

Abstract
The increased use of medical implants has resulted in a concomitant rise in device-related infections. The majority of these infections are caused by Staphylococcus epidermidis biofilms. Immunoprophylaxis and immunotherapy targeting in vivo-expressed, biofilm-associated, bacterial cell surface-exposed proteins are promising new approaches to prevent and treat biofilm-related infections, respectively. Using an in silico procedure, we identified 64 proteins that are predicted to be S. epidermidis surface exposed (Ses), of which 36 were annotated as (conserved) hypothetical. Of these 36 proteins, 5 proteins—3 LPXTG motif-containing proteins (SesL, SesB, and SesC) and 2 of the largest ABC transporters (SesK and SesM)—were selected for evaluation as vaccine candidates. This choice was based on protein size, number of antigenic determinants, or the established role in S. epidermidis biofilm formation of the protein family to which the candidate protein belongs. Anti-SesC antibodies exhibited the greatest inhibitory effect on S. epidermidis biofilm formation in vitro and on colonization and infection in a mouse jugular vein catheter infection model that includes biofilms and organ infections. Active vaccination with a recombinant truncated SesC inhibited S. epidermidis biofilm formation in a rat model of subcutaneous foreign body infection. Antibodies to SesC were shown to be opsonic by an in vitro opsonophagocytosis assay. We conclude that SesC is a promising target for antibody mediated strategies against S. epidermidis biofilm formation.
INTRODUCTION
Staphylococcus epidermidis is considered to be the major cause of device-related infections, especially catheter-related infections. These infections have increased in number, owing to the increased use of such devices (22). The ability to form biofilms on medical implant surfaces is the main virulence factor of S. epidermidis (25). Biofilms are notoriously resistant to both immune and antimicrobial agents (7, 31). Currently, the only completely effective method for curing biofilm infections is to remove the infected device, which is a risky, costly, and stressful procedure.
Different strategies are used against biofilm infections (20). The traditional approach to prevent biofilm formation is administration of bactericidal agents to the patient or the biomaterial (9). Other frequently utilized options involve the modification of biomaterial surface to prevent initiation of bacterial colonization (15, 16, 36, 40). However, these strategies have their disadvantages. There is the ineffectiveness of traditional antibacterial compounds due to the nature of biofilms and high prevalence of antimicrobial resistance, there are the induction, generation, and selection of resistance by the slow release of subinhibitory concentrations of antimicrobials from biomaterials, and there are the problems linked to biochemical and chemical compatibility, increased cost, short time effect, effect on mechanical properties, and cytotoxicity (31, 41).
Immunoprophylaxis and immunotherapy targeting in vivo expressed biofilm-related proteins and cell surface components are promising new approaches for the prevention and treatment of biofilms. Most vaccines now available for human use are whole (killed or attenuated) microorganisms or subunit vaccines. S. epidermidis is a ubiquitous colonizer of human skin, and prior staphylococcal infections do not cause immunological protection (37). However, this does not imply that immunoprophylaxis and immunotherapy against S. epidermidis biofilms and infections would not be possible.
Several recent studies have shown that antibodies against cell surface components of S. epidermidis can affect the rate of biofilm formation or adherence of these bacteria to medical devices in vitro. Cerca et al. (5) showed that antibodies against polysaccharide intercellular adhesion (PIA), which is identical to poly-N-acetylglucosamine (PNAG), readily penetrated the S. epidermidis biofilm and bound to the sessile cells. Sessile bacteria nevertheless exhibited more resistance to opsonic killing than their planktonic counterparts. Using polyclonal antibodies against a fibrinogen-binding protein from S. epidermidis (Fbe), Pei et al. (23) could block adherence of S. epidermidis to fibrogen-coated catheters in vitro. Sun et al. (32) showed that monoclonal antibodies against accumulation-associated protein (Aap) can significantly reduce the accumulation but not initiation phase of S. epidermidis biofilm formation in vitro. Maira-Litran et al. (19) showed that vaccination of rats with purified PIA/PNAG can elicit protective immunity against both CoNS and S. aureus. Hence, surface-expressed components including S. epidermidis surface-exposed “Ses” proteins (4, 12), PIA, and teichoic acids (13, 27) are potential targets for vaccine development.
In the present study, we identified potential surface-exposed proteins of S. epidermidis and investigated the potential use of rabbit polyclonal antibodies raised against five “Ses” proteins and against whole (killed) microorganisms for eradication of S. epidermidis biofilms in vitro. For the most immunogenic protein, SesC, we also tested the effect of immunization with recombinant antigen (active immunization) on in vivo biofilm formation and investigated the immunological effector function of specific rabbit polyclonal anti-SesC IgGs (αSesC-IgGs). This was done by challenging animals in a newly developed central venous catheter murine model with S. epidermidis bacteria preincubated with αSesC-IgGs and by performing an in vitro opsonophagocytosis assay.
MATERIALS AND METHODS
In silico selection of Ses proteins.
The complete sequence of S. epidermidis ATCC 12228 (42) was retrieved from the National Centre of Biotechnology Information (NCBI) GenBank (http://www.ncbi.nlm.nih.gov/GenBank/). N-terminal signal peptides and transmembrane domains in S. epidermidis proteins were predicted with SignalP and TMHMM (http://www.cbs.dtu.dk/services/). Retention domain prediction lipobox motifs, peptidoglycan-binding domains, choline-binding domains, and LPXTG motives were predicted using the PATTINPROT server (http://npsa-pbil.ibcp.fr/) (39). The prediction of protein subcellular localization was reanalyzed using the online tool PSORTb v.2.0.4 (http://www.psort.org/psortb/). The sequences of all identified Ses proteins were subjected to antigenicity analysis using the “Predicting Antigenic Peptides” server (http://imed.med.ucm.es/Tools/antigenic.pl).
Bacterial strains, plasmids, primers, and media.
For biofilm inhibition studies, S. epidermidis strain 10b, which is a strong (PIA-dependent) biofilm-forming strain (38) isolated from a patient with a proven catheter-related infection, was used. For recombinant protein production and PCR screening of isolates, the sequences of the selected ses genes were retrieved via the NCBI GenBank from the complete genome of the non-biofilm-forming S. epidermidis strain ATCC 12228. On the basis of these sequences, all primers were designed and purchased from Eurogentec (Seraing, Belgium). Primers used in the present study are listed in Table 1. Each ses gene was PCR-amplified using genomic DNA isolated from strain 10b as a template and sequenced. For recombinant protein production, amplicons were cloned in pET11c (Stratagene, La Jolla, CA). The recombinant plasmids were transformed into Escherichia coli BL21(DE3). S. epidermidis was grown in brain heart infusion broth (BHI; Oxoid) and E. coli was grown in Luria-Bertani medium supplemented with 100 μg of ampicillin/ml when it was transformed with plasmids. Solid medium consisted of the corresponding liquid medium supplemented with 1 to 2% agar.
Table 1.
Primers used in this study
| Primer | Sequence (5′–3′)a | Useb |
|---|---|---|
| sesCF1 | GTTGATAACCGTCAACAAGG | A |
| sesCR1 | CATGTTGATCTTTTGAATCCC | A |
| sesLF1 | TGGGCCACTCAATACAGTCA | A |
| sesLR1 | TTGGCGTGTTTCTGTCTTTG | A |
| sesMF1 | CAGGTGCCTTGGAATCGC | A |
| sesMR1 | GCGTACCTTGCCAGTAGTC | A |
| sesKF1 | CCAATTACTAGTATTAAATTCAG | A |
| sesKR1 | CTACACTGTTAGACGTGAG | A |
| sesBF1 | GCTATGAAAAATAGTGGTGGC | A |
| sesBR1 | CGTAGTATGAATTGAGCTCAC | A |
| sesCF2 | ACGTGCTAGCGCAGATTCAGAAAGTACATC | B |
| sesCR2 | GAACAGCTACAGCTGATCATCACCATCACCATCACTAGGATCCGCAT | B |
| sesLF2 | CACGTGCTAGCCATCACCATCACCATCACAAAACGCAAGATGAAGCGAAA | B |
| sesLR2 | GGAACTCAAATTATTTATTAAGGATCCGCAT | B |
| sesMF2 | ACTGGCTAGCCATCACCATCACCATCACGGGGGCACCTCAAGTACAG | B |
| sesMR2 | GTTACACCAGAATCTATCTATTAGGATCCGCAT | B |
| sesKF2 | CACGTGCTAGCGCTGAATCAAACACTTCAGTTTCTTCT | B |
| sesKR2 | CTATTACCAAATACAGGTATGCATCACCATCACCATCACTAGGATCCGCAT | B |
| sesBF2 | ACGTGCTAGCGCAGCCGAAGTAACATCTC | B |
| sesBR2 | CTCAATTCATACTACGTAGGTCATCACCATCACCATCACTAGGATCCGCAT | B |
Flanking restriction sites are underlined and in italics, and sequences coding for an N- or C-terminal His6 tag are indicated in boldface.
A, used for PCR screening of ses genes in clinical and commensal isolates; B, used for cloning ses genes in pET11c.
Bacterial isolates and species identification.
A total of 76 S. epidermidis clinical and commensal isolates from hospitalized patients (n = 60), the skin of healthy individuals (n = 11), and five previously described strains (strains 10b and 1457 [18], ATCC 35984/RP62A [12], and ATCC 12228 and TU3298 [1]) were collected. Clinical isolates were recovered from blood cultures of neonates (n = 45) with late-onset sepsis and an intravascular catheter in place at the neonatal intensive care unit of the Erasmus MC–Sophia Children's Hospital in Rotterdam, Netherlands, and from different clinical specimens from patients (n = 15) hospitalized at University Hospital Gasthuisberg (Katholieke Universiteit Leuven) in Leuven, Belgium. Species identification was performed with Vitek 2 (bioMérieux).
PCR screening of ses genes in clinical and commensal isolates.
We performed a duplex PCR, amplifying both a ses gene and the 16S rRNA gene. The primers used for PCR screening of ses genes were sesR1 and sesF1 sets, and primers for the 16S rRNA gene were previously described (35). For each strain, genomic DNA was extracted using a QIAamp DNA minikit (Qiagen) with the addition of 30 μg of lysostaphin/ml at the lysis step.
Cloning, expression, and purification of histidine-tagged fusion proteins.
Recombinant extracellular domains of the Ses proteins were expressed with hexahistidine tags at their N or C termini using the expression vector pET11c (Stratagene, La Jolla, CA). Each ses gene fragment was PCR amplified using reverse and forward (sesF2 and sesR2 set) primers incorporating flanking NheI and BamHI restriction sites and a sequence coding for a N or C-terminal His6 tag. Amplified products were cloned in NheI- and BamHI-digested pET11c, and the resulting plasmids were transformed into E. coli BL21(DE3). Pure plasmid DNA was isolated using a High Pure plasmid isolation kit (Roche Diagnostics), and PCR amplification and sequencing were used for verification. The recombinant plasmids were used for recombinant protein production as previously described in detail (28).
Preparation and purification of polyclonal anti-rSes IgG antibodies.
Polyclonal antibodies were produced by Eurogentec by immunization of rabbits with purified rSes proteins according to standard immunization protocols. In addition, ethanol (80%)-killed S. epidermidis ATCC 12228 was used as whole-cell preparation to raise serum against the complete surface of S. epidermidis. Briefly, specific-pathogen-free rabbits were immunized with 100 μg of rSes proteins or killed bacteria, and boosters were given at 14, 28, and 56 days after the first immunization. Preimmune serum was taken before the first immunization. After 87 days, the total blood of the rabbit was collected, and the serum was stored at −20°C.
Total IgGs from pre- and postimmune sera were purified by absorption to a protein G column (GE Healthcare) according to the manufacturer's instructions. Only for SesC were specific rabbit polyclonal IgGs (αSesC-IgGs) further purified using antigen affinity purification, as previously described in detail (28). The purity and reactivity of the purified IgGs against their respective recombinant proteins and ethanol killed cells were determined by Coomassie blue staining of SDS-PAGE electrophoresis gels, enzyme-linked immunosorbent assay (ELISA), and Western blot according to standard protocols. To evaluate the presence of the respective proteins on the surface of S. epidermidis, an indirect ELISA with the pre- and postimmune sera on whole-cell and lysed S. epidermidis ATCC 12228 was performed. To obtain whole-cell S. epidermidis, the sediment of an overnight culture was resuspended in 1 ml of 0.9% NaCl and heated for 30 min at 56°C. For lysed cells, the sediment of an overnight culture was resuspended in 1 ml of 0.9% NaCl with 100 μg of lysostaphin (Sigma-Aldrich)/ml. Subsequently, the suspension was incubated at 37°C for 4 h in rotating tubes. For coating ELISA plates, the suspensions were diluted to 107 CFU/ml in 0.9% NaCl. Hereafter, 100 μl of the dilution was applied to each well on the plate, effectively coating each well with 106 CFU. Coated plates were incubated overnight at 4°C before the test was performed according to standard protocols (3). The test was considered positive when the optical density at 405 nm (OD405) of postimmune sera was at least twice as high as the OD405 of preimmune sera after subtraction of the average OD405 of the blank wells.
In vitro biofilm inhibition assays.
Using a semiquantitative microtiter plate method (6, 32), the effect of pre- and postimmune IgGs against rSes proteins on in vitro biofilm formation during the first 2 h (primary attachment) and overnight (accumulation and establishment phase) was studied. For quantification of biofilms, the following procedure was used. A 20-μl aliquot of frozen cultures of S. epidermidis strains 10b was inoculated into 5 ml of BHI and grown to the late-exponential/stationary-growth phase in a shaking incubator at 37°C. Cultures were subsequently diluted in BHI to an OD600 of 0.005 (5 × 106 CFU/ml) in fresh BHI.
To evaluate the effect of IgGs on the primary attachment of S. epidermidis strain 10b, these starting cultures with an OD600 of 0.005 were grown at 37°C to an OD600 of 1. Cultures were subsequently mixed with either pre- or postimmune IgGs (10 μg/ml) and, after 2 h of incubation at 4°C, 200-μl portions of the mixtures were pipetted into 96-well polystyrene microtiter plates (BD Biosciences, Heidelberg, Germany), followed by incubation for 2 h at 37°C without shaking.
To study the effect of IgGs on overnight biofilm formation, the cultures diluted to an OD600 of 0.005 were mixed with either pre- or postimmune IgGs (10 μg/ml) and, after 2 h of incubation at 4°C, 200 μl of the mixtures (106 cell per well) was added to each well of 96-well polystyrene microtiter plates, followed by incubation overnight at 37°C without shaking.
After the incubation, the plates were washed three times with phosphate-buffered saline (PBS [pH 6.8], containing 0.5 M NaCl and 10 mM EDTA), and adherent biofilms were stained with 200 μl of 1% (wt/vol) crystal violet (Sigma) for 10 min, after which the plates were washed three times with water and dried. For quantification, 160 μl of 30% (vol/vol) acetic acid was added to each well to dissolve the stain. The OD595 of the dissolved stain was measured in a multipurpose UV/VIS plate reader. The percent inhibition of biofilm formation was calculated by using the following formula: (A595, positive − A595, antibody)/(A595, positive − A595, negative) × 100 (28, 32). The average inhibition for each pre- or postimmune IgG was obtained from at least eight independent measurements generated in at least two independent experiments. S. epidermidis strain 10b in BHI without any added IgG was used as positive control, and BHI without bacteria was used as a negative control.
Biofilm formation in immunized rats.
For the vaccination experiments, ex-germ-free Fisher (EGF) rats (n = 6 per experiment) were divided into two groups of three rats each. Each rat in the first group was immunized twice intraperitoneally with first 100 μg of rSesC (dissolved in normal saline) in complete Freund adjuvant (CFA) and 2 weeks later a second time with 50 μg of rSesC (dissolved in normal saline) in incomplete Freund adjuvant (IFA). The second group of rats was injected with the same volume of normal saline in CFA and IFA but without antigen. Two weeks after the second immunization, the immune response of pre- and postimmunization sera of all rats was tested by ELISA. Subsequently, five catheter (Arrow International, Reading, PA) fragments preincubated with S. epidermidis 10b bacteria (∼104 cells/catheter) were implanted in each rat and 24 h later explanted as previously described (28), and the numbers of sessile cells were quantified by CFU counting as previously described (24). Briefly, after gentle cleaning with 0.9% NaCl, the catheters were placed in a tube containing 1 ml of 0.9% NaCl. Tubes were vortex mixed for 10 s, sonicated for 10 min at 40 kHz in a water bath (Branson 2200; Branson Ultrasonics), and vortex mixed again for 10 s. Thereafter, tube contents were diluted and 50-μl aliquots of 10-fold dilutions were plated on tryptone soy agar (TSA; Oxoid) plates, using a spiral plating system, and the plates were incubated at 37°C overnight. Colonies on all plates were counted, and the number of bacteria was defined as the mean of at least five quantitative cultures. All animal experiments were repeated at least twice and conducted in compliance with the guidelines for animal experimentation. All experimental protocols were approved by the Institutional Animal Care Commission and Ethical Committee of the KU Leuven.
Biofilm formation in jugular vein-catheterized (JVC) mouse model.
In order to investigate the effector function of αSesC-IgGs, we developed and used a previously described central venous catheter murine model (17, 21) that reflects the clinical situation of catheter colonization by contaminated infusions. Briefly, 4-weeks-old Swiss-Webster mice from Taconic (n = 9 mice per experiment, with the experiment repeated twice) were divided into three groups of three mice each. Mice were anesthetized with sodium pentobarbital (nembutal, 40 to 60 mg/kg [body wt]) injected intraperitoneally. The animal was then placed in dorsal recumbency under a dissecting microscope. A small vertical incision was made using a scalpel, and the right jugular vein was identified, mobilized, and exposed with blunt surgical dissection. A single lumen polyethylene catheter (internal diameter, 28 mm; outer diameter, 61 mm; length, 6 cm; insertion length, 1 cm; Intramedic [Becton Dickinson, catalog no. 427400]) was inserted into the right jugular vein and advanced into the superior vena cava via a small incision in the vein made with vein scissors. A ligature was then tied loosely around the catheter, and patency was verified. Once blood flow had been established, the catheter was anchored in place and flushed with 10 μl of saline. Subsequently, a small midline skin incision was made between the scapulae. The catheter port was tunneled back through the scapular incision. The incisions were then closed with stitches. Mice were housed separately and monitored for recovery. An overnight culture of S. epidermidis strain 10b, grown to the late exponential/stationary growth phase in BHI, was pelleted, resuspended, and diluted to an OD600 of 0.3 (∼3E+08) in 0.9% NaCl. Three inoculums were taken, one without any IgG, while two others were mixed with either preimmune or αSesC-IgGs (80 μg/ml) and incubated for 2 h at 4°C. After a 24-h recovery from surgery, 150 μl (∼5E+07) of the mixture was injected through the catheter lumen. Animals in the first, second, and third groups received bacteria, bacteria preincubated with preimmune IgGs, and bacteria preincubated with αSesC-IgGs, respectively. After the blood samples were obtained at day 5 postinfection, the animals were sacrificed. The catheters were aseptically removed, and the portion (1 cm) that was inside the vein was cut, gently washed, and processed for quantitative culturing as explained above. To compare the overall infection rate, the spleens, livers, hearts, veins, and right kidneys were aseptically harvested, mechanically homogenized in 0.9% NaCl, and processed for quantitative cultures as explained above.
Opsonophagocytosis of planktonic and biofilm bacteria by αSesC-IgGs.
To prepare the bacteria for the evaluation of susceptibility to opsonic killing, the overnight culture of S. epidermidis 10b in BHI was pelleted for 5 min at 12,000 × g at 4°C, washed twice with PBS, and subsequently diluted to an OD600 of 1. For evaluation of opsonophagocytosis of planktonic cells, 10 μl of bacterial suspension was used, and for evaluation of opsonophagocytosis of biofilm cells, 5-mm catheter (Arrow International) fragments were added to the bacterial suspension, incubated at 37°C for 2 h, and subsequently washed with 1 ml of PBS. The opsonophagocytosis assay was performed with fresh blood obtained from human healthy volunteers as previously described (29), with some modifications. Briefly, fresh whole blood from two volunteers was collected in vacuum blood collection tubes containing the anticoagulant heparin and then aliquoted into 1.5-ml microcentrifuge tubes (0.5 ml/tube). Catheter fragments preincubated with S. epidermidis strain 10b or planktonic bacteria (10 μl of bacterial suspension with an OD600 of 1) after 1 h preincubation at 4°C with preimmune or αSesC-IgGs (30 μg/ml) or the same volume of PBS or nothing were added to the 1.5-ml microcentrifuge tubes containing 0.5 ml of fresh blood. Tubes were incubated at room temperature with gentle rocking and, after 30 min, the samples containing planktonic bacteria were serially diluted and plated onto TSA plates to determine the number of surviving CFU. For samples containing catheter fragments, catheter fragments were first removed and gently washed with 1 ml of saline and then processed for quantification of the number of surviving cells on the catheter fragments as explained above. In order to determine the input CFU in samples containing planktonic or sessile bacteria attached to the catheter fragments, a portion the of untreated planktonic samples was serially diluted and plated onto TSA plates, or the number of bacteria attached to the untreated catheter fragments was quantified as explained above. All samples were assayed in triplicate, and all experiments were repeated at least twice.
Statistical analysis.
In the present study, for all of the in vitro and in vivo experiments, data were pooled from two independent experiments. For all data from the bacterial adherence and opsonophagocytosis assays, two hypotheses were tested. A significant change in the adherence levels or survival rates for different IgGs (preimmune or anti-Ses IgGs) was tested with a one-way analysis of variance (ANOVA). A significant difference in adherence for an experiment set including pre- and postimmune IgGs from a particular rabbit was tested with a two-way ANOVA. When the one-way ANOVA was significant, two-sided univariate tests with a correction for multiple comparisons were performed (Bonferroni test) to locate the significant differences.
For in vivo experiments, differences between the control and vaccine groups or between groups injected with bacteria treated with preimmune or αSesC-IgGs were examined between the number of bacteria recovered from the catheter fragments (in both in vivo models) or from the organs (only JVC model) using one-way ANOVA. A P value of <0.05 was considered significant.
RESULTS
Selection of Ses proteins.
In order to identify the potential Ses proteins we used our in silico selection procedure summarized in the diagram shown in Fig. 1. In total, from the 2,419 predicted S. epidermidis proteins, 64 proteins were identified as Ses proteins. Of these, 36 were (conserved) proteins with a hypothetical function (see Table S1 in the supplemental material). LPXTG motif-containing and ABC transporter lipoproteins are two major types of cell surface proteins that may play important roles in the pathogenesis of S. epidermidis infections (11, 26). We selected three predicted LPXTG motif-containing proteins and two predicted lipobox-containing ABC transporter proteins of which the function had not yet been characterized. These were based on the protein size, the number of antigenic determinants and the importance of the protein family, to which the candidate protein belongs, in S. epidermidis biofilm formation and pathogenesis (Table 2).
Fig 1.

In silico selection of genes coding for “surface-exposed proteins” of S. epidermidis. SP, signal peptide; TM, transmembrane helix; PBD, peptidoglycan-binding domain; CBD, choline-binding domains.
Table 2.
Ses proteins selected by in silico procedure and used in this study
| Locus | Putative product | Protein accession no. | Protein size (no. of amino acids) | Motif | No. of antigenic determinants |
|---|---|---|---|---|---|
| SE2232 | Conserved hypothetical protein (SesC) | NP_765787.1 | 676 | LPXTG | 20 |
| SE1106 | ABC transporter membrane-spanning protein (SesL) | NP_764661.1 | 564 | Lipobox | 16 |
| SE1981 | Nickel ABC transporter/nickel-binding protein (SesM) | NP_765536.1 | 491 | Lipobox | 18 |
| SE1501 | Hypothetical protein (SesK) | NP_765056.1 | 415 | LPXTG | 11 |
| SE2152 | Hypothetical protein (SesB) | NP_765707.1 | 196 | LPXTG | 7 |
Presence of ses genes in S. epidermidis isolates.
We hypothesized that the ideal targets for immunoprophylaxis or immunotherapy against S. epidermidis biofilms would be surface components that were conserved across the species, in particular those which are highly expressed in the bloodstream and in biofilms, with a possible role in biofilm formation or an essential function. To verify the conserved nature of the selected genes, we investigated the distribution of ses genes in clinical and commensal isolates of S. epidermidis.
Gene-specific PCR amplification of sesM, sesB, and sesC was positive in all tested S. epidermidis isolates (n = 76); for sesL this was 68% (n = 52 of 76 isolates), and for sesK this was 9% (n = 7 of 76 isolates). There was no significant difference in the prevalence of the sesL and sesK genes in clinical or commensal isolates. We also studied the in vitro and in vivo expression of five selected ses genes in planktonic and sessile bacteria, as previously explained (28). Gene expression studies showed that the selected ses genes are expressed at the level of the transcriptome in both planktonic and sessile bacteria in vitro and in vivo. However, the in vitro and in vivo expression patterns of ses genes in planktonic and biofilm cells varied widely (data not shown).
Recognition of Ses proteins by polyclonal antibodies.
The purity and reactivity of the purified IgGs against their respective recombinant proteins and ethanol-killed cells were determined by Coomassie blue staining of SDS-PAGE gels, ELISA, and Western blotting according to standard protocols. Figure 2 shows examples of such evaluations. To evaluate the recognition of selected Ses proteins by the antisera and their expression on the surface, we performed Western blots and ELISAs on purified recombinant proteins and S. epidermidis ATCC 12228 lysate for each anti-Ses serum (Table 3). For antiserum against SesC, SesL, and SesB, both Western blotting and ELISA were positive against the respective recombinant protein and bacterial lysate. ELISA against whole-cell S. epidermidis ATCC 12228 was also positive for these antisera, as was whole-cell antiserum against these three recombinant proteins. For antiserum against SesM, Western blotting and ELISA against SesM recombinant protein and S. epidermidis lysate were positive, but ELISA against whole-cell S. epidermidis was negative. The latter was also true for antiserum against SesK, but additionally, Western blot results against whole-cell S. epidermidis for this antiserum and whole-cell antiserum against rSesK were negative as well. These data suggest that the selected proteins are surface exposed except possibly for SesK and SesM, which are not expressed under the chosen culture conditions for S. epidermidis.
Fig 2.

(A) Coomassie-stained SDS-PAGE gel (12%) of rSes. Lane 1, SDS-PAGE marker; lane 2, total E. coli extract after induction of SesC expression and cell lysis; lane 3, protein extract after binding the His-tagged SesC to the nickel column; lane 4, protein extract after washing the nickel column; lane 5, SesC recombinant protein after elution; lane 6, negative control. (B) Affinity of preimmune (□) and αSesC-IgGs (■) to rSesC. An indirect ELISA was performed using a 96-well ELISA plate coated with rSesC. IgGs were added to each well and incubated for 3 h at 37°C. Bound IgGs were measured at OD405 with an alkaline phosphatase-conjugated anti-rabbit immunoglobulin. x and y axes indicate the log10 IgG concentration (ng/ml) and the OD405 absorbance, respectively.
Table 3.
Western blot and ELISA data obtained using immune sera against recombinant Ses proteins and whole-cell S. epidermidis ATCC 12228a
| Antiserum target | On respective rSes protein |
On S. epidermidis lysate |
On WC S. epidermidis (ELISA) | ||
|---|---|---|---|---|---|
| WB | ELISA | WB | ELISA | ||
| SesC | + | + | + | + | + |
| SesL | + | + | + | + | + |
| SesM | + | + | + | + | – |
| SesK | + | + | – | + | – |
| SesB | + | + | + | + | + |
| WC | +* | + | + | + | + |
WB, Western blot; WC, whole cell.
, Western blot with whole-cell antiserum on recombinant proteins was positive for all proteins except SesK.
Biofilm inhibition by purified polyclonal anti-rSes-IgGs in vitro.
Of the five antibodies, the maximum inhibition of initial attachment and overnight biofilm formation of the bacteria on polystyrene surfaces (Fig. 3A and B), observed in these in vitro experiments, occurred when bacteria were preincubated with IgGs purified from the serum of the rabbit immunized with rSesC. This reduction for overnight biofilm formation was however not statistically significant (P < 0.01 and P > 0.05, one- and two-way ANOVAs).
Fig 3.

Effect of total IgGs purified from preimmune (□) and anti-Ses (L, K, M, B, and C) and anti-whole-cell (WC) sera (■) on primary attachment (A) and overnight biofilm formation (B) of S. epidermidis strain 10b on a polystyrene surface. Overnight cultures were diluted, mixed with IgGs, incubated for 2 h at 4°C, and then transferred into the wells. After 2 h (A) or 14 h (B) of incubation at 37°C, the plates were washed and stained with crystal violet, and the OD595 was measured. The error bars indicate the standard deviations. The data are the averages of eight measurements in two independent experiments. Biofilm inhibition was as defined in the text.
Effect of immunization with rSesC on biofilm formation.
In order to test the potential of SesC as a target for vaccination against S. epidermidis biofilms, rats were actively immunized with rSesC. The number of sessile bacteria attached to the catheter fragments 24 h after implantation in the group immunized with rSesC compared to the normal saline-immunized group was significantly reduced (20-fold; P < 0.01; one-way ANOVA) (Fig. 4).
Fig 4.
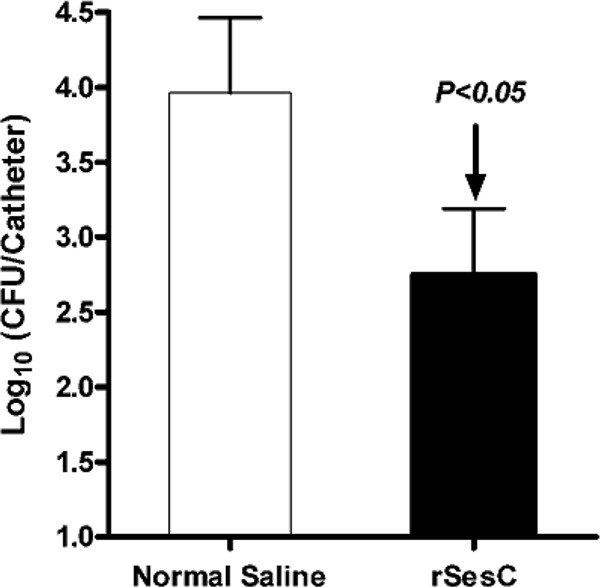
Effect of immunization of EGF rats with rSesC on S. epidermidis 10b biofilm formation. Rats (n = 6) were divided into two groups of three rats each. Rats in the vaccine group were immunized twice intraperitoneally with rSesC. Rats in the control group were injected with the same volume of normal saline. At 2 weeks after the second immunization, the immune response was tested by ELISA. Subsequently, catheter fragments preincubated with S. epidermidis strain 10b (∼104 cells/catheter) were implanted in rats and 24 h later explanted, and the numbers of adherent bacteria on the catheters were quantified. The experiment was repeated twice. Data for each group were obtained from adherent bacteria to 30 catheter fragments implanted in six rats (five catheters per animal, three rats per group in each experiment, two groups, two independent experiments). The error bars indicate the standard errors of the mean.
Effect of αSesC-IgGs on the overall infection rate in the JVC mouse model.
To investigate the effector function of αSesC-IgG antibodies in vivo, the mean numbers of bacteria recovered from the blood, catheters, veins, livers, spleens, hearts, and kidneys of animals in different groups were compared. Preincubation of bacteria with αSesC-IgGs could significantly reduce the number of bacteria recovered from the catheter, vein, spleen, heart, liver, and kidney by 26-, 71.5-, 331-, 327-, 215-, and 52-fold, respectively (P < 0.01, one-way ANOVA), whereas preincubation of bacteria with preimmune IgGs had no effect on the infection rates (Fig. 5). Almost no bacteria were found in the circulation.
Fig 5.

Effect of preincubation of S epidermidis strain 10b with preimmune or αSesC-IgGs on the overall infection rates in the JVC mouse model. A single lumen polyethylene catheter was surgically inserted into the right jugular vein of Swiss-Webster mice. After 24 h, an overnight culture of S. epidermidis strain 10b (∼3E+08) in 0.9% NaCl was mixed with preimmune or αSesC-IgGs (80 μg/ml) or the same volume of PBS, followed by incubation for 2 h at 4°C. After a 24-h recovery from surgery, 150 μl (∼5E+07) of the mixture was injected through the catheter lumen. After 5 days, the overall infection rates in different organs and the catheter colonization were quantified. Data for each group were obtained from bacteria recovered from the catheter or organs of six infected mice (three mice per group in each experiment, three groups, two independent experiments). The error bars indicate the standard errors of the mean.
αSesC-IgGs are opsonic and mediate opsonophagocytosis of planktonic and biofilm cells.
Incubation of bacteria in the planktonic or sessile state with whole blood for 30 min led to a significant reduction (>1 log10) in PBS-treated versus input bacteria (P < 0.001) (Fig. 6). Preincubation of bacteria in both planktonic and sessile forms with αSesC-IgGs significantly enhanced the opsonophagocytic killing of bacteria (P < 0.001, one-way ANOVA) compared to bacteria treated with preimmune IgGs or PBS (Fig. 6). There was no difference between bacteria treated with preimmune IgGs and PBS. αSesC-IgGs showed the same level of enhancement of opsonophagocytic killing of bacteria for both planktonic and sessile bacteria.
Fig 6.
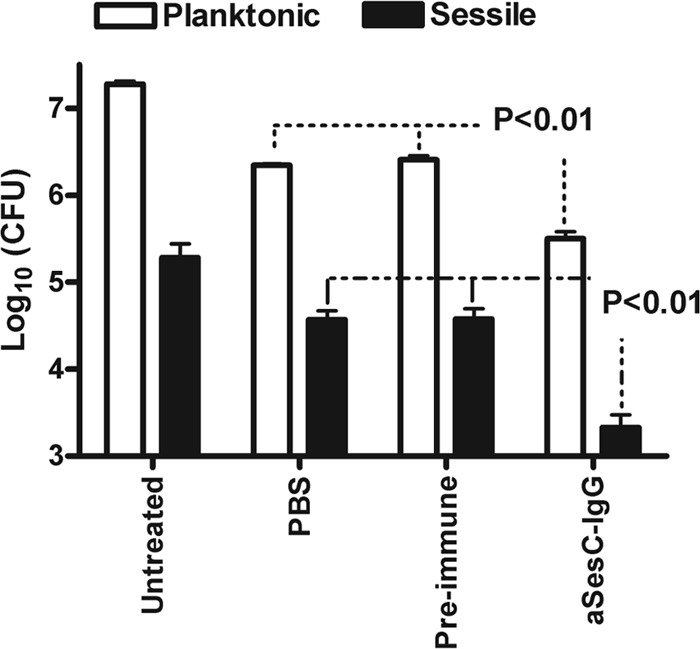
Effect of preincubation of S epidermidis strain 10b with αSesC-IgGs on phagocytosis of planktonic (□) and sessile (■) bacteria by human neutrophils. Bacteria grown in planktonic form or attached to catheter fragments were tested for their ability to survive in human blood after preincubation with preimmune IgGs or αSesC-IgGs. Surviving bacteria were measured by viable counting. The data are presented as the mean log10 of surviving CFU. The data for each group are the average of six measurements in two independent experiments. The error bars represent the standard deviation of the measurements.
DISCUSSION
In this study we tried to identify new potential target(s) for immunoprophylaxis and immunotherapy against S. epidermidis biofilm formation. Using our in silico selection method, we first identified potential Ses proteins. Nine of these proteins were proteins with an LPXTG motif (4, 12). Five LPXTG motif-containing proteins (Aap, Bhp, SdrF, SdrG, and SesI) are known to play important roles in the pathogenesis of S. epidermidis infections (2, 8, 10, 14, 30). In publicly available genomes of S. epidermidis strains RP62A and ATCC 12228, respectively, 11 and 10 genes encoding LPXTG motif-containing proteins have been identified (4), including those already mentioned. Except for the five LPXTG motif-containing proteins mentioned above, the role of other LPXTG motif-containing proteins has not been studied yet. The second major type of cell surface proteins in Gram-positive bacteria are lipoproteins that participate in a wide range of cell envelope functions (33). The major functional category of lipoproteins are the solute-binding proteins of ABC transport systems for the import of a diverse range of substrates (34). Due to the importance of these two families of proteins, five proteins with unknown function, i.e., three hypothetical LPXTG motif-containing proteins (SesC, SesK, and SesB) and two ABC transporters (SesL and SesM), were selected for further studies. This selection was also based on the protein size and the number of antigenic determinants.
We showed by Western blotting that the recombinant proteins of the selected proteins except for SesK were recognized by whole-cell antisera, suggesting these proteins, except possibly for SesK, are most likely surface exposed. Anti-SesK and anti-SesM antisera did not recognize whole cells, suggesting that SesK and SesM are not (sufficiently) expressed under the culture conditions chosen. In addition, sesK is in only present in <10% of tested isolates.
Total IgGs isolated from the hyperimmune sera of rabbits immunized with rSesC showed the highest inhibition effect on primary attachment and biofilm formation in vitro, indicating a possible role for this protein in S. epidermidis biofilm formation. The lower effect of IgGs isolated from the sera of animals immunized with other Ses proteins or whole cells can be due to either a low expression level of selected Ses proteins on the surface during biofilm formation or to the fact that the concentration of antibodies used was too low. Also, some of the selected proteins may not be accessible to antibodies in the biofilm structure.
Based on the in vitro biofilm inhibition studies, SesC seemed to be the most promising target for prevention and treatment of S. epidermidis biofilms. We therefore selected SesC for further studies. Unfortunately, attempts to create sesC knockout mutants have been unsuccessful, and natural S. epidermidis sesC mutants have not been found yet. However, there are indications that SesC might be a potential fibrinogen-binding protein, playing a role in the attachment to abiotic surfaces (28). Further studies on the function of SesC in biofilm formation are therefore warranted.
Our in vivo rat model, although closely resembling the subcutaneous models for catheter-related infections and mimicking intraoperative contamination with skin flora, does not mimic conditions found in the human intravascular system. Intravascular devices are nevertheless the most frequently used medical devices. In addition, the immune response at the site of infection in our subcutaneous rat model may not reflect the immune response to the intravascular device-related infections in peripheral blood. Hence, we developed and used our JVC model to investigate the immune response and effector function of antibodies.
The primary functions of antibodies are neutralization and opsonization. The effect of αSesC-IgGs on S. epidermidis biofilms in vitro, in the absence of immune system components, suggested a neutralizing effect of αSesC-IgGs or blocking of the function of SesC. However, in vivo, in addition to neutralization, antibodies can opsonize their ligand, thus facilitating its uptake and destruction by natural killer cells, activating complement, and enhancing phagocytosis. The effect of αSesC-IgG antibodies on opsonophagocytosis of planktonic cells in vitro indicates their potential opsonic activity, whereas the reduction of the number of sessile bacteria on catheters in this experiment can be due to both neutralization and the opsonic activity of αSesC-IgG. It is possible that binding by αSesC-IgG of SesC on the surface of sessile bacteria triggers their detachment from catheter fragments and subsequent phagocytosis by neutrophils.
The 20-fold reduction of attached bacteria in the vaccinated group compared to the control group suggests a significant expression of SesC in sessile bacteria and is consistent with a role of this protein in S. epidermidis biofilm formation. This is also in line with our in vitro data and a previous report that subcutaneous injection of anti-SesC-IgGs at the place of implantation of catheter fragments reduced the number of attached bacteria to the catheter fragments 60-fold compared to the control group treated with preimmune IgGs (28). The JVC mouse model is a more physiological model for investigating the mechanism of action of antibodies on S. epidermidis biofilms and organ infections, since the reduction in the overall infection rate in JVC murine model can be due to both neutralization and the opsonization activity of αSesC-IgG antibodies. The clear effect of the αSesC-IgG antibodies on the organ infections suggest, on the one hand, a significant role of sesC in tissue infections and, on the other hand, confirm the hypothesis that αSesC-IgG antibodies work in preventing the adherence of the bacteria.
In conclusion, antibodies against recombinant SesC were shown to be both neutralizing and opsonic to S. epidermidis and can inhibit biofilm formation in vitro and in vivo. Active vaccination with SesC and preincubation of bacteria with αSesC-IgGs showed a reduction of biofilm formation and infection rates in vivo. The findings of the present study are consistent with those of Shahrooei et al. (28), who reported that SesC might play an important role in S. epidermidis biofilms and be a promising target for immunoprophylaxis and immunotherapy of S. epidermidis biofilms. The precise role of SesC in S. epidermidis biofilm formation remains to be identified.
Supplementary Material
Footnotes
Published ahead of print 16 July 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Allgaier H, Jung G, Werner RG, Schneider U, Zahner H. 1985. Elucidation of the structure of epidermin, a ribosomally synthesized, tetracyclic heterodetic polypeptide antibiotic. Angew. Chem. Int. Ed. 24:1051–1053 [Google Scholar]
- 2. Arrecubieta C, Lee MH, Macey A, Foster TJ, Lowy FD. 2007. SdrF, a Staphylococcus epidermidis surface protein, binds type I collagen. J. Biol. Chem. 282:18767–18776 [DOI] [PubMed] [Google Scholar]
- 3. Bogaert D, et al. 2005. Pneumococcal conjugate vaccination does not induce a persisting mucosal IgA response in children with recurrent acute otitis media. Vaccine 23:2607–2613 [DOI] [PubMed] [Google Scholar]
- 4. Bowden MG, et al. 2005. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology 151:1453–1464 [DOI] [PubMed] [Google Scholar]
- 5. Cerca N, Jefferson MK, Oliveira R, Pier GB, Azeredo J. 2006. Comparative antibody-mediated phagocytosis of Staphylococcus epidermidis cells grown in a biofilm or in the planktonic state. Infect. Immun. 74:4849–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Christensen GD, et al. 1985. Adherence of coagulase-negative staphylococci to plastic tissue-culture plates: a quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 22:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 8. Cucarella C, et al. 2001. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 183:2888–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danese PN. 2002. Antibiofilm approaches: prevention of catheter colonization. Chem. Biol. 9:873–880 [DOI] [PubMed] [Google Scholar]
- 10. Davis SL, Gurusiddappa S, Mccrea KW, Perkins S, Hook M. 2001. SdrG, a fibrinogen-binding bacterial adhesin of the microbial surface components recognizing adhesive matrix molecules subfamily from Staphylococcus epidermidis, targets the thrombin cleavage site in the B beta chain. J. Biol. Chem. 276:27799–27805 [DOI] [PubMed] [Google Scholar]
- 11. Desvaux M, Dumas E, Chafsey I, Hebraud M. 2006. Protein cell surface display in Gram-positive bacteria: from single protein to macromolecular protein structure. FEMS Microbiol. Lett. 256:1–15 [DOI] [PubMed] [Google Scholar]
- 12. Gill SR, et al. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gotz F. 2004. Staphylococci in colonization and disease: prospective targets for drugs and vaccines. Curr. Opin. Microbiol. 7:477–487 [DOI] [PubMed] [Google Scholar]
- 14. Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. 1997. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect. Immun. 65:519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Illingworth B, Bianco RW, Weisberg S. 2000. In vivo efficacy of silver-coated fabric against Staphylococcus epidermidis. J. Heart Valve Dis. 9:135–141 [PubMed] [Google Scholar]
- 16. Kockro RA, et al. 2000. Use of scanning electron microscopy to investigate the prophylactic efficacy of rifampin-impregnated CSF shunt catheters. J. Med. Microbiol. 49:441–450 [DOI] [PubMed] [Google Scholar]
- 17. Lazzell AL, et al. 2009. Treatment and prevention of Candida albicans biofilms with caspofungin in a novel central venous catheter murine model of candidiasis. J. Antimicrob. Chemother. 64:567–570 [DOI] [PubMed] [Google Scholar]
- 18. Mack D, Siemssen N, Laufs R. 1992. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infect. Immun. 60:2048–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maira-Litran T, Kropec A, Goldmann D, Pier GB. 2004. Biologic properties and vaccine potential of the staphylococcal poly-N-acetyl glucosamine surface polysaccharide. Vaccine 22:872–879 [DOI] [PubMed] [Google Scholar]
- 20. Mccann MT, Gilmore BF, Gorman SP. 2008. Staphylococcus epidermidis device-related infections: pathogenesis and clinical management. J. Pharm. Pharmacol. 60:1551–1571 [DOI] [PubMed] [Google Scholar]
- 21. Oosterlinck W, Vanderper A, Flameng W, Herijgers P. 2011. Glucose tolerance and left ventricular pressure-volume relationships in frequently used mouse strains. J. Biomed. Biotechnol. 2011:281312 doi:10.1155/2011/281312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otto M. 2009. Staphylococcus epidermidis: the “accidental” pathogen. Nat. Rev. Microbiol. 7:555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pei L, Flock JI. 2001. Functional study of antibodies against a fibrogenin-binding protein in Staphylococcus epidermidis adherence to polyethylene catheters. J. Infect. Dis. 184:52–55 [DOI] [PubMed] [Google Scholar]
- 24. Pintens V, et al. 2008. The role of sigma(B) in persistence of Staphylococcus epidermidis foreign body infection. Microbiology 154:2827–2836 [DOI] [PubMed] [Google Scholar]
- 25. Raad I, Alrahwan A, Rolston K. 1998. Staphylococcus epidermidis: emerging resistance and need for alternative agents. Clin. Infect. Dis. 26:1182–1187 [DOI] [PubMed] [Google Scholar]
- 26. Scott JR, Barnett TC. 2006. Surface proteins of Gram-positive bacteria and how they get there. Annu. Rev. Microbiol. 60:397–423 [DOI] [PubMed] [Google Scholar]
- 27. Sellman BR, Howell AP, Kelly-Boyd C, Baker SM. 2005. Identification of immunogenic and serum binding proteins of Staphylococcus epidermidis. Infect. Immun. 73:6591–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shahrooei M, et al. 2009. Inhibition of Staphylococcus epidermidis biofilm formation by rabbit polyclonal antibodies against the SesC protein. Infect. Immun. 77:3670–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. 2011. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect. Immun. 79:3801–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soderquist B. 2007. Surgical site infections in cardiac surgery: microbiology. APMIS 115:1008–1011 [DOI] [PubMed] [Google Scholar]
- 31. Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138 [DOI] [PubMed] [Google Scholar]
- 32. Sun DQ, Accavitti MA, Bryers JD. 2005. Inhibition of biofilm formation by monoclonal antibodies against Staphylococcus epidermidis RP62A accumulation-associated protein. Clin. Diagn. Lab. Immunol. 12:93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sutcliffe IC, Harrington DJ. 2002. Pattern searches for the identification of putative lipoprotein genes in Gram-positive bacterial genomes. Microbiology 148:2065–2077 [DOI] [PubMed] [Google Scholar]
- 34. Sutcliffe IC, Harrington DJ. 2004. Putative lipoproteins of Streptococcus agalactiae identified by bioinformatic genome analysis. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 85:305–315 [DOI] [PubMed] [Google Scholar]
- 35. Vandecasteele SJ, Peetermans WE, Merckx R, Van Eldere J. 2001. Quantification of expression of Staphylococcus epidermidis housekeeping genes with TaqMan quantitative PCR during in vitro growth and under different conditions. J. Bacteriol. 183:7094–7101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van der Borden AJ, van der Werf H, van der Mei HC, Busscher HJ. 2004. Electric current-induced detachment of Staphylococcus epidermidis biofilms from surgical stainless steel. Appl. Environ. Microbiol. 70:6871–6874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Mellaert L, Shahrooei M, Hofmans D, Van Eldere J. 2012. Immunoprophylaxis and immunotherapy of Staphylococcus epidermidis infections: challenges and prospects. Expert Rev. Vaccines 11:319–334 [DOI] [PubMed] [Google Scholar]
- 38. Van Wijngaerden E, et al. 1999. Foreign body infection: a new rat model for prophylaxis and treatment. J. Antimicrob. Chemother. 44:669–674 [DOI] [PubMed] [Google Scholar]
- 39. Wegmann U, et al. 2007. Complete genome sequence of the prototype lactic acid bacterium Lactococcus lactis subsp cremoris MG1363. J. Bacteriol. 189:3256–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu B, Davis EM, Hodges RS, Irvin RT, Li DY. 2008. Surface nanocrystallization of stainless steel for reduced biofilm adherence. Nanotechnology 19(33):335101 doi:10.1088/0957-4484/19/33/335101 [DOI] [PubMed] [Google Scholar]
- 41. Yu B, Lesiuk A, Davis E, Irvin RT, Li DY. 2010. Surface nanocrystallization for bacterial control. Langmuir 26:10930–10934 [DOI] [PubMed] [Google Scholar]
- 42. Zhang YQ, et al. 2003. Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49:1577–1593 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.