

Abstract
Yersinia pestis and many other Gram-negative pathogenic bacteria use the chaperone/usher (CU) pathway to assemble virulence-associated surface fibers termed pili or fimbriae. Y. pestis has two well-characterized CU pathways: the caf genes coding for the F1 capsule and the psa genes coding for the pH 6 antigen. The Y. pestis genome contains additional CU pathways that are capable of assembling pilus fibers, but the roles of these pathways in the pathogenesis of plague are not understood. We constructed deletion mutations in the usher genes for six of the additional Y. pestis CU pathways. The wild-type (WT) and usher deletion strains were compared in the murine bubonic (subcutaneous) and pneumonic (intranasal) plague infection models. Y. pestis strains containing deletions in CU pathways y0348-0352, y1858-1862, and y1869-1873 were attenuated for virulence compared to the WT strain by the intranasal, but not subcutaneous, routes of infection, suggesting specific roles for these pathways during pneumonic plague. We examined binding of the Y. pestis WT and usher deletion strains to A549 human lung epithelial cells, HEp-2 human cervical epithelial cells, and primary human and murine macrophages. Y. pestis CU pathways y0348-0352 and y1858-1862 were found to contribute to adhesion to all host cells tested, whereas pathway y1869-1873 was specific for binding to macrophages. The correlation between the virulence attenuation and host cell binding phenotypes of the usher deletion mutants identifies three of the additional CU pathways of Y. pestis as mediating interactions with host cells that are important for the pathogenesis of plague.
INTRODUCTION
Yersinia pestis is a facultative intracellular, Gram-negative bacterial pathogen that causes the deadly disease plague. Y. pestis is maintained in a number of natural reservoirs, including rats, rabbits, prairie dogs, and other small mammals (53). The bacteria are primarily spread through the bite of infected arthropods, including, most notably, the rat flea, Xenopsylla cheopis (37, 38, 74). Y. pestis introduced into a human host through the bite of an infected flea can enter the bloodstream and spread to a regional lymph node. In the lymph node, the bacteria will begin to replicate and initiate an inflammatory response. This inflammation causes the lymph node to swell and transform into a painful black bubo, the hallmark of bubonic plague (53). Y. pestis can re-enter the bloodstream from the lymph node, leading to septicemic plague and dissemination to secondary organs. Colonization of the lungs by this route results in a secondary pneumonic plague, which can then lead to direct human-to-human spread of the bacteria via respiratory droplets. Direct inhalation of Y. pestis results in a primary pneumonic plague. Humans infected with all forms of plague have a poor survival rate if left untreated, with a near 100% mortality for pneumonic plague in the absence of antibiotic intervention (14, 53).
Y. pestis evolved from the enteric pathogen Y. pseudotuberculosis 1,500 to 20,000 years ago (2). Key to its adaptation to the vector-borne lifestyle was the acquisition of two Y. pestis-specific plasmids, pMT1 and pPCP1 (9). The pPCP1 plasmid encodes the Pla plasminogen activating protease, which is important for the ability of Y. pestis to proliferate and persist within the lungs in pneumonic form and for its dissemination from the site of infection in bubonic form (9, 45, 62). Pla also contributes to the bacteria's ability to adhere to and invade host cells, making it one of the adhesins expressed by Y. pestis (4, 18). The second pestis-specific plasmid, pMT1, contributes to both transmission through fleas and increased virulence in the human host. The caf genes present on pMT1 code for expression of the fraction 1 (F1) capsule. The F1 capsule, which is expressed at 37°C, is a major protective antigen of Y. pestis and forms a dense coating around the bacteria that prevents phagocytosis by host cells (10, 13, 17, 24, 49, 60, 67). The F1 capsule also has roles in transmission from the flea vector and contributes to pathogenesis in the mammalian host (61, 73). Y. pestis also contains a third plasmid, pCD1, that is shared with enteropathogenic Yersinia spp. (Y. pseudotuberculosis and Y. enterocolitica) and is critical for virulence. The pCD1 virulence plasmid encodes the type three secretion system (T3SS) and Yersinia outer proteins (Yops). The T3SS is induced at 37°C under specific growth conditions and allows the direct injection of Yops into the cytoplasm of target host cells, causing a variety of effects, including modulation of the host immune response, blocking phagocytosis, and triggering host cell death (5, 7, 15, 16, 32, 65).
Adhesins are critical virulence factors of pathogenic bacteria, mediating interactions with host cells and allowing colonization of specific sites within the host (55). For the pathogenic Yersinia, adhesin-mediated host cell binding is also important to facilitate the delivery of Yop effector proteins via the pCD1-encoded T3SS. Interestingly, Y. pestis lacks two of the major adhesins expressed by the enteropathogenic Yersinia: YadA and Inv (18, 41, 59, 63). Alternate adhesins have been identified in Y. pestis, including the outer membrane protein Ail and the autotransporter proteins YapC and YapE (26, 27, 46). In addition, the Y. pestis genome contains 10 gene clusters belonging to the chaperone/usher (CU) pathway (Fig. 1) (21, 52). The CU pathway is a conserved secretion system present in Gram-negative bacteria that is dedicated to the assembly of virulence-associated surface fibers, generally involved in adhesion, termed pili or fimbriae (66, 71, 77). The functions of two of the Y. pestis CU pathways have been extensively characterized: the plasmid pMT1-encoded caf system that expresses the F1 capsule as described above and the psa genes coding for the pH 6 antigen. The pH 6 antigen is typically expressed at 37°C under low-pH conditions and forms thin, flexible fibers on the bacterial surface (48, 49). The pH 6 antigen functions in adhesion to host cells and has been shown to bind phosphatidylcholine on lung epithelial cells but also plays a role in evasion of phagocytosis (25, 33, 40, 49). The pH 6 antigen is a virulence factor of Y. pestis and contributes to the pathogenesis of both bubonic and pneumonic plague (47, 73).
Fig 1.

CU pathways of Y. pestis. The CU gene clusters are depicted, with the gene designations indicated on the left and the usher genes noted within each cluster. The caf (F1) and psa (pH 6) CU gene clusters are shown at the bottom of the figure. Usher gene y1543 is disrupted by an insertion sequence (IS). The usher for pathway y4060-4063 is disrupted by a frameshift mutation into two open reading frames (y4061 and y4062).
Biogenesis of pili by the CU pathway relies on a periplasmic chaperone and an integral outer membrane protein termed the usher, which provides a platform for pilus assembly and secretion (54, 66, 71, 77). The usher genes for two of the Y. pestis CU pathways (y1539-1544 and y4060-4063) are disrupted by an insertion sequence or premature stop codon (Fig. 1), and thus these pathways are not expected to be functional. The caf and psa genes belong to the FGL subfamily of CU pathways that assemble thin, flexible fibers (77). In contrast, the additional CU pathways of Y. pestis belong to the FGS subfamily that assembles rigid, rod-like adhesive pili. In agreement with this, Y. pestis was shown to express pilus fibers distinct from the F1 capsule and pH 6 antigen (60). Moreover, a recent study by Felek et al. demonstrated that heterologous expression of the six intact additional Y. pestis CU pathways in Escherichia coli resulted in the assembly pilus-like structures on the bacterial surface (25). In support of a role for the Y. pestis CU pathways as adhesins, expression of locus y0561-0563 in E. coli conferred binding to HEp-2 human epithelial and THP-1 human macrophage-like cells, and expression of pathways y0348-0352, y0561-0563, y1858-1862, and y3478-3480 enhanced biofilm formation by E. coli (25). However, when deletion mutations of the CU pathways were constructed in the Y. pestis KIM5 strain, only loss of the psa locus, coding for the pH 6 antigen, resulted in decreased adhesion to host cells and decreased biofilm formation (25). Finally, Felek et al. found that a Y. pestis KIM5 strain containing a deletion of CU pathway y1858-1862 was attenuated for virulence in mice via the intravenous route of infection compared to the parental wild-type (WT) strain (25). Taken together, these studies show that the additional Y. pestis CU pathways are capable of assembling adhesive pili and suggest that these pili may contribute to virulence. However, the functions of the Y. pestis CU pathways in host-pathogen interactions and in the pathogenesis of plague remain to be established.
In the present study, we tested the roles of the CU pathways of Y. pestis in virulence, using the murine bubonic (subcutaneous) and pneumonic (intranasal) plague infection models. Deletions of the usher gene for each of the six intact additional CU pathways were constructed in the fully virulent Y. pestis KIM5+ strain background. Comparison of these mutants with WT KIM5+ revealed that CU loci y0348-0352, y1858-1862, and y1869-1873 contribute to virulence via the intranasal, but not subcutaneous, routes of infection. We found no differences between WT Y. pestis and usher deletion mutants for biofilm formation or autoaggregation in vitro. However, Y. pestis KIM6+ usher mutants containing deletions in CU loci y0348-0352, y1858-1862, and y1869-1873 were defective for adhesion to host cells compared to the WT strain. The Y. pestis CU pathways found to function in host cell binding were the same as found to contribute to virulence during intranasal infection, establishing a function for at least three of the additional CU pathways in Y. pestis-host interactions and the pathogenesis of plague.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used in the present study are listed in Table 1. The Y. pestis strains are derived from KIM, a biovar 2.MED strain (1). Y. pestis KIM6+ is an attenuated pgm-positive strain that lacks the pCD1 virulence plasmid. Y. pestis KIM5+Ap is fully virulent and contains both the pgm locus and the pCD1 virulence plasmid marked with an ampicillin resistance (Ampr) gene (34). Specific growth conditions for Y. pestis are noted for each experiment. E. coli strains were grown in LB medium at 37°C with aeration. When needed, the bacterial growth medium was supplemented with ampicillin (Amp) at 100 μg/ml, kanamycin (Kan) at 50 μg/ml, or chloramphenicol (Cml) at 25 μg/ml, except where noted otherwise. IPTG (isopropyl-β-d-thiogalactopyranoside) was added to 50 μM final concentration to induce expression of plasmid-encoded genes when necessary.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Cloning strain; hsdR recA endA | 36 |
| BW25141 | Strain for maintenance of conditional replicative oriRγ plasmids | 20 |
| S17λpir | Strain used for conjugation | 62a |
| Y. pestis | ||
| KIM6+ | Attenuated biovar 2.MED strain; pgm+, pCD1− | 68 |
| KIM6+ Δy0350 | Deletion of usher gene y0350 in KIM6+ | This study |
| KIM6+ Δy0562 | Deletion of usher gene y0562 in KIM6+ | This study |
| KIM6+ Δy1858 | Deletion of usher gene y1858 in KIM6+ | This study |
| KIM6+ Δy1871 | Deletion of usher gene y1871 in KIM6+ | This study |
| KIM6+ Δy2390 | Deletion of usher gene y2390 in KIM6+ | This study |
| KIM6+ Δy3480 | Deletion of usher gene y3480 in KIM6+ | This study |
| KIM5+Ap | Fully virulent biovar 2.MED strain; KIM6+/pCD1Ap | 34 |
| KIM5+Ap Δy0350 | Deletion of usher gene y0350 in KIM5+ | This study |
| KIM5+Ap Δy0562 | Deletion of usher gene y0562 in KIM5+ | This study |
| KIM5+Ap Δy1858 | Deletion of usher gene y1858 in KIM5+ | This study |
| KIM5+Ap Δy1871 | Deletion of usher gene y1871 in KIM5+ | This study |
| KIM5+Ap Δy2390 | Deletion of usher gene y2390 in KIM5+ | This study |
| KIM5+Ap Δy3480 | Deletion of usher gene y3480 in KIM5+ | This study |
| Plasmids | ||
| pGEM-T Easy | Cloning vector; Ampr | Promega |
| pMMB91 | Expression vector, IPTG-inducible promoter; Kanr | 23 |
| py0350 | Usher gene y0350 in pMMB91 | This study |
| py1858 | Usher gene y1858 in pMMB91 | This study |
| py1871 | Usher gene y1871 in pMMB91 | This study |
| pKD4 | FRT sites; Kanr Ampr | 20 |
| pKOBEG-sacB | λ phage redγβα, arabinose inducible; Clmr | 12, 50a |
| pFLP2 | sacB FLP-λpR; Ampr | 39 |
| pCD1Ap | pCD1 virulence plasmid with bla; Ampr cassette | 34 |
| pGFP | gfp3.1 in vector pMMB207, IPTG inducible; Ampr Clmr | 35 |
Clmr, chloramphenicol resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance.
The usher genes were deleted in Y. pestis KIM6+ using the pKOBEG-sacB system (20, 22). Briefly, the pKOBEG-sacB plasmid was first introduced into KIM6+ by electroporation. A DNA fragment containing a Kanr cassette with flanking ends homologous to the target usher gene was amplified by PCR with the primer pairs y(usher gene)_kanF and y(usher gene)_kanR (Table 2). Approximately 1 to 2 μg of purified PCR product was used to electroporate KIM6+/pKOBEG-sacB cells. Kanr transformants harboring the mutated allele were isolated and screened by PCR. Plasmid pKOBEG was cured by subculturing the bacteria at 26°C on medium containing sucrose and then checking for Clms clones. The KIM6+ (usher gene)::kan strain was then transformed with plasmid pFLP2 by conjugation with E. coli strain S17λpir, and the Kanr cassette was excised using the flanking FRT sites. To cure pFLP2, bacteria were subcultured at 26°C in the presence of sucrose and plated onto LB agar to select for Amps clones. These final strains were designated KIM6+ Δ(usher gene) (Table 1). All usher gene deletions were confirmed by PCR using appropriate primers.
Table 2.
Primers used in this study
| Primer function and name | Sequence (5′–3′) |
|---|---|
| Primers used for usher deletions in Y. pestis | |
| y0350_kanF | TGCCGGGAATTATGATGCTGTAGATGTTTTGGTTAATAACAAATT |
| GTTCATGTAGGCTGGAGCTGCTTCG | |
| y0350_kanR | CGGCACCCAGTGGTAACCCCTCACCTTCGGCTTGAATTAACACCG |
| CATAGATGGGAATTAGCCATGGTCC | |
| y0562_kanF | ATTATTAATGCCAGCAAGACCTACCGTATTGCAGCGACCTTCGAT |
| AGCGTGTAGGCTGGAGCTGCTTC | |
| y0562_kanR | AATCGGCAGCATTTCACCATCACTACCACGGATGGTTAGCAGCA |
| ATTGGCGATGGGAATTAGCCATGGTCC | |
| y1858_kanF | CAACAACCGTTATTACCTTTCTCAATCACAGCTAAGAACGATTGG |
| GCTTCTGTAGGCTGGAGCTGCTTCG | |
| y1858_kanR | GTAAGTCTTGGCCTGATGCGTCTGTAATTCTAATATTCACTGCGG |
| AAATTATGGGAATTAGCCATGGTCC | |
| y1871_kanF | CCCCGGTGAGCGTCAGGTTGATATCTATTTAAATGATCAATGGAT |
| TGGTCTGTAGGCTGGAGCTGCTTCG | |
| y1871_kanR | CGCCATCTTTTACTTCCGCCCCAAACGGCAAGGGGGTGTTATCTG |
| CCAGAATGGGAATTAGCCATGGTCC | |
| y2390_kanF | GTATATGAACAACAAATTTGTTGATCGCCTAAAAATGCTGTTTGT |
| TGATATGTAGGCTGGAGCTGCTTCG | |
| y2390_kanR | CGTTAGTGCCCTTGCCGTTGTAATTATAAACGACGGTTCCCATAG |
| GCAATATGGGAATTAGCCATGGTCC | |
| y3480_kanF | TCCCGAGCAGGTGATCGCTTTTTATGCCCGTGATGATGAACCGAA |
| TAGCATGTAGGCTGGAGCTGCTTCG | |
| y3480_kanR | GCGGATGGCTACTGTCGGTCAGGCTGATGGTTGCCAGTACCTTCT |
| CCCCTATGGGAATTAGCCATGGTCC | |
| Primers used to confirm usher deletions in Y. pestis | |
| y0350TESTF | CCAACGGACATCAAGTGTACC |
| y0350TESTR | GCATCAGGTTTGACCCAGC |
| y0562TESTF | GTGGTCGCCCGTTGTATTAATC |
| y0562TESTR | CTATATTCAAAAAGTCTTCACATTTGGTC |
| y1858TESTF | GTTACCGTCACTGATACCCTTG |
| y1858TESTR | GTTACCGTCACTGATACCCTTG |
| y1871TESTF | CGCTATTTGTGGTATTGGTGGC |
| y1871TESTR | GCTTGTTGA TTCACTCGGAC |
| y2390TESTF | CGTCAGCGGCGGATAATAAC |
| y2390TESTR | GTAGATAGGCTTGGCTCG |
| y3480TESTF | CCTTTGGCACTGATGACCA |
| y3480TESTR | CTGATGCAATGGATGCTAACG |
| Primers used to amplify usher genes for complementation plasmids | |
| y350FExp | GAATTCATAGACCGCAGTGGTAGACT |
| y350RExp | GGATCCAAGATGCCGGTAGCTATCGT |
| y1858FExp | GAATTCGACCTACTCTGTTGTTGCCT |
| y1858RExp | GGATCCCAGTAACTAGCCCATACCAG |
| y1871FExp | GGATCCGGCCTCGATATAATGCTTAGTC |
| y1871RExp | CACCTGCGCGCAGGTACATGTTGATA |
For construction of complementing plasmids py0350, py1858, and py1871 (Table 1), the usher genes were amplified from KIM6+ by PCR using Taq polymerase (Invitrogen) and the primer pairs listed in Table 2. The PCR products were ligated into plasmid pGEM-T Easy (Promega), the resulting plasmids were then digested with EcoRI and BamHI or BamHI and SalI, and the fragments encoding the usher genes were purified using a MinElute PCR purification kit (Qiagen). The usher gene fragments were then ligated into plasmid pMMB91 that had been similarly digested and purified. Ligation products were transformed into E. coli DH5α for selection of Kanr colonies. Final plasmids, containing the usher genes downstream of the IPTG-inducible Ptac promoter, were confirmed by sequencing. Purified plasmids were then transformed into their cognate KIM6+ usher deletion strains.
To generate the usher deletion mutations in the fully virulent Y. pestis KIM5+ background, plasmid pCD1Ap was introduced by electroporation into the KIM6+ usher deletion strains under biosafety level 3 (BSL3) conditions. The resultant KIM5+Ap strains (Table 1) were selected by plating onto Yersinia Selective Media (YSM) agar containing 30 μg/ml Amp.
Transmission electron microscopy.
Y. pestis was grown at 28°C or 37°C overnight in either Bacto heart infusion broth (HIB; Becton Dickinson) or TMH-Hi (65) with aeration, harvested by centrifugation, washed, resuspended into phosphate-buffered saline (PBS), and then adsorbed to polyvinyl formal-carbon-coated grids (E. F. Fullman) for 2 min. The grids were fixed with 1% glutaraldehyde for 1 min, washed twice with PBS, washed twice with water, and then negatively stained with 0.5% phosphotungstic acid (Ted Pella) for 35 s. The grids were examined on a TECNAI 12 BioTwin G02 microscope (FEI) at an 80-kV accelerating voltage. Digital images were captured with an AMT XR-60 charge-coupled device digital camera system (Advanced Microscopy Techniques).
Mouse infection experiments.
Mouse infections were performed at the University of Medicine and Dentistry of New Jersey Regional Biocontainment Laboratory (Newark, NJ) under BSL3 conditions. All animal research protocols were approved by the Institutional Animal Care and Use Committees of Stony Brook University and the Regional Biocontainment Laboratory.
Six- to eight-week-old female C57BL/6 mice (Jackson Laboratories) were utilized for the infections. Inoculation via the subcutaneous and intranasal routes were used to mimic bubonic and pneumonic plague, respectively. Y. pestis strains were grown overnight at 28°C in HIB, resuspended in PBS, and diluted in PBS to achieve the desired infectious dose. Groups of five mice were injected subcutaneously with 50 μl containing 200 to 250 CFU or inoculated intranasally with 25 μl containing 2,000 to 2,500 CFU (approximately 4 to 5 50% lethal doses [LD50] for the respective routes). The actual infectious doses were determined by retrospective CFU counts. The mice were observed twice daily and monitored for survival for 21 days.
Biofilm formation assay.
Crystal violet staining was used to determine biofilm formation and cell attachment to polystyrene as described by O'Toole et al. (51). Briefly, overnight cultures of Y. pestis were resuspended in HIB to an optical density at 600 nm (OD600) of ∼1.0. A 100-μl portion of bacterial culture was put into a flat-bottom 96-well polystyrene culture plate (Falcon) and incubated statically at either 28 or 37°C for 24 h. The OD600 of the cultures was read in a microplate reader (SpectraMax). Bacterial cultures were then washed twice with 100 μl of PBS, and 0.01% crystal violet was added. The bacteria were incubated with the crystal violet for 15 min at room temperature. The bacteria were then washed three times with 150 μl of distilled water, and the bound crystal violet was solubilized with 80% ethanol and 20% acetone. The absorbance of the crystal violet was read by the microplate reader at 595 nm. The results were normalized to bacterial culture density.
Autoaggregation assay.
Y. pestis strains were grown overnight at either 28 or 37°C in HIB. The overnight cultures were resuspended to an OD600 of approximately 1.1 to 1.6 in 1 ml of HIB in cuvettes. The cultures were then incubated statically at either 28 or 37°C, and the OD600 was read every 20 min.
Tissue culture.
A549 human lung epithelial cells were grown in Dulbecco modified Eagle medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; HyClone). HEp-2 human epithelial cells were grown in MEM (Gibco) containing 10% FBS. Murine bone marrow-derived macrophages (muBMDM) were obtained as described previously (57, 58). Briefly, cells from the femurs of female WT C57BL/6 mice were grown in bone marrow medium (49% DMEM [Gibco], 30% L-cell supernatant, 20% FBS, 1% sodium pyruvate [Gibco]). When muBMDM were seeded for infection, they were grown in infection medium (79% DMEM [Gibco], 15% L-cell supernatant, 5% FBS, 1% sodium pyruvate). Human monocyte-derived macrophages (huMDM) were isolated as described previously (30) from healthy human donors and directly seeded at 1.5 × 105 cells/well in 24-well plates (Corning) on coverslips. The cells were allowed to differentiate for 5 days in RPMI medium (Gibco) containing 10% FBS and 10 ng of macrophage colony-stimulating factor (Sigma)/ml.
Bacterial adhesion and invasion assays.
Mammalian cells were seeded onto coverslips in a 24-well plate (Corning) at concentrations of 1.5 × 105 cells per well, followed by incubation overnight at 37°C in 5% CO2 in the growth conditions described above. Overnight cultures of WT or usher deletion mutant Y. pestis KIM6+ strains or the same strains containing the pGFP plasmid were diluted 1:20 into fresh HIB and grown at 37°C with aeration until the OD600 was 0.7. Y. pestis strains containing the pGFP plasmid (for invasion studies) or deletion strains containing complementation plasmids were induced for expression of the plasmid-encoded genes by the addition of IPTG at 2 h of growth. For adhesion experiments, the bacteria were resuspended to a multiplicity of infection (MOI) of 50 in the appropriate medium for the host cell type used. For invasion experiments, bacteria were resuspended to an MOI of 50 for the epithelial cells or an MOI of 10 for the macrophages. For adhesion experiments, the muBMDM and huMDM were pretreated with 5 μg of cytochalasin/ml D for 1 h prior to infection. After the mammalian cells were washed three times with PBS, 1 ml of bacteria was added to each well, and the plate was centrifuged (50 × g, 4 min, room temperature) to facilitate bacterial contact. After 2 h at 37°C in 5% CO2 (or 20 min for the macrophage invasion studies), the cells were washed once with PBS and fixed for 30 min in 2.5% paraformaldehyde at room temperature. All subsequent steps were completed at room temperature. The cells were blocked with 3% bovine serum albumin (BSA) in PBS for 20 min and then incubated with rabbit anti-Yersinia antiserum SB349 (6) diluted 1:1,000 in 3% BSA in PBS for 30 min. The cells were then washed three times with PBS. Next, a secondary goat anti-rabbit antibody conjugated to Alexa Fluor 594 (Invitrogen) was added at a 1:2,000 dilution in 3% BSA in PBS, followed by incubation for 30 min. The cells were then washed again three times with PBS. The coverslips were mounted on glass slides using ProLong Gold antifade reagent (Invitrogen), and the slides were examined on a Zeiss Axioplan2 microscope using a ×40 objective lens. Phase-contrast and epifluorescence images were captured using a Spot camera (Diagnostic Instruments) and processed using Adobe Photoshop. For each experimental replicate, 10 random fields per bacterial strain were photographed. The total numbers of host cells and the total numbers of bacteria bound to the host cells were quantified for each field to calculate the number of bacteria/cell. The values obtained for the 10 fields were then averaged to obtain the number of bacteria/cell for the experimental replicate.
Bacterial survival during macrophage infection.
muBMDM were seeded on coverslips in a 24-well plate (Corning) at concentrations of 1.5 × 105 cells per well, followed by incubation overnight at 37°C in 5% CO2 in the growth conditions described above. Overnight cultures of WT or usher deletion mutant Y. pestis KIM6+ strains were diluted 1:20 into fresh HIB and grown at 37°C with aeration until the OD600 was 0.7. The muBMDM were washed three times with PBS, 1 ml of bacteria with an MOI of 5 was added to each well, and the plate was centrifuged (50 × g, 4 min, room temperature) to facilitate bacterial contact. After 20 min at 37°C, 5% CO2 the cells were washed once with PBS, and fresh infection media containing 8 μg of gentamicin/ml were added to each well. This was noted as the 0-h time point. After 1 h of incubation, the wells were washed three times with 1 ml of PBS. Fresh medium with or without 2 μg of gentamicin/ml was then added to each well, and the infected macrophages were incubated for an additional 23 h.
At the 0- and 24-h time points, the infected macrophages were washed three times with 1 ml of PBS and then lysed in 0.5 ml of 0.1% Triton X-100 in PBS for 10 min at 37°C. The lysates were removed, the wells were washed with 0.5 ml of PBS, and the lysates and wash were pooled in a 1.5-ml microcentrifuge tube. Serial 10-fold dilutions were then plated on LB agar, followed by incubation at 28°C for 2 days, and the CFU were enumerated.
Outer membrane isolation and analysis.
KIM6+ WT or the usher deletion Δy0350, Δy1858, or Δy1871 mutants were grown in 10 ml of HIB at 37°C with aeration until an OD600 of ∼0.7 was reached. Bacteria were harvested, washed, resuspended in 1 ml of 20 mM Tris-HCl (pH 8) containing Complete protease inhibitor cocktail (Roche), and lysed by sonication for 2 min (15 s on, 15 s off) in an ice-water bath. Whole bacteria were removed by centrifugation (8,000 × g, 2 min, 4°C). Sarkosyl (sodium-N-lauroylsarcosinate; Fisher) was added to the supernatant fraction to a 0.5% final concentration, and the mixture was incubated for 5 min at room temperature to selectively solubilize the cytoplasmic membrane. The outer membrane was then pelleted by centrifugation (15,000 × g, 30 min, 4°C) and resuspended in 0.1 ml of 20 mM Tris (pH 8)–0.3 M NaCl. An equal volume of 2× sodium dodecyl sulfate (SDS) sample buffer was added, and the sample was incubated for 10 min at 95°C prior to separation by SDS-polyacrylamide gel electrophoresis (PAGE). The expression levels of outer membrane proteins were determined by inspection of Coomassie blue-stained SDS-PAGE gels or by immunoblotting with anti-F1 antibody (60) at 1:1,000, anti-Pla antibody (72) at 1:500, anti-PsaA antibody (47) at 1:1,500, or anti-Ail antibody (76) at 1:500. Immunoblots were developed with alkaline phosphatase-conjugated secondary antibodies and BCIP (5-bromo-4-chloro-3-indolylphosphate)/NBT (nitroblue tetrazolium) substrate (KPL).
Statistical analysis.
Mouse survival curves were compared using the log-rank test with data obtained from three independent experiments with five mice per experiment per strain. The results for bacterial adhesion to host cells were analyzed for significance using data obtained from three to six independent experiments with three replicates each. Statistical significance was calculated by one-way analysis of variance and Tukey's multiple-comparison post test. Statistical calculations were performed using GraphPad Prism.
RESULTS
Construction and initial phenotypic characterization of Y. pestis usher deletion mutants.
To examine functions of the CU pathways of Y. pestis in addition to the well-studied caf and psa systems, we constructed a set of deletion mutations in the KIM6+ background (pgm-positive, pCD1-negative) using the lambda Red recombination system (20, 22). We deleted the usher gene for each of the six intact additional CU pathways (Fig. 1), creating strains KIM6+ Δy0350, Δy0562, Δy1858, Δy1871, Δy2390, and Δy3480. The KIM6+ usher deletion mutants showed no growth defects compared to the parental WT strain when grown in HIB at either 28 or 37°C (data not shown). In addition, no differences were observed between the outer membrane protein profiles of the WT strain and the usher deletion mutants, nor were differences detected in levels of the F1 capsule, pH 6 antigen, Ail, or Pla (data not shown). Y. pestis KIM6+ was previously shown to express pilus-like fibers on its cell surface that were distinct from the F1 capsule and pH 6 antigen (25, 60). To determine whether one of six CU pathways is responsible for biogenesis of these fibers, we examined the KIM6+ usher deletion mutants for the presence of pili by transmission electron microscopy. Each deletion mutant expressed pilus fibers similar to the WT strain (data not shown), suggesting either that more than one of the CU pathways encodes the pilus fibers or that the fibers are encoded by another, as-yet-unidentified, pathway.
Y. pestis forms biofilms in the flea midgut to allow for effective transmission of the bacteria from the flea vector to the host (19). Biofilm formation generally depends on the hms system, which is found in the pgm locus (8, 37, 53). CU pili contribute to biofilm formation in a number of pathogenic bacteria and ectopic expression of four of the Y. pestis CU loci (y0348-0352, y0561-0563, y1858-1862, and y3478-3480) was previously shown to promote biofilm formation by E. coli (25, 56, 69). However, disruption of these CU pathways did not alter hms-independent biofilm formation by the Y. pestis KIM5 (Δpgm) strain (25). To test whether the CU pathways might contribute to biofilm formation in the presence of an intact hms system, we examined our panel of KIM6+ (pgm-positive) usher deletion mutants. The mutant bacteria formed biofilms similar to WT KIM6+ when grown in HIB at 28 or 37°C (data not shown), indicating that none of the six CU pathways is required for biofilm formation under the experimental conditions tested. In addition to biofilm formation, Y. pestis bacteria autoaggregate during growth in culture media at 28°C, and there is evidence that autoaggregation may correlate with the virulence of Yersinia spp. (28, 43). We compared the Y. pestis usher deletion mutants and parental KIM6+ strain grown in HIB at 28°C for autoaggregation. All bacteria behaved similarly under both growth conditions (data not shown), suggesting that none of the CU pathways is required for Y. pestis autoaggregation. As previously reported, the bacteria did not autoaggregate at 37°C, likely due to expression of the F1 antigen (28).
Contributions of the additional CU pathways of Y. pestis in the murine model of plague.
To analyze the roles of the Y. pestis CU pathways in the host during infection, we first converted the KIM6+ usher deletion mutants to the fully virulent KIM5+ background by adding in the pCD1 virulence plasmid by electroporation. As for the KIM6+ usher deletion mutants, the KIM5+ mutants showed no growth defects compared to the WT strain when grown in HIB at either 28 or 37°C, and no differences were detected in biofilm formation or autoaggregation (data not shown). Approximately 4 to 5 LD50 of the parental KIM5+ strain or equal doses of the six usher deletion mutants were delivered to mice via subcutaneous injection (200 to 250 CFU) or intranasal inoculation (2,000 to 2,500 CFU) to mimic bubonic or pneumonic plague, respectively. No statistically significant differences between the WT strain and any single usher deletion mutant were observed in the time to death for mice infected by the subcutaneous route (Fig. 2). However, there was a trend toward increased survival times for the usher deletion mutants compared to WT KIM5+, with mutants Δy0350, Δy0562, Δy1858, and Δy1871 having P values just above significance (0.052, 0.059, 0.084, and 0.083, respectively). All mice succumbed to infection between days 3 and 7, which is typical for this route of infection with fully virulent Y. pestis (11, 50, 67). The mean time to death for all groups was ∼3 days postinfection. Thus, the CU pathways tested do not provide critical functions during the murine model of bubonic plague.
Fig 2.

Subcutaneous infection of C57BL/6 mice with Y. pestis usher deletion mutants. Mice were infected with WT KIM5+ or usher deletion mutants via the subcutaneous route with 4 to 5 LD50 (200 to 250 CFU), and the time to death was recorded. Mice were monitored for signs of illness or death for 21 days. Each graph represents the combined data from three separate experiments with 5 mice each for a total of 15 mice per strain. There were no significant differences for any of the deletion mutants compared to the WT strain.
For the intranasal route of infection, mimicking pneumonic plague, three of the single usher deletion mutants had significant shifts in their mouse survival curves compared to the parental KIM5+ strain. Deletion of ushers y0350 or y1858 caused small, yet significant (P = 0.0137) attenuation in Y. pestis virulence, with a higher percentage of mice surviving past day 3 postinfection (Fig. 3). For the Δy1858 mutant, 7% (1 of 15) of the mice survived the entire 21-day course of the infection. Mice infected with fully virulent Y. pestis via the intranasal route typically die between days 3 and 5, with the majority of mice dying on or before day 3 (3, 11, 44). Most significantly (P = 0.0006), infection with usher deletion mutant KIM5+ Δy1871 resulted in 13% (2 of 15) of the mice surviving the course of the infection and an increase in the mean time of death to 4 days, compared to 3 days for mice infected with WT KIM5+ (Fig. 3). Mice infected with the other usher deletion mutants all succumbed to infection with a mean time to death of 3 days and had survival curves similar to infection with the WT strain. We conclude from these findings that CU pathways y0348-0352, y1858-1862, and y1869-1873 function within the host and contribute to the virulence of Y. pestis during pneumonic plague.
Fig 3.

Intranasal infection of C57BL/6 mice with Y. pestis usher deletion mutants. Mice were infected with WT KIM5+ or usher deletion mutants via the intranasal route with 4 to 5 LD50 (2,000 to 2,500 CFU), and the time to death was recorded. Mice were monitored for signs of illness or death for 21 days. Each graph represents the combined data from three separate experiments with 5 mice each for a total of 15 mice per strain. Mice infected with deletion mutants Δy0350, Δy1858, or Δy1871 were significantly attenuated (*, P < 0.05; ***, P < 0.001) compared to mice infected with the WT strain.
Y. pestis CU pathways y0348-0352 and y1858-1862 mediate adhesion to human epithelial cells.
The virulence attenuation of the Y. pestis usher deletion mutants suggests that pili assembled by the additional CU pathways may mediate interactions with host cells during infection. We therefore used a microscopy-based assay to compare WT KIM6+ to the usher deletion mutants for association with different host cells. We first examined the interactions of the bacteria with A549 cells, a human lung epithelial cell line, and HEp-2 cells, a human cervical epithelial cell line. Bacterial binding to the host cells was detected using a polyclonal anti-Y. pestis antibody in the absence of permeabilization, so only surface-bound bacteria were visualized (Fig. 4A). For A549 cells, the parental KIM6+ strain, pregrown at 37°C and added at an MOI of 50, adhered at a level of approximately two bacteria per host cell. No differences were detected in adhesion of the KIM6+ Δy0562, Δy1871, Δy2390, or Δy3480 usher deletion mutants compared to the WT strain; however, usher deletion mutants Δy0350 and Δy1858 both exhibited ∼2-fold decreased adherence (Fig. 4B). Binding of the KIM6+ Δy0350 and Δy1858 deletion mutants to A549 cells was restored back to WT levels upon expression of the deleted usher gene in trans, demonstrating the specificity of the usher deletion mutations (Fig. 4B).
Fig 4.
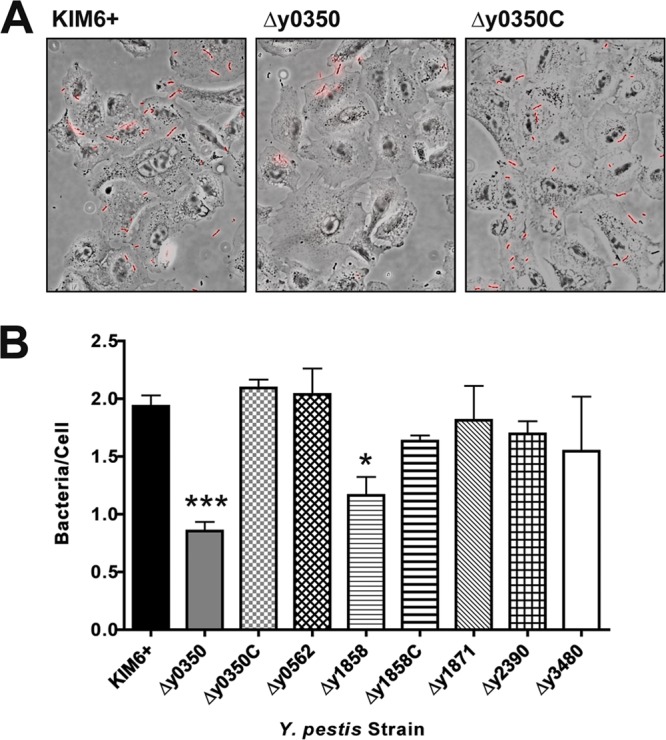
Binding of Y. pestis usher deletion mutants to A549 cells. A549 cells were infected with KIM6+ WT, usher deletion mutants, or complemented strains (denoted with a “C”) at an MOI of 50 for 2 h. The cells were then fixed and stained with a rabbit anti-Yersinia primary antibody and a goat anti-rabbit secondary antibody conjugated to Alexa Fluor 594 (red). For each infection, phase-contrast and epifluorescence images from 10 random fields were captured using a ×40 objective, and the average number of bacteria/cell was calculated. (A) Representative overlay images of phase-contrast and fluorescence microscopy pictures for the KIM6+ WT, Δy0350, and Δy0350 complemented strains. (B) Adhesion data for all strains. The results (bacteria/cell) were calculated from three independent experiments with three replicates per experiment. Bars represent means ± the standard errors of the mean (SEM) (*, P < 0.05; ***, P < 0.001 [for comparison of each strain with the WT]).
A similar result was obtained for binding of the KIM6+ WT and usher deletion mutants to HEp-2 cells. The overall level of bacterial association with the HEp-2 cells was lower than for the A549 cells, with ∼0.5 bacteria binding per HEp-2 cell for the parental KIM6+ strain. Despite this lower overall binding, 2-fold decreases in adhesion were again observed for the KIM6+ Δy0350 and Δy1858 deletion mutants, although the binding defect of the Δy1858 mutant did not reach statistical significance (Fig. 5). No differences in binding were detected for the other KIM6+ usher deletion mutants, and the complemented Δy0350 and Δy1858 strains bound to the HEp-2 cells at levels indistinguishable from WT (Fig. 5).
Fig 5.
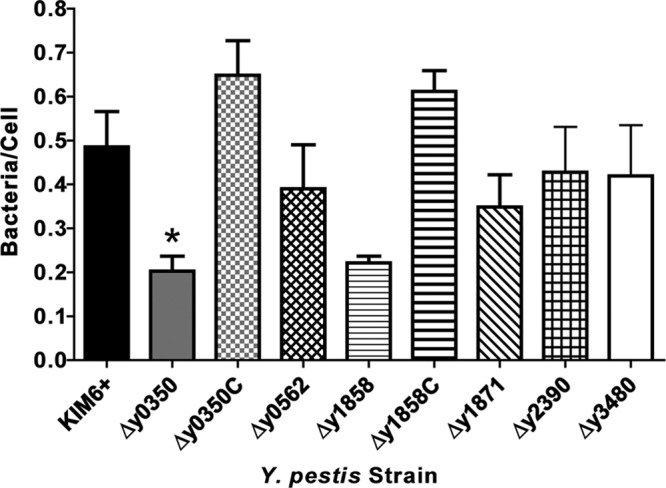
Binding of Y. pestis usher deletion mutants to HEp-2 cells. HEp-2 cells were infected with KIM6+ WT, usher deletion mutants, or complemented strains as described in Fig. 4. The results (bacteria/cell) were calculated from three independent experiments with three replicates per experiment. Bars represent means ± the SEM (*, P < 0.05 [for comparison of each strain with the WT]).
To examine whether loss of the CU pathways affected invasion of Y. pestis inside the A549 and HEp-2 cells, similar experiments were performed with Y. pestis strains expressing green fluorescent protein (GFP). In these assays, intracellular bacteria appeared green due to the GFP and extracellular bacteria appeared both green from the GFP and red due to antibody labeling. The level of uptake for all Y. pestis strains into the A549 and HEp-2 cells was very low, with no invasion seen for the A549 cells and approximately one intracellular bacterium detected for every 10 HEp-2 cells (data not shown). This low level of uptake is in keeping with the epithelial nature of the A549 and HEp-2 cell lines, and matches previously reported data (26, 42, 49). Furthermore, no differences in uptake into either host cell type were detected for the parental KIM6+ strain and usher deletion mutants. Given the low-to-absent levels of bacterial uptake, we conclude that the decreased association of the Y. pestis Δy0350 and Δy1858 deletion mutants with the A549 and HEp-2 cells is due to loss of adhesion to the host cell surface. These data identify CU pathways y0348-0352 and y1858-1862 as important for Y. pestis binding to epithelial cells and suggest that loss of these interactions may underlie the virulence attenuation of the corresponding usher deletion mutants in the intranasal infections.
Y. pestis CU pathways y0348-0352, y1858-1862, and y1869-1873 mediate adhesion to primary murine and human macrophages.
We next examined interactions of the Y. pestis strains with primary murine and human macrophages, muBMDM and huMDM, respectively. Macrophages are professional phagocytes and using GFP-expressing bacteria, we found that ca. 80% of cell-associated Y. pestis were internalized after 20 min of coincubation at an MOI of 50 (data not shown). As with the A549 and HEp-2 cells, no differences in uptake into the macrophages were detected for the parental KIM6+ strain and usher deletion mutants. We also found no differences in the intracellular or extracellular survival of the bacteria over a 24-h infection period (data not shown). To specifically measure binding of the Y. pestis strains to the macrophage cell surface, we treated the muBMDM and huMDM with cytochalasin D prior to infection to block phagocytosis. Under these conditions, WT KIM6+ and usher deletion mutants Δy0562, Δy2390, and Δy3480 adhered at levels of approximately 1.5 and 1.0 bacteria per muBMDM and huMDM, respectively (Fig. 6). The Δy0350 and Δy1858 usher deletion mutants, which had decreased binding to the epithelial cells, also exhibited significantly decreased binding to both the human and murine the macrophages, with levels approximately 2- to 3-fold lower than to the WT strain (Fig. 6). Interestingly, usher deletion mutant Δy1871 also showed a similar two-to-3-fold reduction in binding to the muBMDM and huMDM (Fig. 6). Binding of the three usher deletion mutants was fully restored back to WT levels upon complementation with the deleted usher gene in trans, again demonstrating the specificity of the usher deletion mutations (Fig. 6). Taken together, these results show that CU pathways y0348-0352 and y1858-1862 confer binding to receptors present on epithelial cells and macrophages of both human and murine origin and that CU pathway y1869-1873 mediates binding to a factor present on human and murine macrophages but absent from human epithelial cells. Of note is the fact that the KIM5+ Δy1871 usher deletion mutant had the strongest virulence attenuation observed in the murine pneumonic plague model, suggesting that Y. pestis-macrophage interactions mediated by pathway y1869-1873 may be especially important during pathogenesis.
Fig 6.
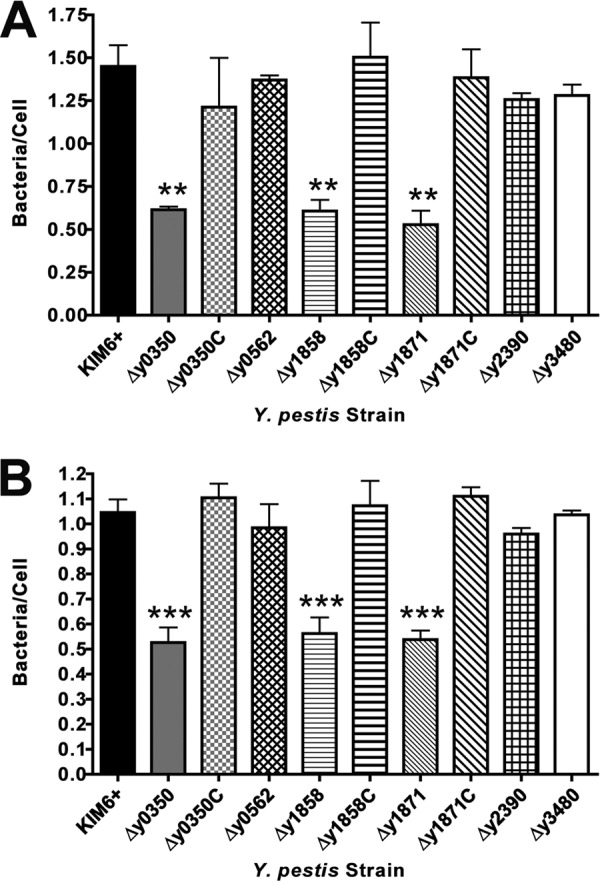
Binding of Y. pestis usher deletion mutants to murine bone marrow-derived macrophages. muBMDM (A) or huMDM (B) were treated with 5 μg of cytochalasin D/ml for 1 h and then infected with KIM6+ WT, usher deletion mutants, or complemented strains as described in Fig. 4. The results (bacteria/cell) were calculated from three independent experiments with three replicates per experiment. Bars represent means ± the SEM (**, P < 0.01; ***, P < 0.001 [for comparison of each strain with the WT]).
DISCUSSION
The Y. pestis genome encodes 10 gene clusters belonging to the CU secretion system that assembles adhesive pili associated with virulence (Fig. 1) (21, 52, 60). Two of these gene clusters, the caf and psa systems encoding for the F1 capsule and pH 6 antigen, respectively, have been well studied, and both make important contributions to the pathogenesis of plague (24, 47–49, 61, 73). Of the eight remaining additional Y. pestis CU pathways, two contain disruptions to their usher genes and thus are not expected to be functional (pathways y1539-1544 and y4060-4063; Fig. 1). We focused here on characterizing roles of the remaining six CU pathways in Y. pestis virulence and host-pathogen interactions. Analysis of single usher deletion mutants in the fully virulent KIM5+ background identified CU pathways y0348-0352, y1858-1862, and y1869-1873 as contributing to the virulence of Y. pestis in mice via the intranasal, but not the subcutaneous, route of infection. Cell culture infection studies of the single usher deletion mutants in the KIM6+ background (lacking the pCD1 virulence plasmid) identified the same three CU pathways as important for binding to host cells. These results show that at least three of the additional CU pathways are virulence factors of Y. pestis and function in adhesion to host cells.
In agreement with our findings, Felek et al. previously identified CU locus y1858-1862 as contributing to the virulence of Y. pestis in mice infected by the intravenous route (25). This suggests that pili assembled by pathway y1858-1862 may be important for host interactions during the systemic phase of infection. However, our Δy1858 mutant was attenuated by the pneumonic, but not the bubonic, routes, arguing against a general systemic role for pathway y1858-1862. Future studies will need to address the specific host receptors recognized by the y1858-1862 pili, as well as the other Y. pestis CU pili, and determine at what points in the infectious process these pili function. In contrast to our findings, Felek et al. did not identify roles for CU pathways y0348-0352 or y1869-1873 in virulence (25). There are several differences between the studies that may explain these results, including that the previous study used the attenuated KIM5 (Δpgm) strain of Y. pestis and only examined the intravenous route of infection. Although we did not detect a role for the y2388-2392 gene cluster in our studies, it is worth noting that this cluster is located in the pgm locus, and thus Δpgm strains of Y. pestis lack this CU pathway.
The individual usher deletion mutations did not alter biofilm formation or autoaggregation by Y. pestis, suggesting that the in vivo virulence attenuation we observed is due to some other mechanism. The lack of biofilm or autoaggregation phenotypes is consistent with the known role of the hms system as the major biofilm-forming pathway of Y. pestis and the function of the pgmA gene in mediating autoaggregation (8, 28, 37, 53). Importantly, we obtained significant phenotypes for adhesion of the KIM6+ usher deletion mutants to host cells. Specifically, compared to the WT strain, deletions of the usher genes for CU pathways y0348-0352 and y1858-1862 resulted in 2- to 3-fold decreased binding to A549 human lung epithelial cells, HEp-2 human cervical epithelial cells, and primary human and murine macrophages. Deletion of the usher gene for CU pathway y1869-1873 also resulted in 2- to 3-fold decreased binding, but this was specific for the macrophages. The correlation between the CU pathways identified as important for binding to host cells and those found to contribute to virulence in mice strongly supports roles for these three CU pathways in interactions with host cells during pathogenesis. In contrast to our results, Felek et al. did not detect any loss in binding to host cells for Y. pestis KIM5 strains containing deletions of the same CU pathways (25). Although both studies examined binding to HEp-2 epithelial cells and macrophages, the prior study did not examine binding to A549 cells and used macrophage-like cell lines (RAW 264.2 and THP-1) rather than primary macrophages. In addition, Felek et al. used an MOI of 1 to 3 and measured binding by a CFU plating assay, as opposed to an MOI of 50 and the microscopy-based method used here. We have found the microscopy-based assay to be more sensitive and reproducible for detecting differences in bacterial association with host cells (M. Hatkoff and D. G. Thanassi, unpublished data); this, combined with the higher MOI, may have allowed us to detect the 2- to 3-fold changes in binding observed for our Y. pestis CU pathway mutants. Finally, the KIM5 strain used by Felek et al. contains the pCD1 virulence plasmid, and it is possible that under tissue culture conditions the engagement of host cells by the T3SS apparatus may have masked binding defects caused by loss of the CU pathways.
The decreased binding to host cells for the Δy0350, Δy1858, and Δy1871 usher deletion mutants compared to WT Y. pestis was significant but modest (3-fold at most). The in vivo attenuation in virulence for these usher deletion strains was also relatively small; the Δy1871 strain had the strongest phenotype, with a 1-day increase in mean time to death and 13% of mice surviving the course of the infection. These results likely reflect redundancy among the multiple CU pathways and other adhesins expressed by Y. pestis. Although it lacks the major YadA and Inv adhesins expressed by enteropathogenic Yersinia spp. (59, 63), Y. pestis is known to express a number of other adhesins, including Pla, Ail, YapC, YapE, and the pH 6 antigen (4, 9, 18, 26, 27, 29, 31, 46, 49). Similar to our results and consistent with redundant functions, deletion of any one adhesin has not been found to completely abolish binding to a given host cell type (18, 25–27, 29, 31, 46, 49). Studies on the functions of the multiple CU pathways present in the genomes of other bacterial pathogens have also found redundancy and have documented cross talk among the various pathways, such that loss of one pathway may be compensated for by expression of another (64, 70, 75). Thus, it may be necessary to delete multiple CU pathways to obtain a strong virulence phenotype and reveal the critical functions of the encoded pili during pathogenesis (64, 70).
In conclusion, we have identified roles for three of the additional Y. pestis CU pathways in binding to host cells and virulence. Our results support a model whereby loci y0348-0352 and y1858-1862 assemble adhesive pili that bind to a general receptor common to both epithelial cells and macrophages, whereas pili assembled by pathway y1869-1873 bind to a receptor expressed only by macrophages. The three CU pathways appear to make specific contributions during pneumonic plague, of which binding to macrophages conferred by pathway y1869-1873 may be particularly important. Adhesion mediated by the pili could aid in colonization of specific sites within the lung and facilitate the delivery of Yops into host cells by the Yersinia T3SS. Adhesion to mobile host cells such as macrophages could also enhance dissemination of Y. pestis from the lung to secondary sites of infection, promoting the development of systemic disease.
ACKNOWLEDGMENTS
We thank Steven Park, David S. Perlin, and the staff of the University of Medicine and Dentistry of New Jersey Regional Biocontainment Laboratory for performing the Y. pestis mouse infection experiments. We thank Susan Van Horn at the Central Microscopy Imaging Center, Stony Brook University, for help with electron microscopy. We thank Galina Romanov (Stony Brook University) for isolation of muBMDM. We thank Shan Lu (University of Massachusetts), Susan Straley (University of Kentucky), and Ralph Isberg (Tufts University) for providing the anti-Pla, anti-PsaA, and anti-Ail antibodies, respectively. We thank Eric Krukonis (University of Michigan) for helpful discussions and critical reading of the manuscript.
This study was supported by Public Health Service grant AI055621 from the National Institute of Allergy and Infectious Diseases (NIAID). M.H. was supported by grant T32 AI007539 from the NIAID. The Regional Biocontainment Laboratory is supported by Funding from the Northeast Biodefense Center U54-AI057158-Lipkin and the Northeast Biodefense Center Animal Core (Perlin).
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Achtman M, et al. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 101:17837–17842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Achtman M, et al. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96:14043–14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agar SL, et al. 2008. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology 154:1939–1948 [DOI] [PubMed] [Google Scholar]
- 4. Benedek O, Nagy G, Emody L. 2004. Intracellular signalling and cytoskeletal rearrangement involved in Yersinia pestis plasminogen activator (Pla) mediated HeLa cell invasion. Microb. Pathog. 37:47–54 [DOI] [PubMed] [Google Scholar]
- 5. Bergsbaken T, Cookson BT. 2009. Innate immune response during Yersinia infection: critical modulation of cell death mechanisms through phagocyte activation. J. Leukoc. Biol. 86:1153–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Black DS, Bliska JB. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37:515–527 [DOI] [PubMed] [Google Scholar]
- 7. Bliska JB. 2000. Yop effectors of Yersinia spp. and actin rearrangements. Trends Microbiol. 8:205–208 [DOI] [PubMed] [Google Scholar]
- 8. Bobrov AG, Kirillina O, Forman S, Mack D, Perry RD. 2008. Insights into Yersinia pestis biofilm development: topology and co-interaction of Hms inner membrane proteins involved in exopolysaccharide production. Environ. Microbiol. 10:1419–1432 [DOI] [PubMed] [Google Scholar]
- 9. Brubaker RR. 1991. Factors promoting acute and chronic diseases caused by yersiniae. Clin. Microbiol. Rev. 4:309–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brubaker RR. 1972. The genus Yersinia: biochemistry and genetics of virulence. Curr. Top. Microbiol. Immunol. 57:111–158 [DOI] [PubMed] [Google Scholar]
- 11. Bubeck SS, Dube PH. 2007. Yersinia pestis CO92 ΔyopH is a potent live, attenuated plague vaccine. Clin. Vaccine Immunol. 14:1235–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chaveroche MK, Ghigo JM, d'Enfert C. 2000. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28:E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chichester JA, et al. 2009. A single component two-valent LcrV-F1 vaccine protects non-human primates against pneumonic plague. Vaccine 27:3471–3474 [DOI] [PubMed] [Google Scholar]
- 14. Cole ST, Buchrieser C. 2001. Bacterial genomics: a plague o' both your hosts. Nature 413:.467:469–470 [DOI] [PubMed] [Google Scholar]
- 15. Cornelis GR. 2002. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat. Rev. Mol. Cell. Biol. 3:742–752 [DOI] [PubMed] [Google Scholar]
- 16. Cornelis GR, et al. 1998. The virulence plasmid of Yersinia, an antihost genome. Microbiol. Mol. Biol. Rev. 62:1315–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cornelius C, Quenee L, Anderson D, Schneewind O. 2007. Protective immunity against plague. Adv. Exp. Med. Biol. 603:415–424 [DOI] [PubMed] [Google Scholar]
- 18. Cowan C, Jones HA, Kaya YH, Perry RD, Straley SC. 2000. Invasion of epithelial cells by Yersinia pestis: evidence for a Y. pestis-specific invasin. Infect. Immun. 68:4523–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darby C. 2008. Uniquely insidious: Yersinia pestis biofilms. Trends Microbiol. 16:158–164 [DOI] [PubMed] [Google Scholar]
- 20. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng W, et al. 2002. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184:4601–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Derbise A, Lesic B, Dacheux D, Ghigo JM, Carniel E. 2003. A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol. Med. Microbiol. 38:113–116 [DOI] [PubMed] [Google Scholar]
- 23. Dodson KW, Jacob-Dubuisson F, Striker RT, Hultgren SJ. 1993. Outer membrane PapC usher discriminately recognizes periplasmic chaperone-pilus subunit complexes. Proc. Natl. Acad. Sci. U. S. A. 90:3670–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Du Y, Rosqvist R, Forsberg A. 2002. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect. Immun. 70:1453–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Felek S, et al. 2011. Contributions of chaperone/usher systems to cell binding, biofilm formation, and Yersinia pestis virulence. Microbiology 157:805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Felek S, Krukonis ES. 2009. The Yersinia pestis Ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77:825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Felek S, Lawrenz MB, Krukonis ES. 2008. The Yersinia pestis autotransporter YapC mediates host cell binding, autoaggregation and biofilm formation. Microbiology 154:1802–1812 [DOI] [PubMed] [Google Scholar]
- 28. Felek S, et al. 2010. Phosphoglucomutase of Yersinia pestis is required for autoaggregation and polymyxin B resistance. Infect. Immun. 78:1163–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Felek S, Tsang TM, Krukonis ES. 2010. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 78:4134–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Forestal CA, et al. 2008. A conserved and immunodominant lipoprotein of Francisella tularensis is proinflammatory but not essential for virulence. Microb. Pathog. 44:512–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forman S, et al. 2008. yadBC of Yersinia pestis, a new virulence determinant for bubonic plague. Infect. Immun. 76:578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forsberg A, Rosqvist R, Wolf-Watz H. 1994. Regulation and polarized transfer of the Yersinia outer proteins (Yops) involved in antiphagocytosis. Trends Microbiol. 2:14–19 [DOI] [PubMed] [Google Scholar]
- 33. Galvan EM, Chen H, Schifferli DM. 2007. The Psa fimbriae of Yersinia pestis interact with phosphatidylcholine on alveolar epithelial cells and pulmonary surfactant. Infect. Immun. 75:1272–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gong S, Bearden SW, Geoffroy VA, Fetherston JD, Perry RD. 2001. Characterization of the Yersinia pestis Yfu ABC inorganic iron transport system. Infect. Immun. 69:2829–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grabenstein JP, Fukuto HS, Palmer LE, Bliska JB. 2006. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect. Immun. 74:3727–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U. S. A. 87:4645–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hinnebusch BJ, Perry RD, Schwan TG. 1996. Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science 273:367–370 [DOI] [PubMed] [Google Scholar]
- 38. Hinnebusch BJ, et al. 2002. Role of Yersinia murine toxin in survival of Yersinia pestis in the midgut of the flea vector. Science 296:733–735 [DOI] [PubMed] [Google Scholar]
- 39. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 40. Huang XZ, Lindler LE. 2004. The pH 6 antigen is an antiphagocytic factor produced by Yersinia pestis independent of Yersinia outer proteins and capsule antigen. Infect. Immun. 72:7212–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hudson KJ, Bliska JB, Bouton AH. 2005. Distinct mechanisms of integrin binding by Yersinia pseudotuberculosis adhesins determine the phagocytic response of host macrophages. Cell Microbiol. 7:1474–1489 [DOI] [PubMed] [Google Scholar]
- 42. Kolodziejek AM, et al. 2007. Phenotypic characterization of OmpX, an Ail homologue of Yersinia pestis KIM. Microbiology 153:2941–2951 [DOI] [PubMed] [Google Scholar]
- 43. Laird WJ, Cavanaugh DC. 1980. Correlation of autoagglutination and virulence of yersiniae. J. Clin. Microbiol. 11:430–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786–17791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513 [DOI] [PubMed] [Google Scholar]
- 46. Lawrenz MB, Lenz JD, Miller VL. 2009. A novel autotransporter adhesin is required for efficient colonization during bubonic plague. Infect. Immun. 77:317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lindler LE, Klempner MS, Straley SC. 1990. Yersinia pestis pH 6 antigen: genetic, biochemical, and virulence characterization of a protein involved in the pathogenesis of bubonic plague. Infect. Immun. 58:2569–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindler LE, Tall BD. 1993. Yersinia pestis pH 6 antigen forms fimbriae and is induced by intracellular association with macrophages. Mol. Microbiol. 8:311–324 [DOI] [PubMed] [Google Scholar]
- 49. Liu F, Chen H, Galvan EM, Lasaro MA, Schifferli DM. 2006. Effects of Psa and F1 on the adhesive and invasive interactions of Yersinia pestis with human respiratory tract epithelial cells. Infect. Immun. 74:5636–5644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lukaszewski RA, et al. 2005. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect. Immun. 73:7142–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50a. Murphy KC. 1998. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 180:2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Toole GA, et al. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91–109 [DOI] [PubMed] [Google Scholar]
- 52. Parkhill J, et al. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413:523–527 [DOI] [PubMed] [Google Scholar]
- 53. Perry RD, Fetherston JD. 1997. Yersinia pestis: etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Phan G, et al. 2011. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 474:49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pizarro-Cerda J, Cossart P. 2006. Bacterial adhesion and entry into host cells. Cell 124:715–727 [DOI] [PubMed] [Google Scholar]
- 56. Pratt LA, Kolter R. 1998. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis, and type I pili. Mol. Microbiol. 30:285–293 [DOI] [PubMed] [Google Scholar]
- 57. Pujol C, Bliska JB. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 71:5892–5899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pujol C, Grabenstein JP, Perry RD, Bliska JB. 2005. Replication of Yersinia pestis in interferon gamma-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc. Natl. Acad. Sci. U. S. A. 102:12909–12914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rosqvist R, Skurnik M, Wolf-Watz H. 1988. Increased virulence of Yersinia pseudotuberculosis by two independent mutations. Nature 334:522–524 [DOI] [PubMed] [Google Scholar]
- 60. Runco LM, Myrczek S, Bliska JB, Thanassi DG. 2008. Biogenesis of the fraction 1 capsule and analysis of the ultrastructure of Yersinia pestis. J. Bacteriol. 190:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sebbane F, Jarrett C, Gardner D, Long D, Hinnebusch BJ. 2009. The Yersinia pestis caf1M1A1 fimbrial capsule operon promotes transmission by flea bite in a mouse model of bubonic plague. Infect. Immun. 77:1222–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. U. S. A. 103:5526–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62a. Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1:784–791 [Google Scholar]
- 63. Simonet M, Riot B, Fortineau N, Berche P. 1996. Invasin production by Yersinia pestis is abolished by insertion of an IS200-like element within the inv gene. Infect. Immun. 64:375–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spurbeck RR, et al. 2011. Fimbrial profiles predict virulence of uropathogenic Escherichia coli strains: contribution of Ygi and Yad fimbriae. Infect. Immun. 79:4753–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Straley SC, Bowmer WS. 1986. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect. Immun. 51:445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thanassi DG, Saulino ET, Hultgren SJ. 1998. The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr. Opin. Microbiol. 1:223–231 [DOI] [PubMed] [Google Scholar]
- 67. Titball RW, Williamson ED. 2001. Vaccination against bubonic and pneumonic plague. Vaccine 19:4175–4184 [DOI] [PubMed] [Google Scholar]
- 68. Une T, Brubaker RR. 1984. In vivo comparison of avirulent Vwa- and Pgm- or Pstr phenotypes of yersiniae. Infect. Immun. 43:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. 2001. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 98:6911–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. van der Velden AW, Baumler AJ, Tsolis RM, Heffron F. 1998. Multiple fimbrial adhesins are required for full virulence of Salmonella typhimurium in mice. Infect. Immun. 66:2803–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Waksman G, Hultgren SJ. 2009. Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat. Rev. Microbiol. 7:765–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang S, et al. 2008. Relative immunogenicity and protection potential of candidate Yersinia pestis antigens against lethal mucosal plague challenge in BALB/c mice. Vaccine 26:1664–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weening EH, et al. 2011. The dependence of the Yersinia pestis capsule on pathogenesis is influenced by the mouse background. Infect. Immun. 79:644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wimsatt J, Biggins DE. 2009. A review of plague persistence with special emphasis on fleas. J. Vector Borne Dis. 46:85–99 [PubMed] [Google Scholar]
- 75. Xia Y, Gally D, Forsman-Semb K, Uhlin BE. 2000. Regulatory cross-talk between adhesin operons in Escherichia coli: inhibition of type 1 fimbriae expression by the PapB protein. EMBO J. 19:1450–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang Y, Merriam JJ, Mueller JP, Isberg RR. 1996. The psa locus is responsible for thermoinducible binding of Yersinia pseudotuberculosis to cultured cells. Infect. Immun. 64:2483–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zav'yalov V, Zavialov A, Zav'yalova G, Korpela T. 2010. Adhesive organelles of Gram-negative pathogens assembled with the classical chaperone/usher machinery: structure and function from a clinical standpoint. FEMS Microbiol. Rev. 34:317–378 [DOI] [PubMed] [Google Scholar]