is important to understand how genetic variants in multiple genes interact with each other. For example, TNF induces the expression of multiple genes in the innate immune response (259), but a clear understanding of the epistatic interactions among these genes is lacking. Results from GWAS may yield clues to relevant candidate genes, but testing gene-gene interactions typically fails because of the exponentially expanding statistical problem of multiple-hypothesis testing, unless the underlying biological mechanisms are well understood. A systems biology approach, as outlined above, will assist in identifying two or more genes where validated polymorphisms are likely to potentiate or mitigate each other's effects. Few examples of such a systematic approach are available to date in any area, let alone in TB research. Logistic regression can be used as a preferable statistical method, as has been used in gene-gene interactions in TB (62) and other diseases (52).
(v) We note that many of the polymorphisms associated with TB represent regulatory variants rather than nonsynonymous SNPs that alter the amino acid sequence (e.g., TNF, NRAMP1, and TLR2) (1, 18, 265). It is perhaps intuitively obvious that regulation of gene expression could be a primary target of evolutionary pressure, since such changes are gradual and account for the balance that needs to be struck between, for example, immunity against TB and autoimmune disorders (1). In addition, regulatory polymorphisms are context dependent, namely, they can be active in a target cell (e.g., macrophage) and not in other cells or they can be selectively responsive to external stimuli. Again, few studies focus on these causative relationships that could shed light on a role in TB pathogenesis. For example, hypoxia-inducible factor 1 (HIF1) was shown to regulate NRAMP1 expression levels by interacting with a microsatellite repeat region in the promoter of NRAMP1, known to be under evolutionary selection pressure (18). This critical insight provides a window into the cellular pathways potentially operative under conditions of infection. Much more work is needed along this line of investigation.
(vi) Some compounding factors, such as population substructure and linkage disequilibrium differences among populations, can complicate the results of candidate gene association studies. Dense SNP mapping, particularly in the case of African descent, is needed in order to capture linkage disequilibrium in the study population (215).
(vii) It is critical to integrate host gene-gene interactions with the genetic characteristics of M. tuberculosis strains and with environmental factors. If one considers each of these factors in concert with the others, one will be able to discern ethnic, cultural, and geographic differences that can inform health policies on a global scale. Certainly the tools are at hand for integration.
(viii) Finally, understanding the genetic and epigenetic underpinnings of TB will enable the development of new tools for the diagnosis and control of TB. For example, detection of functional polymorphisms will provide next-generation genetic biomarkers that can be used for predicting the risk of infection and assessing TB risk in M. tuberculosis-infected individuals. Such data can inform health policy decisions and preventive measures. A better understanding of the genetic factors determining disease progression can also lead to the development of novel TB therapies (e.g., knowledge of an SNP that causes a nonfunctional protein and protection from TB can be used to express the target gene in a model system for screening inhibitor compounds).
In summary, the stage has been set to understand the genetics and evolution of TB with a view of both the pathogen and the host, the latter being the focus of this review in the context of innate immunity. It is likely that a number of critical genetic factors will converge to yield a comprehensive and dynamic representation of TB pathogenesis, with sufficient insight to successfully tackle the global problems caused by this age-old scourge of humankind.
Biographies

Abul Azad started his research career as a microbiologist in mycobacterial genetics and subsequently moved to studies of mycobacterial pathogenesis. The knowledge gained in tuberculosis pathogenesis studies relevant to humans sparked a more recent interest in the study of human population genetics. He developed an intense interest in the study of genetic variations in the human genome and their relationship to the susceptibility and resistance of the human host to tuberculosis. He has been in this area of investigation for about 6 years, and he is currently an adjunct assistant professor in the Department of Microbial Infection and Immunity.

Wolfgang Sadee received a doctorate degree in Pharmaceutical Chemistry from the FU Berlin in 1968 and served on the pharmacy faculties of USC and UCSF until 2002. He is now Felts Mercer Professor of Medicine and Pharmacology, Chair, Department of Pharmacology, and Director, Program in Pharmacogenomics, with appointments in Psychiatry, Pharmacy, and Public Health, the Davis Heart & Lung Research Institute, and OSU Comprehensive Cancer Center. Dr. Sadee's research focuses on pharmacogenomics, and he participates in the NIH Pharmacogenomics Research Network. Specific areas of interest include expression genetics and human evolution as important drivers of human phenotypic variability, including disease risk, genetic factors in infections, and drug response. Based on his areas of expertise, he formed a partnership with the Schlesinger research team approximately 5 years ago to study the importance of frequent polymorphisms in genes of the innate immune system that are relevant to tuberculosis and other infectious diseases.

Larry Schlesinger is the Saslaw Professor of Medicine, Chair, Department of Microbial Infection and Immunity, and founding Director, OSU Center for Microbial Interface Biology (cmib.osu.edu). He has been studying pathogenic mechanisms for tuberculosis and diseases due to intracellular pathogens that subvert lung innate immune mechanisms for more than 20 years, focusing on pathogen-human phagocyte interactions. He has made major contributions related to phagocytosis and trafficking, mycobacterial glycolipids and host adaptation, and surfactant collectins in microbial immune responses. He has devoted his career to understanding the human immune response to pathogens. During his career, he has gained a keen appreciation for the marked variability in immune responses seen between donors in experiments. Thus, he has become interested in the application of novel approaches to tackle the underexplored area of frequent polymorphisms in innate immunity genes among healthy individuals and formed a partnership with the Sadee research team approximately 5 years ago.
Footnotes
Published ahead of print 23 July 2012
REFERENCES
- 1. Aguillon JC, et al. 2006. Could single-nucleotide polymorphisms (SNPs) affecting the tumour necrosis factor promoter be considered as part of rheumatoid arthritis evolution? Immunobiology 211:75–84 [DOI] [PubMed] [Google Scholar]
- 2. Alagarasu K, Selvaraj P, Swaminathan S, Narendran G, Narayanan PR. 2009. 5′ Regulatory and 3′ untranslated region polymorphisms of vitamin D receptor gene in south Indian HIV and HIV-TB patients. J. Clin. Immunol. 29:196–204 [DOI] [PubMed] [Google Scholar]
- 3. Alagarasu K, et al. 2007. Mannose binding lectin gene variants and susceptibility to tuberculosis in HIV-1 infected patients of South India. Tuberculosis (Edinb.) 87:535–543 [DOI] [PubMed] [Google Scholar]
- 4. Alter A, et al. 2010. Genetic and functional analysis of common MRC1 exon 7 polymorphisms in leprosy susceptibility. Hum. Genet. 127:337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amirzargar AA, et al. 2006. Cytokine single nucleotide polymorphisms in Iranian patients with pulmonary tuberculosis. Eur. Cytokine Netw. 17:84–89 [PubMed] [Google Scholar]
- 6. Andraos C, Koorsen G, Knight JC, Bornman L. 2011. Vitamin D receptor gene methylation is associated with ethnicity, tuberculosis, and TaqI polymorphism. Hum. Immunol. 72:262–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ansari A, et al. 2009. Cytokine gene polymorphisms across tuberculosis clinical spectrum in Pakistani patients. PLoS One 4:e4778 doi:10.1371/journal.pone.0004778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ates O, et al. 2009. NRAMP1 (SLC11A1) gene polymorphisms that correlate with autoimmune versus infectious disease susceptibility in tuberculosis and rheumatoid arthritis. Int. J. Immunogenet. 36:15–19 [DOI] [PubMed] [Google Scholar]
- 9. Ates O, Musellim B, Ongen G, Topal-Sarikaya A. 2008. Interleukin-10 and tumor necrosis factor-alpha gene polymorphisms in tuberculosis. J. Clin. Immunol. 28:232–236 [DOI] [PubMed] [Google Scholar]
- 10. Austin CM, Ma X, Graviss EA. 2008. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J. Infect. Dis. 197:1713–1716 [DOI] [PubMed] [Google Scholar]
- 11. Awomoyi AA, et al. 2005. Polymorphism in IL1B: IL1B-511 association with tuberculosis and decreased lipopolysaccharide-induced IL-1beta in IFN-gamma primed ex-vivo whole blood assay. J. Endotoxin Res. 11:281–286 [DOI] [PubMed] [Google Scholar]
- 12. Awomoyi AA, et al. 2002. Interleukin-10, polymorphism in SLC11A1 (formerly NRAMP1), and susceptibility to tuberculosis. J. Infect. Dis. 186:1808–1814 [DOI] [PubMed] [Google Scholar]
- 13. Azad AK, Torrelles JB, Schlesinger LS. 2008. Mutation in the DC-SIGN cytoplasmic triacidic cluster motif markedly attenuates receptor activity for phagocytosis and endocytosis of mannose-containing ligands by human myeloid cells. J. Leukoc. Biol. 84:1594–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker AR, et al. 2011. Genetic susceptibility to tuberculosis associated with cathepsin Z haplotype in a Ugandan household contact study. Hum. Immunol. 72:426–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barnes I, Duda A, Pybus OG, Thomas MG. 2011. Ancient urbanization predicts genetic resistance to tuberculosis. Evolution 65:842–848 [DOI] [PubMed] [Google Scholar]
- 16. Barreiro LB, et al. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 5:e1000562 doi:10.1371/journal.pgen.1000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barreiro LB, et al. 2006. Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med. 3:e20 doi:10.1371/journal.pmed.0030020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bayele HK, et al. 2007. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood 110:3039–3048 [DOI] [PubMed] [Google Scholar]
- 19. Beharka AA, et al. 2002. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J. Immunol. 169:3565–3573 [DOI] [PubMed] [Google Scholar]
- 20. Bekpen C, Xavier RJ, Eichler EE. 2010. Human IRGM gene “to be or not to be.” Semin. Immunopathol. 32:437–444 [DOI] [PubMed] [Google Scholar]
- 21. Bellamy R, et al. 2000. Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc. Natl. Acad. Sci. U. S. A. 97:8005–8009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bellamy R, et al. 1999. Tuberculosis and chronic hepatitis B virus infection in Africans and variation in the vitamin D receptor gene. J. Infect. Dis. 179:721–724 [DOI] [PubMed] [Google Scholar]
- 23. Bellamy R, et al. 1998. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N. Engl. J. Med. 338:640–644 [DOI] [PubMed] [Google Scholar]
- 24. Ben-Ali M, Barbouche MR, Bousnina S, Chabbou A, Dellagi K. 2004. Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin. Diagn. Lab Immunol. 11:625–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ben-Ali M, et al. 2007. Promoter and neck region length variation of DC-SIGN is not associated with susceptibility to tuberculosis in Tunisian patients. Hum. Immunol. 68:908–912 [DOI] [PubMed] [Google Scholar]
- 26. Ben-Selma W, et al. 2011. Polymorphisms in the RANTES gene increase susceptibility to active tuberculosis in Tunisia. DNA Cell Biol. 30:789–800 [DOI] [PubMed] [Google Scholar]
- 27. Ben-Selma W, Harizi H, Boukadida J. 2011. Association of TNF-alpha and IL-10 polymorphisms with tuberculosis in Tunisian populations. Microbes Infect. 13:837–843 [DOI] [PubMed] [Google Scholar]
- 28. Ben-Selma W, Harizi H, Boukadida J. 2011. MCP-1-2518 A/G functional polymorphism is associated with increased susceptibility to active pulmonary tuberculosis in Tunisian patients. Mol. Biol. Rep. 38:5413–5419 [DOI] [PubMed] [Google Scholar]
- 29. Berrington WR, Hawn TR. 2007. Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol. Rev. 219:167–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackwell JM, et al. 2001. SLC11A1 (formerly NRAMP1) and disease resistance. Cell Microbiol. 3:773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blackwell JM, Jamieson SE, Burgner D. 2009. HLA and infectious diseases. Clin. Microbiol. Rev. 22:370–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boily-Larouche G, Zijenah LS, Mbizvo M, Ward BJ, Roger M. 2007. DC-SIGN and DC-SIGNR genetic diversity among different ethnic populations: potential implications for pathogen recognition and disease susceptibility. Hum. Immunol. 68:523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bornman L, et al. 2004. Vitamin D receptor polymorphisms and susceptibility to tuberculosis in West Africa: a case-control and family study. J. Infect. Dis. 190:1631–1641 [DOI] [PubMed] [Google Scholar]
- 34. Brooks MN, et al. 2011. NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell. Microbiol. 13:402–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brown GD, Gordon S. 2001. Immune recognition. A new receptor for beta-glucans. Nature 413:36–37 [DOI] [PubMed] [Google Scholar]
- 36. Brown GD, et al. 2003. Dectin-1 mediates the biological effects of beta-glucans. J. Exp. Med. 197:1119–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buijtels PC, et al. 2008. Polymorphism in CC-chemokine ligand 2 associated with tuberculosis in Zambia. Int. J. Tuberc. Lung Dis. 12:1485–1488 [PubMed] [Google Scholar]
- 38. Campbell MC, Tishkoff SA. 2008. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet. 9:403–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Capparelli R, et al. 2009. Role played by human mannose-binding lectin polymorphisms in pulmonary tuberculosis. J. Infect. Dis. 199:666–672 [DOI] [PubMed] [Google Scholar]
- 40. Carneiro LA, Magalhaes JG, Tattoli I, Philpott DJ, Travassos LH. 2008. Nod-like proteins in inflammation and disease. J. Pathol. 214:136–148 [DOI] [PubMed] [Google Scholar]
- 41. Caws M, et al. 2008. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 4:e1000034 doi:10.1371/journal.ppat.1000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan ED, Chan J, Schluger NW. 2001. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25:606–612 [DOI] [PubMed] [Google Scholar]
- 43. Chan ED, et al. 2001. Induction of inducible nitric oxide synthase-NO• by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-kB signaling pathways. Infect. Immun. 69:2001–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chensue SW, et al. 1999. Differential expression and cross-regulatory function of RANTES during mycobacterial (type 1) and schistosomal (type 2) antigen-elicited granulomatous inflammation. J. Immunol. 163:165–173 [PubMed] [Google Scholar]
- 45. Chu SF, et al. 2007. Association between RANTES functional polymorphisms and tuberculosis in Hong Kong Chinese. Genes Immun. 8:475–479 [DOI] [PubMed] [Google Scholar]
- 46. Comas I, et al. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Comas I, Gagneux S. 2009. The past and future of tuberculosis research. PLoS Pathog. 5:e1000600 doi:10.1371/journal.ppat.1000600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Comstock GW. 1978. Tuberculosis in twins: a re-analysis of the Prophit survey. Am. Rev. Respir. Dis. 117:621–624 [DOI] [PubMed] [Google Scholar]
- 49. Cooke GS, et al. 2008. Mapping of a novel susceptibility locus suggests a role for MC3R and CTSZ in human tuberculosis. Am. J. Respir. Crit. Care Med. 178:203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cooke GS, et al. 2004. Interleukin-8 polymorphism is not associated with pulmonary tuberculosis in The Gambia. J. Infect. Dis. 189:1545–1546 [DOI] [PubMed] [Google Scholar]
- 51. Cooper AM, Solache A, Khader SA. 2007. Interleukin-12 and tuberculosis: an old story revisited. Curr. Opin. Immunol. 19:441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cordell HJ. 2009. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 10:392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Correa PA, Gomez LM, Cadena J, Anaya JM. 2005. Autoimmunity and tuberculosis. Opposite association with TNF polymorphism. J. Rheumatol. 32:219–224 [PubMed] [Google Scholar]
- 54. Coulombe F, et al. 2009. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J. Exp. Med. 206:1709–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dalgic N, et al. 2011. Arg753Gln polymorphism of the human Toll-like receptor 2 gene from infection to disease in pediatric tuberculosis. Hum. Immunol. 72:440–445 [DOI] [PubMed] [Google Scholar]
- 56. Davila S, et al. 2008. Genetic association and expression studies indicate a role of Toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 4:e1000218 doi:10.1371/journal.pgen.1000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Delgado JC, Baena A, Thim S, Goldfeld AE. 2002. Ethnic-specific genetic associations with pulmonary tuberculosis. J. Infect. Dis. 186:1463–1468 [DOI] [PubMed] [Google Scholar]
- 58. Deng G, et al. 2008. Regulatory polymorphisms in the promoter of CXCL10 gene and disease progression in male hepatitis B virus carriers. Gastroenterology 134:716–726 [DOI] [PubMed] [Google Scholar]
- 59. Denholm JT, McBryde ES, Eisen DP. 2010. Mannose-binding lectin and susceptibility to tuberculosis: a meta-analysis. Clin. Exp. Immunol. 162:84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deretic V. 2011. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240:92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Deretic V, et al. 2009. Autophagy in immunity against mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy. Curr. Top. Microbiol. Immunol. 335:169–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Wit E, van der Merwe L, Van Helden PD, Hoal EG. 2011. Gene-gene interaction between tuberculosis candidate genes in a South African population. Mamm. Genome 22:100–110 [DOI] [PubMed] [Google Scholar]
- 63. Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519–550 [DOI] [PubMed] [Google Scholar]
- 64. Dinarello CA, et al. 1998. Overview of interleukin-18: more than an interferon-gamma inducing factor. J. Leukoc. Biol. 63:658–664 [PubMed] [Google Scholar]
- 65. Dommett RM, Klein N, Turner MW. 2006. Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 68:193–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dubos R, Dubos J. 1952. The white plague: tuberculosis, man and society. Little, Brown & Co., Boston, MA [Google Scholar]
- 67. El Baghdadi J, et al. 2003. Variants of the human NRAMP1 gene and susceptibility to tuberculosis in Morocco. Int. J. Tuberc. Lung Dis. 7:599–602 [PubMed] [Google Scholar]
- 68. El Sahly HM, Reich RA, Dou SJ, Musser JM, Graviss EA. 2004. The effect of mannose binding lectin gene polymorphisms on susceptibility to tuberculosis in different ethnic groups. Scand. J. Infect. Dis. 36:106–108 [DOI] [PubMed] [Google Scholar]
- 69. Fairbairn IP, Stober CB, Kumararatne DS, Lammas DA. 2001. ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X(7)-dependent process inducing bacterial death by phagosome-lysosome fusion. J. Immunol. 167:3300–3307 [DOI] [PubMed] [Google Scholar]
- 70. Fenton MJ, Riley LW, Schlesinger LS. 2005. Receptor-mediated recognition of Mycobacterium tuberculosis by host cells, p 405–426 In Cole ST, Eisenach KD, McMurray DN, Jacobs WR., Jr. (ed), Tuberculosis and the tubercle bacillus. ASM Press, Washington, DC [Google Scholar]
- 71. Ferguson JS, et al. 2006. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect. Immun. 74:7005–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. 1999. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163:312–321 [PubMed] [Google Scholar]
- 73. Fernando SL, et al. 2007. A polymorphism in the P2X7 gene increases susceptibility to extrapulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 175:360–366 [DOI] [PubMed] [Google Scholar]
- 74. Ferwerda B, Kibiki GS, Netea MG, Dolmans WM, van der Ven AJ. 2007. The toll-like receptor 4 Asp299Gly variant and tuberculosis susceptibility in HIV-infected patients in Tanzania. AIDS 21:1375–1377 [DOI] [PubMed] [Google Scholar]
- 75. Figdor CG, Van Kooyk Y, Adema GJ. 2002. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2:77–84 [DOI] [PubMed] [Google Scholar]
- 76. Fitness J, et al. 2004. Large-scale candidate gene study of leprosy susceptibility in the Karonga district of northern Malawi. Am. J. Trop. Med. Hyg. 71:330–340 [PubMed] [Google Scholar]
- 77. Flesch IE, Hess JH, Kaufmann SH. 1994. NADPH diaphorase staining suggests a transient and localized contribution of nitric oxide to host defence against an intracellular pathogen in situ. Int. Immunol. 6:1751–1757 [DOI] [PubMed] [Google Scholar]
- 78. Flores-Villanueva PO, et al. 2005. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J. Exp. Med. 202:1649–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Floros J, et al. 2000. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J. Infect. Dis. 182:1473–1478 [DOI] [PubMed] [Google Scholar]
- 80. Flynn JL, Chan J. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129 [DOI] [PubMed] [Google Scholar]
- 81. Flynn JL, et al. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2:561–572 [DOI] [PubMed] [Google Scholar]
- 82. Franchi L, Warner N, Viani K, Nunez G. 2009. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227:106–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Freidin MB, Rudko AA, Kolokolova OV, Strelis AK, Puzyrev VP. 2006. Association between the 1188 A/C polymorphism in the human IL12B gene and Th1-mediated infectious diseases. Int. J. Immunogenet. 33:231–232 [DOI] [PubMed] [Google Scholar]
- 84. Fulton SA, Johnsen JM, Wolf SF, Sieburth DS, Boom WH. 1996. Interleukin-12 production by human monocytes infected with Mycobacterium tuberculosis: role of phagocytosis. Infect. Immun. 64:2523–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gao L, Tao Y, Zhang L, Jin Q. 2010. Vitamin D receptor genetic polymorphisms and tuberculosis: updated systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 14:15–23 [PubMed] [Google Scholar]
- 86. Geijtenbeek TB, et al. 2003. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197:7–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Glaziou P, Floyd K, Raviglione M. 2009. Global burden and epidemiology of tuberculosis. Clin. Chest Med. 30:621–636 [DOI] [PubMed] [Google Scholar]
- 88. Gomez LM, et al. 2006. Analysis of DC-SIGN (CD209) functional variants in patients with tuberculosis. Hum. Immunol. 67:808–811 [DOI] [PubMed] [Google Scholar]
- 89. Gomez LM, et al. 2007. A polymorphism in the inducible nitric oxide synthase gene is associated with tuberculosis. Tuberculosis (Edinb.) 87:288–294 [DOI] [PubMed] [Google Scholar]
- 90. Gomez LM, et al. 2006. Analysis of IL1B, TAP1, TAP2 and IKBL polymorphisms on susceptibility to tuberculosis. Tissue Antigens 67:290–296 [DOI] [PubMed] [Google Scholar]
- 91. Gruenheid S, Pinner E, Desjardins M, Gros P. 1997. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 185:717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Haataja R, Hallman M. 2002. Surfactant proteins as genetic determinants of multifactorial pulmonary diseases. Ann. Med. 34:324–333 [DOI] [PubMed] [Google Scholar]
- 93. Han M, et al. 2011. Relationship between single nucleotide polymorphism of interleukin-18 and susceptibility to pulmonary tuberculosis in the Chinese Han population. Microbiol. Immunol. 55:388–393 [DOI] [PubMed] [Google Scholar]
- 94. Harishankar M, Selvaraj P, Rajeswari DN, Anand SP, Narayanan PR. 2007. Promoter polymorphism of IL-18 gene in pulmonary tuberculosis in South Indian population. Int. J. Immunogenet. 34:317–320 [DOI] [PubMed] [Google Scholar]
- 95. Hawn TR, et al. 2006. A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J. Infect. Dis. 194:1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Heldwein KA, Fenton MJ. 2002. The role of Toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 4:937–944 [DOI] [PubMed] [Google Scholar]
- 97. Henao MI, Montes C, Paris SC, Garcia LF. 2006. Cytokine gene polymorphisms in Colombian patients with different clinical presentations of tuberculosis. Tuberculosis (Edinb.) 86:11–19 [DOI] [PubMed] [Google Scholar]
- 98. Henning LN, et al. 2008. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 180:7847–7858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hoal EG, et al. 2004. SLC11A1 (NRAMP1) but not SLC11A2 (NRAMP2) polymorphisms are associated with susceptibility to tuberculosis in a high-incidence community in South Africa. Int. J. Tuberc. Lung Dis. 8:1464–1471 [PubMed] [Google Scholar]
- 100. Inohara N, Nunez G. 2003. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3:371–382 [DOI] [PubMed] [Google Scholar]
- 101. Jabado N, et al. 2000. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J. Exp. Med. 192:1237–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jamieson SE, et al. 2004. Evidence for a cluster of genes on chromosome 17q11-q21 controlling susceptibility to tuberculosis and leprosy in Brazilians. Genes Immun. 5:46–57 [DOI] [PubMed] [Google Scholar]
- 103. Jin J, et al. 2009. SLC11A1 (formerly NRAMP1) gene polymorphisms associated with pediatric tuberculosis in China. Clin. Infect. Dis. 48:733–738 [DOI] [PubMed] [Google Scholar]
- 104. Jo EK. 2008. Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr. Opin. Infect. Dis. 21:279–286 [DOI] [PubMed] [Google Scholar]
- 105. Jo EK, Yang CS, Choi CH, Harding CV. 2007. Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from Toll-like receptors. Cell Microbiol. 9:1087–1098 [DOI] [PubMed] [Google Scholar]
- 106. Kang BK, et al. 2005. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202:987–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kang BK, Schlesinger LS. 1998. Characterization of mannose receptor-dependent phagocytosis mediated by Mycobacterium tuberculosis lipoarabinomannan. Infect. Immun. 66:2769–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kang YA, et al. 2009. Association between the -159C/T CD14 gene polymorphism and tuberculosis in a Korean population. FEMS Immunol. Med. Microbiol. 57:229–235 [DOI] [PubMed] [Google Scholar]
- 109. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 110. Khor CC, et al. 2007. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 39:523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kobayashi KS, et al. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307:731–734 [DOI] [PubMed] [Google Scholar]
- 112. Kusner DJ, Adams J. 2000. ATP-induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J. Immunol. 164:379–388 [DOI] [PubMed] [Google Scholar]
- 113. Kusuhara K, Yamamoto K, Okada K, Mizuno Y, Hara T. 2007. Association of IL12RB1 polymorphisms with susceptibility to and severity of tuberculosis in Japanese: a gene-based association analysis of 21 candidate genes. Int. J. Immunogenet. 34:35–44 [DOI] [PubMed] [Google Scholar]
- 114. Ladel CH, et al. 1997. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 65:4843–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Larcombe LA, et al. 2008. Functional gene polymorphisms in Canadian aboriginal populations with high rates of tuberculosis. J. Infect. Dis. 198:1175–1179 [DOI] [PubMed] [Google Scholar]
- 116. Lee HW, et al. 2005. Lack of an association between interleukin-12 receptor beta1 polymorphisms and tuberculosis in Koreans. Respiration 72:365–368 [DOI] [PubMed] [Google Scholar]
- 117. Lee SH, et al. 2011. Association between the interleukin-18 promoter polymorphism and pulmonary tuberculosis in a Korean population. Int. J. Tuberc. Lung Dis. 15:1246–1251 [DOI] [PubMed] [Google Scholar]
- 118. Leung KH, et al. 2007. Sex- and age-dependent association of SLC11A1 polymorphisms with tuberculosis in Chinese: a case control study. BMC Infect. Dis. 7:19 doi:10.1186/1471-2334-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li CM, et al. 2002. Association of a polymorphism in the P2X7 gene with tuberculosis in a Gambian population. J. Infect. Dis. 186:1458–1462 [DOI] [PubMed] [Google Scholar]
- 120. Li D, et al. 2011. Genetic study of two single nucleotide polymorphisms within corresponding microRNAs and susceptibility to tuberculosis in a Chinese Tibetan and Han population. Hum. Immunol. 72:598–602 [DOI] [PubMed] [Google Scholar]
- 121. Li HT, Zhang TT, Zhou YQ, Huang QH, Huang J. 2006. SLC11A1 (formerly NRAMP1) gene polymorphisms and tuberculosis susceptibility: a meta-analysis. Int. J. Tuberc. Lung Dis. 10:3–12 [PubMed] [Google Scholar]
- 122. Li X, et al. 2011. SLC11A1 (NRAMP1) polymorphisms and tuberculosis susceptibility: updated systematic review and meta-analysis. PLoS One 6:e15831 doi:10.1371/journal.pone.0015831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lin Y, Gong J, Zhang M, Xue W, Barnes PF. 1998. Production of monocyte chemoattractant protein 1 in tuberculosis patients. Infect. Immun. 66:2319–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu PT, et al. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773 [DOI] [PubMed] [Google Scholar]
- 125. Liu W, et al. 2004. VDR and NRAMP1 gene polymorphisms in susceptibility to pulmonary tuberculosis among the Chinese Han population: a case-control study. Int. J. Tuberc. Lung Dis. 8:428–434 [PubMed] [Google Scholar]
- 126. Liu W, et al. 2006. Sequence variations in the MBL gene and their relationship to pulmonary tuberculosis in the Chinese Han population. Int. J. Tuberc. Lung Dis. 10:1098–1103 [PubMed] [Google Scholar]
- 127. Lombard Z, Dalton DL, Venter PA, Williams RC, Bornman L. 2006. Association of HLA-DR, -DQ, and vitamin D receptor alleles and haplotypes with tuberculosis in the Venda of South Africa. Hum. Immunol. 67:643–654 [DOI] [PubMed] [Google Scholar]
- 128. Lopez-Maderuelo D, et al. 2003. Interferon-gamma and interleukin-10 gene polymorphisms in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 167:970–975 [DOI] [PubMed] [Google Scholar]
- 129. Lu B, et al. 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 187:601–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ma X, et al. 2002. 5′ dinucleotide repeat polymorphism of NRAMP1 and susceptibility to tuberculosis among Caucasian patients in Houston, Texas. Int. J. Tuberc. Lung Dis. 6:818–823 [PubMed] [Google Scholar]
- 131. Ma X, et al. 2007. Full-exon resequencing reveals Toll-like receptor variants contribute to human susceptibility to tuberculosis disease. PLoS One 2:e1318 doi:10.1371/journal.pone.0001318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ma X, et al. 2003. Association between interleukin-8 gene alleles and human susceptibility to tuberculosis disease. J. Infect. Dis. 188:349–355 [DOI] [PubMed] [Google Scholar]
- 133. MacMicking JD, et al. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Madan T, Saxena S, Murthy KJ, Muralidhar K, Sarma PU. 2002. Association of polymorphisms in the collagen region of human SP-A1 and SP-A2 genes with pulmonary tuberculosis in Indian population. Clin. Chem. Lab. Med. 40:1002–1008 [DOI] [PubMed] [Google Scholar]
- 135. Mahasirimongkol S, et al. 2012. Genome-wide association studies of tuberculosis in Asians identify distinct at-risk locus for young tuberculosis. J. Hum. Genet. 57:363–367 [DOI] [PubMed] [Google Scholar]
- 136. Mahasirimongkol S, et al. 2009. Genome-wide SNP-based linkage analysis of tuberculosis in Thais. Genes Immun. 10:77–83 [DOI] [PubMed] [Google Scholar]
- 137. Malik S, et al. 2005. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc. Natl. Acad. Sci. U. S. A. 102:12183–12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Malik S, et al. 2006. Variants of the SFTPA1 and SFTPA2 genes and susceptibility to tuberculosis in Ethiopia. Hum. Genet. 118:752–759 [DOI] [PubMed] [Google Scholar]
- 139. McGreal EP, Martinez-Pomares L, Gordon S. 2004. Divergent roles for C-type lectins expressed by cells of the innate immune system. Mol. Immunol. 41:1109–1121 [DOI] [PubMed] [Google Scholar]
- 140. Means TK, et al. 1999. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 163:3920–3927 [PubMed] [Google Scholar]
- 141. Merza M, et al. 2009. The NRAMPI, VDR and TNF-alpha gene polymorphisms in Iranian tuberculosis patients: the study on host susceptibility. Braz. J. Infect. Dis. 13:252–256 [DOI] [PubMed] [Google Scholar]
- 142. Miao R, Li J, Sun Z, Xu F, Shen H. 2011. Meta-analysis on the association of TIRAP S180L variant and tuberculosis susceptibility. Tuberculosis (Edinb.) 91:268–272 [DOI] [PubMed] [Google Scholar]
- 143. Miller CL. 2009. The evolution of schizophrenia: a model for selection by infection, with a focus on NAD. Curr. Pharm. Des. 15:100–109 [DOI] [PubMed] [Google Scholar]
- 144. Miller EN, et al. 2004. Genome-wide scans for leprosy and tuberculosis susceptibility genes in Brazilians. Genes Immun. 5:63–67 [DOI] [PubMed] [Google Scholar]
- 145. Mokrousov I, Sapozhnikova N, Narvskaya O. 2008. Mycobacterium tuberculosis co-existence with humans: making an imprint on the macrophage P2X(7) receptor gene? J. Med. Microbiol. 57:581–584 [DOI] [PubMed] [Google Scholar]
- 146. Moller M, de Wit E, Hoal EG. 2010. Past, present and future directions in human genetic susceptibility to tuberculosis. FEMS Immunol. Med. Microbiol. 58:3–26 [DOI] [PubMed] [Google Scholar]
- 147. Moller M, et al. 2010. A functional haplotype in the 3′untranslated region of TNFRSF1B is associated with tuberculosis in two African populations. Am. J. Respir. Crit. Care Med. 181:388–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Moller M, Hoal EG. 2010. Current findings, challenges and novel approaches in human genetic susceptibility to tuberculosis. Tuberculosis (Edinb.) 90:71–83 [DOI] [PubMed] [Google Scholar]
- 149. Moller M, et al. 2007. Host susceptibility to tuberculosis: CARD15 polymorphisms in a South African population. Mol. Cell Probes 21:148–151 [DOI] [PubMed] [Google Scholar]
- 150. Moller M, et al. 2009. Investigation of chromosome 17 candidate genes in susceptibility to TB in a South African population. Tuberculosis (Edinb.) 89:189–194 [DOI] [PubMed] [Google Scholar]
- 151. Morahan G, et al. 2007. Association of variants in the IL12B gene with leprosy and tuberculosis. Tissue Antigens 69(Suppl 1):234–236 [DOI] [PubMed] [Google Scholar]
- 152. Morris GA, et al. 2011. Interleukin 12B (IL12B) genetic variation and pulmonary tuberculosis: a study of cohorts from The Gambia, Guinea-Bissau, United States and Argentina. PLoS One 6:e16656 doi:10.1371/journal.pone.0016656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Mortellaro A, Robinson L, Ricciardi-Castagnoli P. 2009. Spotlight on mycobacteria and dendritic cells: will novel targets to fight tuberculosis emerge? EMBO Mol. Med. 1:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Mosaad YM, Soliman OE, Tawhid ZE, Sherif DM. 2010. Interferon-gamma +874 T/A and interleukin-10-1082 A/G single nucleotide polymorphism in Egyptian children with tuberculosis. Scand. J. Immunol. 72:358–364 [DOI] [PubMed] [Google Scholar]
- 155. Motsinger-Reif AA, et al. 2010. Polymorphisms in IL-1beta, vitamin D receptor Fok1, and Toll-like receptor 2 are associated with extrapulmonary tuberculosis. BMC Med. Genet. 11:37 doi:10.1186/1471-2350-11-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nejentsev S, et al. 2008. Analysis of association of the TIRAP (MAL) S180L variant and tuberculosis in three populations. Nat. Genet. 40:261–262 [DOI] [PubMed] [Google Scholar]
- 157. Newport MJ, et al. 2004. The Toll-like receptor 4 Asp299Gly variant: no influence on LPS responsiveness or susceptibility to pulmonary tuberculosis in The Gambia. Tuberculosis (Edinb.) 84:347–352 [DOI] [PubMed] [Google Scholar]
- 158. Newport MJ, Finan C. 2011. Genome-wide association studies and susceptibility to infectious diseases. Brief. Funct. Genomics 10:98–107 [DOI] [PubMed] [Google Scholar]
- 159. Neyrolles O, Gicquel B, Quintana-Murci L. 2006. Towards a crucial role for DC-SIGN in tuberculosis and beyond. Trends Microbiol. 14:383–387 [DOI] [PubMed] [Google Scholar]
- 160. Nicholson S, et al. 1996. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183:2293–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Nino-Moreno P, et al. 2007. P2X7 and NRAMP1/SLC11 A1 gene polymorphisms in Mexican mestizo patients with pulmonary tuberculosis. Clin. Exp. Immunol. 148:469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ogus AC, et al. 2004. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur. Respir. J. 23:219–223 [DOI] [PubMed] [Google Scholar]
- 163. Oh JH, et al. 2007. Polymorphisms of interleukin-10 and tumour necrosis factor-alpha genes are associated with newly diagnosed and recurrent pulmonary tuberculosis. Respirology 12:594–598 [DOI] [PubMed] [Google Scholar]
- 164. Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. 1998. Regulation of interferon-gamma production by IL-12 and IL-18. Curr. Opin. Immunol. 10:259–264 [DOI] [PubMed] [Google Scholar]
- 165. O'Leary S, O'Sullivan MP, Keane J. 2011. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. 45:172–180 [DOI] [PubMed] [Google Scholar]
- 166. Olesen R, et al. 2007. DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes Immun. 8:456–467 [DOI] [PubMed] [Google Scholar]
- 167. Reference deleted.
- 168. Oral HB, et al. 2006. Interleukin-10 (IL-10) gene polymorphism as a potential host susceptibility factor in tuberculosis. Cytokine 35:143–147 [DOI] [PubMed] [Google Scholar]
- 169. Pacheco AG, Cardoso CC, Moraes MO. 2008. IFNG +874T/A, IL10 -1082G/A and TNF -308G/A polymorphisms in association with tuberculosis susceptibility: a meta-analysis study. Hum. Genet. 123:477–484 [DOI] [PubMed] [Google Scholar]
- 170. Parwati I, van Crevel R, van Soolingen D. 2010. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect. Dis. 10:103–111 [DOI] [PubMed] [Google Scholar]
- 171. Perkins TT, et al. 2009. A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet. 5:e1000569 doi:10.1371/journal.pgen.1000569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Png E, et al. 2012. A genome wide association study of pulmonary tuberculosis susceptibility in Indonesians. BMC Med. Genet. 13:5 doi:10.1186/1471-2350-13-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Prabhu Anand S, Selvaraj P, Jawahar MS, Adhilakshmi AR, Narayanan PR. 2007. Interleukin-12B & interleukin-10 gene polymorphisms in pulmonary tuberculosis. Indian J. Med. Res. 126:135–138 [PubMed] [Google Scholar]
- 174. Prigozy TI, et al. 1997. The mannose receptor delivers lipoglycan antigens to endosomes for presentation to T cells by CD1b molecules. Immunity 6:187–197 [DOI] [PubMed] [Google Scholar]
- 175. Puig-Kroger A, et al. 2004. Regulated expression of the pathogen receptor dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin in THP-1 human leukemic cells, monocytes, and macrophages. J. Biol. Chem. 279:25680–25688 [DOI] [PubMed] [Google Scholar]
- 176. Pulido I, et al. 2010. The TLR4 ASP299GLY polymorphism is a risk factor for active tuberculosis in Caucasian HIV-infected patients. Curr. HIV Res. 8:253–258 [DOI] [PubMed] [Google Scholar]
- 177. Quesniaux V, et al. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6:946–959 [DOI] [PubMed] [Google Scholar]
- 178. Quesniaux VJ, et al. 2004. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 172:4425–4434 [DOI] [PubMed] [Google Scholar]
- 179. Rajaram MV, et al. 2010. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185:929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Rajaram MV, et al. 2011. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc. Natl. Acad. Sci. U. S. A. 108:17408–17413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Remus N, et al. 2004. Association of IL12RB1 polymorphisms with pulmonary tuberculosis in adults in Morocco. J. Infect. Dis. 190:580–587 [DOI] [PubMed] [Google Scholar]
- 182. Riedel DD, Kaufmann SH. 1997. Chemokine secretion by human polymorphonuclear granulocytes after stimulation with Mycobacterium tuberculosis and lipoarabinomannan. Infect. Immun. 65:4620–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Roach DR, et al. 2001. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 193:239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Rosas-Taraco AG, et al. 2007. CD14 C(-159)T polymorphism is a risk factor for development of pulmonary tuberculosis. J. Infect. Dis. 196:1698–1706 [DOI] [PubMed] [Google Scholar]
- 185. Rosenberg PS, Che A, Chen BE. 2006. Multiple hypothesis testing strategies for genetic case-control association studies. Stat. Med. 25:3134–3149 [DOI] [PubMed] [Google Scholar]
- 186. Rothfuchs AG, et al. 2007. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J. Immunol. 179:3463–3471 [DOI] [PubMed] [Google Scholar]
- 187. Ryu S, et al. 2000. 3′UTR polymorphisms in the NRAMP1 gene are associated with susceptibility to tuberculosis in Koreans. Int. J. Tuberc. Lung Dis. 4:577–580 [PubMed] [Google Scholar]
- 188. Sabat R, et al. 2010. Biology of interleukin-10. Cytokine Growth Factor Rev. 21:331–344 [DOI] [PubMed] [Google Scholar]
- 189. Sadek MI, Sada E, Toossi Z, Schwander SK, Rich EA. 1998. Chemokines induced by infection of mononuclear phagocytes with mycobacteria and present in lung alveoli during active pulmonary tuberculosis. Am. J. Respir. Cell Mol. Biol. 19:513–521 [DOI] [PubMed] [Google Scholar]
- 190. Saiga H., Shimada Y., Takeda K. 2011. Innate immune effectors in mycobacterial infection. Clin. Dev. Immunol. 2011:347594 doi:10.1155/2011/347594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Salam N, et al. 2008. Protective immunity to Mycobacterium tuberculosis infection by chemokine and cytokine conditioned CFP-10 differentiated dendritic cells. PLoS One 3:e2869 doi:10.1371/journal.pone.0002869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Sanchez-Castanon M, et al. 2009. Polymorphisms in CCL5 promoter are associated with pulmonary tuberculosis in northern Spain. Int. J. Tuberc. Lung Dis. 13:480–485 [PubMed] [Google Scholar]
- 193. Sasindran J, Torrelles JB. 2011. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front. Microbiol. 2:2 doi:10.3389/fmicb.2011.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Saukkonen JJ, et al. 2002. Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infect. Immun. 70:1684–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Schindler R, et al. 1990. Correlations and interactions in the production of interleukin-6 (IL-6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood 75:40–47 [PubMed] [Google Scholar]
- 196. Schlesinger LS, et al. 2008. Determinants of phagocytosis, phagosome biogenesis and autophagy for Mycobacterium tuberculosis, p 1–22 In Kaufmann SHE, Britton WJ. (ed), Handbook of tuberculosis: immunology and cell biology. Wiley-VCH, Weinheim, Germany [Google Scholar]
- 197. Schlesinger LS, Hull SR, Kaufman TM. 1994. Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J. Immunol. 152:4070–4079 [PubMed] [Google Scholar]
- 198. Schneemann M, Schoeden G. 2007. Macrophage biology and immunology: man is not a mouse. J. Leukoc. Biol. 81:579. [DOI] [PubMed] [Google Scholar]
- 199. Searle S, Blackwell JM. 1999. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. J. Med. Genet. 36:295–299 [PMC free article] [PubMed] [Google Scholar]
- 200. Selvaraj P. 2004. Host genetics and tuberculosis susceptibility. Curr. Science 86:115–121 [Google Scholar]
- 201. Selvaraj P, et al. 2008. Cytokine gene polymorphisms and cytokine levels in pulmonary tuberculosis. Cytokine 43:26–33 [DOI] [PubMed] [Google Scholar]
- 202. Selvaraj P, Alagarasu K, Swaminathan S, Harishankar M, Narendran G. 2009. CD209 gene polymorphisms in South Indian HIV and HIV-TB patients. Infect. Genet. Evol. 9:256–262 [DOI] [PubMed] [Google Scholar]
- 203. Selvaraj P, Narayannan PR, Reetha AM. 1999. Association of functional mutant homozygotes of the mannose binding protein gene with susceptibility to pulmonary tuberculosis in India. Tuber. Lung Dis. 79:221–227 [DOI] [PubMed] [Google Scholar]
- 204. Serrano-Gomez D, et al. 2004. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin mediates binding and internalization of Aspergillus fumigatus conidia by dendritic cells and macrophages. J. Immunol. 173:5635–5643 [DOI] [PubMed] [Google Scholar]
- 205. Sharma S, et al. 2010. Association of P2X7 receptor +1513 (A–>C) polymorphism with tuberculosis in a Punjabi population. Int. J. Tuberc. Lung Dis. 14:1159–1163 [PubMed] [Google Scholar]
- 206. Shaw TC, Thomas LH, Friedland JS. 2000. Regulation of IL-10 secretion after phagocytosis of Mycobacterium tuberculosis by human monocytic cells. Cytokine 12:483–486 [DOI] [PubMed] [Google Scholar]
- 207. Shin HD, et al. 2005. Common interleukin 10 polymorphism associated with decreased risk of tuberculosis. Exp. Mol. Med. 37:128–132 [DOI] [PubMed] [Google Scholar]
- 208. Shiratsuchi H, Johnson JL, Ellner JJ. 1991. Bidirectional effects of cytokines on the growth of Mycobacterium avium within human monocytes. J. Immunol. 146:3165–3170 [PubMed] [Google Scholar]
- 209. Singla N, Gupta D, Joshi A, Batra N, Singh J. 2012. Genetic polymorphisms in the P2X7 gene and its association with susceptibility to tuberculosis. Int. J. Tuberc. Lung Dis. 16:224–229 [DOI] [PubMed] [Google Scholar]
- 210. Singla N, et al. 2012. Association of mannose-binding lectin gene polymorphism with tuberculosis susceptibility and sputum conversion time. Int. J. Immunogenet. 39:10–14 [DOI] [PubMed] [Google Scholar]
- 211. Soborg C, et al. 2007. Influence of candidate susceptibility genes on tuberculosis in a high endemic region. Mol. Immunol. 44:2213–2220 [DOI] [PubMed] [Google Scholar]
- 212. Soborg C, et al. 2003. Mannose-binding lectin polymorphisms in clinical tuberculosis. J. Infect. Dis. 188:777–782 [DOI] [PubMed] [Google Scholar]
- 213. Stahl PD, Ezekowitz RA. 1998. The mannose receptor is a pattern recognition receptor involved in host defense. Curr. Opin. Immunol. 10:50–55 [DOI] [PubMed] [Google Scholar]
- 214. Stead WW, Senner JW, Reddick WT, Lofgren JP. 1990. Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N. Engl. J. Med. 322:422–427 [DOI] [PubMed] [Google Scholar]
- 215. Stein CM. 2011. Genetic epidemiology of tuberculosis susceptibility: impact of study design. PLoS Pathog. 7:e1001189 doi:10.1371/journal.ppat.1001189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Stein CM, et al. 2007. Linkage and association analysis of candidate genes for TB and TNFalpha cytokine expression: evidence for association with IFNGR1, IL-10, and TNF receptor 1 genes. Hum. Genet. 121:663–673 [DOI] [PubMed] [Google Scholar]
- 217. Stein CM, et al. 2008. Genome scan of M. tuberculosis infection and disease in Ugandans. PLoS One 3:e4094 doi:10.1371/journal.pone.0004094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Stockton JC, et al. 2004. Polymorphism in NOD2, Crohn's disease, and susceptibility to pulmonary tuberculosis. FEMS Immunol. Med. Microbiol. 41:157–160 [DOI] [PubMed] [Google Scholar]
- 219. Sugawara I, et al. 1999. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 67:2585–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Tailleux L, et al. 2005. DC-SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. 2:e381 doi:10.1371/journal.pmed.0020381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Tailleux L, et al. 2003. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 197:121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Takahashi K, Ezekowitz RA. 2005. The role of the mannose-binding lectin in innate immunity. Clin. Infect. Dis. 41(Suppl 7):S440–S444 [DOI] [PubMed] [Google Scholar]
- 223. Takahashi K, et al. 2008. SLC11A1 (formerly NRAMP1) polymorphisms associated with multidrug-resistant tuberculosis. Tuberculosis (Edinb.) 88:52–57 [DOI] [PubMed] [Google Scholar]
- 224. Takeuchi O, et al. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169:10–14 [DOI] [PubMed] [Google Scholar]
- 225. Tang NL, et al. 2009. Genetic association between a chemokine gene CXCL-10 (IP-10, interferon gamma inducible protein 10) and susceptibility to tuberculosis. Clin. Chim. Acta 406:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Thuong NT, et al. 2007. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun. 8:422–428 [DOI] [PubMed] [Google Scholar]
- 227. Thye T, et al. 2009. MCP-1 promoter variant -362C associated with protection from pulmonary tuberculosis in Ghana, West Africa. Hum. Mol. Genet. 18:381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Thye T, et al. 2011. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS One 6:e20908 doi:10.1371/journal.pone.0020908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Thye T, et al. 2010. Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat. Genet. 42:739–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Torrelles JB, Azad AK, Schlesinger LS. 2006. Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J. Immunol. 177:1805–1816 [DOI] [PubMed] [Google Scholar]
- 231. Torrelles JB, Azad AK, Henning LN, Carlson TK, Schlesinger LS. 2008. Role of C-type lectins in mycobacterial infections. Curr. Drug Targets 9:102–112 [DOI] [PubMed] [Google Scholar]
- 232. Trajkov D, et al. 2009. Association of 22 cytokine gene polymorphisms with tuberculosis in Macedonians. Indian J. Tuberc. 56:117–131 [PubMed] [Google Scholar]
- 233. Trinchieri G. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 13:251–276 [DOI] [PubMed] [Google Scholar]
- 234. Trinchieri G, Pflanz S, Kastelein RA. 2003. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 19:641–644 [DOI] [PubMed] [Google Scholar]
- 235. Trowsdale J. 2011. The MHC, disease and selection. Immunol. Lett. 137:1–8 [DOI] [PubMed] [Google Scholar]
- 236. Tso HW, Lau YL, Tam CM, Wong HS, Chiang AK. 2004. Associations between IL12B polymorphisms and tuberculosis in the Hong Kong Chinese population. J. Infect. Dis. 190:913–919 [DOI] [PubMed] [Google Scholar]
- 237. Turner DM, et al. 1997. An investigation of polymorphism in the interleukin-10 gene promoter. Eur. J. Immunogenet. 24:1–8 [DOI] [PubMed] [Google Scholar]
- 238. Turner J, et al. 2002. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 169:6343–6351 [DOI] [PubMed] [Google Scholar]
- 239. Uciechowski P, et al. 2011. Susceptibility to tuberculosis is associated with TLR1 polymorphisms resulting in a lack of TLR1 cell surface expression. J. Leukoc. Biol. 90:377–388 [DOI] [PubMed] [Google Scholar]
- 240. Underhill DM. 2007. Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immunol. Rev. 219:75–87 [DOI] [PubMed] [Google Scholar]
- 241. van Crevel R, et al. 2009. Infection with Mycobacterium tuberculosis Beijing genotype strains is associated with polymorphisms in SLC11A1/NRAMP1 in Indonesian patients with tuberculosis. J. Infect. Dis. 200:1671–1674 [DOI] [PubMed] [Google Scholar]
- 242. van der Eijk EA, Van DV, Vandenbroucke JP, Van Dissel JT. 2007. Heredity versus environment in tuberculosis in twins: the 1950s United Kingdom Prophit survey: Simonds and Comstock revisited. Am. J. Respir. Crit. Care Med. 176:1281–1288 [DOI] [PubMed] [Google Scholar]
- 243. van de Veerdonk FL, et al. 2010. Mycobacterium tuberculosis induces IL-17A responses through TLR4 and dectin-1 and is critically dependent on endogenous IL-1. J. Leukoc. Biol. 88:227–232 [DOI] [PubMed] [Google Scholar]
- 244. Vankayalapati R, et al. 2001. T cells enhance production of IL-18 by monocytes in response to an intracellular pathogen. J. Immunol. 166:6749–6753 [DOI] [PubMed] [Google Scholar]
- 245. van Kooyk Y. 2008. C-type lectins on dendritic cells: key modulators for the induction of immune responses. Biochem. Soc. Trans. 36:1478–1481 [DOI] [PubMed] [Google Scholar]
- 246. Vannberg FO, Chapman SJ, Hill AV. 2011. Human genetic susceptibility to intracellular pathogens. Immunol. Rev. 240:105–116 [DOI] [PubMed] [Google Scholar]
- 247. Vannberg FO, et al. 2008. CD209 genetic polymorphism and tuberculosis disease. PLoS One 3:e1388 doi:10.1371/journal.pone.0001388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Vejbaesya S, Chierakul N, Luangtrakool P, Sermduangprateep C. 2007. NRAMP1 and TNF-alpha polymorphisms and susceptibility to tuberculosis in Thais. Respirology 12:202–206 [DOI] [PubMed] [Google Scholar]
- 249. Velez DR, et al. 2009. Association of SLC11A1 with tuberculosis and interactions with NOS2A and TLR2 in African-Americans and Caucasians. Int. J. Tuberc. Lung Dis. 13:1068–1076 [PMC free article] [PubMed] [Google Scholar]
- 250. Velez DR, et al. 2009. NOS2A, TLR4, and IFNGR1 interactions influence pulmonary tuberculosis susceptibility in African-Americans. Hum. Genet. 126:643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Velez DR, et al. 2010. Variants in toll-like receptors 2 and 9 influence susceptibility to pulmonary tuberculosis in Caucasians, African-Americans, and West Africans. Hum. Genet. 127:65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Vesosky B, Rottinghaus EK, Stromberg P, Turner J, Beamer G. 2010. CCL5 participates in early protection against Mycobacterium tuberculosis. J. Leukoc. Biol. 87:1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Vidal SM, Malo D, Vogan K, Skamene E, Gros P. 1993. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73:469–485 [DOI] [PubMed] [Google Scholar]
- 254. Vishnoi A, Srivastava A, Roy R, Bhattacharya A. 2008. MGDD: Mycobacterium tuberculosis genome divergence database. BMC Genomics 9:373 doi:10.1186/1471-2164-9-373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. 2005. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genomics 15:693–704 [PubMed] [Google Scholar]
- 256. Wang Q, Zhan P, Qiu LX, Qian Q, Yu LK. 2012. TNF-308 gene polymorphism and tuberculosis susceptibility: a meta-analysis involving 18 studies. Mol. Biol. Rep. 39:3393–3400 [DOI] [PubMed] [Google Scholar]
- 257. Wang X, Xiao H, Lan H, Mao C, Chen Q. 2011. Lack of association between the P2X7 receptor A1513C polymorphism and susceptibility to pulmonary tuberculosis: a meta-analysis. Respirology 16:790–795 [DOI] [PubMed] [Google Scholar]
- 258. WHO 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response. WHO/HTM/TB/2010.3 WHO Press, Geneva, Switzerland [Google Scholar]
- 259. Wiens GD, Glenney GW. 2011. Origin and evolution of TNF and TNF receptor superfamilies. Dev. Comp. Immunol. 35:1324–1335 [DOI] [PubMed] [Google Scholar]
- 260. Wilbur AK, Kubatko LS, Hurtado AM, Hill KR, Stone AC. 2007. Vitamin D receptor gene polymorphisms and susceptibility to M. tuberculosis in native Paraguayans. Tuberculosis (Edinb.) 87:329–337 [DOI] [PubMed] [Google Scholar]
- 261. Wright JR. 2005. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5:58–68 [DOI] [PubMed] [Google Scholar]
- 262. Xiao J, et al. 2009. Lack of association between polymorphisms in the P2X7 gene and tuberculosis in a Chinese Han population. FEMS Immunol. Med. Microbiol. 55:107–111 [DOI] [PubMed] [Google Scholar]
- 263. Xiao J, et al. 2010. Metaanalysis of P2X7 gene polymorphisms and tuberculosis susceptibility. FEMS Immunol. Med. Microbiol. 60:165–170 [DOI] [PubMed] [Google Scholar]
- 264. Yadav M, Schorey JS. 2006. The {beta}-glucan receptor Dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Yim JJ, Adams AA, Kim JH, Holland SM. 2006. Evolution of an intronic microsatellite polymorphism in Toll-like receptor 2 among primates. Immunogenetics 58:740–745 [DOI] [PubMed] [Google Scholar]
- 266. Yim JJ, Selvaraj P. 2010. Genetic susceptibility in tuberculosis. Respirology 15:241–256 [DOI] [PubMed] [Google Scholar]
- 267. Yoshikai Y. 2006. Immunological protection against mycobacterium tuberculosis infection. Crit. Rev. Immunol. 26:515–526 [DOI] [PubMed] [Google Scholar]
- 268. Zakham F, et al. 2011. Mycobacterial species as case-study of comparative genome analysis. Cell Mol. Biol. (Noisy-le-Grand) 57(Suppl):OL1462–OL1469 [PubMed] [Google Scholar]
- 269. Zenaro E, Donini M, Dusi S. 2009. Induction of Th1/Th17 immune response by Mycobacterium tuberculosis: role of dectin-1, mannose receptor, and DC-SIGN. J. Leukoc. Biol. 86:1393–1401 [DOI] [PubMed] [Google Scholar]
- 270. Zhang J, et al. 2011. Interleukin-10 polymorphisms and tuberculosis susceptibility: a meta-analysis. Int. J. Tuberc. Lung Dis. 15:594–601 [DOI] [PubMed] [Google Scholar]
- 271. Zhang W, et al. 2005. Variants of the natural resistance-associated macrophage protein 1 gene (NRAMP1) are associated with severe forms of pulmonary tuberculosis. Clin. Infect. Dis. 40:1232–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Zhang X, et al. 2012. Polymorphic allele of human MRC1 confers protection against tuberculosis in a Chinese population. Int. J. Biol. Sci. 8:375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Zhang Y, et al. 1995. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. J. Clin. Invest. 95:586–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. 2005. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J. Biol. Chem. 280:32618–32624 [DOI] [PubMed] [Google Scholar]
- 275. Zheng R, et al. 2011. Relationship between polymorphism of DC-SIGN (CD209) gene and the susceptibility to pulmonary tuberculosis in an eastern Chinese population. Hum. Immunol. 72:183–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Zhu XW, Friedland JS. 2006. Multinucleate giant cells and the control of chemokine secretion in response to Mycobacterium tuberculosis. Clin. Immunol. 120:10–20 [DOI] [PubMed] [Google Scholar]