

Abstract
Clostridium perfringens iota-toxin is composed of an enzymatic component (Ia) and a binding component (Ib). Ib binds to a cell surface receptor, undergoes oligomerization in lipid rafts, and binds Ia. The resulting complex is then endocytosed. Here, we show the intracellular trafficking of iota-toxin. After the binding of the Ib monomer with cells at 4°C, oligomers of Ib formed at 37°C and later disappeared. Immunofluorescence staining of Ib revealed that the internalized Ib was transported to early endosomes. Some Ib was returned to the plasma membrane through recycling endosomes, whereas the rest was transported to late endosomes and lysosomes for degradation. Degraded Ib was delivered to the plasma membrane by an increase in the intracellular Ca2+ concentration caused by Ib. Bafilomycin A1, an endosomal acidification inhibitor, caused the accumulation of Ib in endosomes, and both nocodazole and colchicine, microtubule-disrupting agents, restricted Ib's movement in the cytosol. These results indicated that an internalized Ia and Ib complex was delivered to early endosomes and that subsequent delivery of Ia to the cytoplasm occurs mainly in early endosomes. Ib was either sent back to the plasma membranes through recycling endosomes or transported to late endosomes and lysosomes for degradation. Degraded Ib was transported to plasma membranes.
INTRODUCTION
Clostridium perfringens type E, which produces an iota-toxin consisting of an enzyme component (Ia) and a binding component (Ib), causes enterotoxemia in calves, lambs, and piglets (24, 26, 28). Ia ADP-ribosylates skeletal muscle α-actin and nonmuscle β/γ-actin (1, 3), and Ib binds to the cell, forming oligomers (15, 18, 30). The components lack toxic activity when each is injected alone, but together they have cytotoxic, lethal, and dermonecrotic effects (1, 24, 26). Iota-toxin belongs to a family of binary actin-ADP-ribosylating toxins that includes Clostridium botulinum C2 toxin, Clostridium spiroforme iota-like toxin, Clostridium difficile ADP-ribosyltransferase, and vegetative insecticidal protein from Bacillus cereus (1, 26).
Crystallography of Ia revealed that it is divided into two domains, the N domain (residues 1 to 210), which is responsible for interaction with Ib, and the C domain (residues 211 to 413), which is involved in the catalytic activity of ADP-ribosyltransferase (25, 32). We described the structure of a Michaelis complex with iota-toxin, actin, and a nonhydrolyzable NAD+ analogue (33). Based on this structure, we provided some insight into substrate recognition. In particular, we were able to show that Tyr62 on loop I and Arg248 on loop II in Ia play an essential role at the actin-toxin interface. We also proposed a common reaction mechanism for the actin-targeting mono-ADP-ribosylating toxins whereby an oxocarbenium intermediate is formed following the cleavage of the nicotinamide moiety from NAD+. Rotation then allows for the release of the conformational strain and the formation of a second cationic intermediate. Finally, the nucleophilic attack on Arg177 of the target actin leaves the ADP-ribose group covalently bound to this target protein (33).
Ib binds to cells, forming oligomers to create ion-permeable channels (15, 18, 30). Recently, we reported that Ib alone caused the death of A431 and A549 cells (21). Ib bound and formed oligomers in the membranes of A431 cells. However, Ib did not enter A431 cells. Ib also induced cell swelling and the rapid depletion of cellular ATP in both A431 and A549 cells but not in insensitive cell lines. Moreover, Ib induced permeabilization by propidium iodide without DNA fragmentation in A431 cells. Ultrastructural studies revealed that A431 cells undergo necrosis after treatment with Ib. We demonstrated that Ib by itself produces cytotoxic activity through necrosis (21). On the other hand, Ib enters MDCK cells via endocytosis and did not cause cell death, indicating that internalization of Ib is required for cellular survival and suggesting a role for endocytosis as an innate cellular defense mechanism against small membrane pores (21).
Papatheodorou et al. (22) reported that iota-toxin enters target cells via the lipolysis-stimulated lipoprotein receptor (LSR). Iota-toxin enters host cells and induces toxicity by exploiting the cell's endogenous pathways as follows (3, 4, 8, 26). Ib specifically binds to a receptor on the cytoplasmic membrane of cells via the C-terminal domain and accumulates in lipid rafts, and the Ia bound to the oligomers of Ib formed on the rafts then enters the cell (11, 19). Ia and Ib are transported to the early endosome, where acidification promotes cytosolic entry of Ia (8, 26). Ia then binds to G-actin in the cytosol and ADP-ribosylates it, thereby blocking the polymerization of actin and eventually intoxicating cells (1, 3, 26). Although the mechanism of cell rounding induced by iota-toxin has been investigated in detail, the endosomal trafficking pathway of internalized Ib is unknown. MDCK cells provide a good model system to study the binding and internalization of Ib (19, 21, 29). In this study, we have analyzed the intracellular trafficking of Ib using MDCK cells.
MATERIALS AND METHODS
Materials.
Rabbit anti-Ib antibody was prepared as described previously (23). Bafilomycin A1 (BAF), nocodazole, colchicine, cytochalasin D, cholera toxin subunit B (CTB), mouse anti-Golgi 58K antibody, and anti-mouse IgG–fluorescein isothiocyanate (FITC) were obtained from Sigma (St. Louis, MO). Mouse anti-early endosome antigen 1α (anti-EEA1α) and anti-β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and mouse anti-lysosome-associated membrane protein 2 (anti-Lamp2) antibody was obtained from AbD Serotec (Oxford, United Kingdom). Horseradish peroxidase-labeled goat anti-rabbit IgG, horseradish peroxidase-labeled sheep anti-mouse IgG, and an ECL Western blotting kit were purchased from GE Healthcare (Tokyo, Japan). Dulbecco's modified Eagle's medium (DMEM) and Hanks' balanced salt solution (HBSS) were obtained from Gibco BRL (New York, NY). The expression vector for GFP-Rab11 was prepared as described previously (31). Alexa Fluor 488-conjugated cholera subunit B, Alexa Fluor 568-conjugated goat anti-rabbit IgG, Alexa Fluor 488-conjugated goat anti-mouse IgG, CellLights lysosome-green fluorescent protein (GFP), CellLights endoplasmic reticulum (ER)-GFP, and 4′,6′-diamino-2-phenylindole (DAPI) were obtained from Molecular Probes (Eugene, OR). Fura-2 acetoxymethyl (AM) ester was obtained from Dojindo Laboratories (Kumamoto, Japan). Ten to 20 percent Tricine-SDS gels, Tricine-SDS sample buffer, and Tricine-SDS running buffer were purchased from Invitrogen (Tokyo, Japan).
Expression and purification of Ia and Ib.
Recombinant Ia was purified from culture supernatants of Bacillus subtilis ISW1214 carrying a plasmid containing the Ia gene, as described previously (17). Ib was expressed, fused with glutathione S-transferase (GST) in Escherichia coli BL21, as described previously (18).
Cell culture and assay of cytotoxicity.
Madin-Darby canine kidney (MDCK) cells were obtained from Riken Cell Bank (Tsukuba, Japan). Cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS), 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 2 mM glutamine (FCS-DMEM). All incubation steps were carried out at 37°C in a 5% CO2 atmosphere. The test for cytotoxicity was done on MDCK cells. The cells were cultivated in FCS-DMEM. For cytotoxicity assays, the cells were inoculated in 48-well tissue culture plates (Falcon, Oxnard, CA). Various concentrations of Ia and Ib were mixed in FCS-DMEM and inoculated onto cell monolayers. The cells were observed for morphological alterations at 4 h after inoculation, as described previously (18). To measure the effects of BAF, nocodazole, and colchicine on the cytotoxicity of iota-toxin, MDCK cells were preincubated with these agents at 37°C for 1 h and then incubated with Ia and Ib at 37°C for 4 h.
Immunoblot analysis.
The samples were incubated in 2% SDS sample buffer at 37°C for 5 min and subjected to SDS-PAGE. Alternatively, the protein samples were analyzed by 10 to 20% Tricine-SDS-PAGE (Invitrogen), according to the manufacturer's instructions. For Western blot analysis, the proteins on SDS-polyacrylamide gels and Tricine-SDS-polyacrylamide gels were transferred to polyvinylidene difluoride membranes (Immobilon P; Millipore). The membranes were blocked with Tris-buffered saline (TBS) containing 2% Tween 20 and 5% skim milk and incubated first with a primary antibody against Ib or β-actin in TBS containing 1% skim milk, then with a horseradish peroxidase-conjugated secondary antibody, and finally with the antibody from an enhanced chemiluminescence analysis kit (GE Healthcare).
Immunofluorescence analysis.
Cells were plated on a polylysine-coated glass-bottom dish (Matsunami, Osaka, Japan) and incubated at 37°C in a 5% CO2 incubator overnight in FCS-DMEM. To study the internalization of Ib, Ib (1 μg/ml) was incubated with cells at 4°C for 1 h in FCS-DMEM. After three washes in cold FCS-DMEM, cells were transferred to FCS-DMEM prewarmed to 37°C and incubated at the same temperature for various periods. They were washed four times with cold phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde at room temperature. For antibody labeling, the dishes were then incubated at room temperature for 15 min in 50 mM NH4Cl in PBS and in PBS containing 0.1% Triton X-100 at room temperature for 20 min. After being washed with PBS containing 0.02% Triton X-100, the dishes were incubated at room temperature for 1 h with PBS containing 4% bovine serum albumin (BSA), followed by primary antibody (rabbit anti-Ib antibody, mouse anti-EEA1 antibody, mouse anti-Lamp2 antibody or mouse anti-Golgi 58K antibody) in PBS containing 4% BSA at room temperature for 1 h. They were then washed with PBS containing 0.02% Triton X-100, incubated with secondary antibody (Alexa Fluor 568-conjugated anti-rabbit IgG or anti-mouse IgG-FITC) in PBS containing 4% BSA at room temperature for 1 h, washed extensively with PBS containing 0.02% Triton X-100, and analyzed under a Nikon A1 laser scanning confocal microscope (Tokyo, Japan) (20, 21). Nuclei were stained with DAPI.
To study the localization of lysosome-GFP and ER-GFP, MDCK cells were seeded and grown at 37°C for 12 h on polylysine-coated glass-bottom dishes before transfection with CellLights lysosome-GFP or CellLights ER-GFP according to the manufacturer's instructions. Twenty-four hours later, they were treated with Ib, fixed, permeabilized, and blocked as described above. For experiments with GFP-Rab11, cells were transfected with Nucleofector (Amaxa, Koln, Germany) according to the following program: MDCK cells, kit L, program A24. Briefly, 1 × 106 MDCK cells were pelleted and resuspended in 0.1 ml of solution L and then electroporated with 2 μg of pGFP-Rab11 plasmid. The electroporated cells were resuspended in 0.35 ml of complete medium. Of this solution, 0.1 ml was seeded on polylysine-coated glass-bottom dishes and incubated at 37°C. After 24 h, they were treated with Ib, fixed, permeabilized, and blocked as described above. All images represent a single section through the focal plane. Images shown in the figures are representative of at least three independent experiments and were produced with Adobe Photoshop (20, 21).
Measurement of intracellular Ca2+ concentrations.
MDCK cells were seeded in a 35-mm poly-l-lysine-coated glass-bottom dish and cultured at 37°C for 20 h. The cells were washed three times in wash buffer (Ca2+-free Hanks'-HEPES buffer containing 2.5 mM probenecid and 1% [wt/vol] bovine serum albumin) and then loaded with the intracellular Ca2+-sensitive fluorescent indicator fura-2 AM (4.5 μM) (Dojindo, Kumamoto, Japan) for 30 min at 37°C in dye-loading buffer (Hanks'-HEPES buffer containing 2.5 mM probenecid, 1% [vol/vol] FBS, and 0.05% Pluronic F-127). After being washed three times with wash buffer, cells were incubated in assay buffer (Hanks'-HEPES buffer containing 2.5 mM probenecid and 1% [wt/vol] bovine serum albumin) for an additional 15 min at 37°C in the measurement equipment to allow for hydrolysis of the acetoxymethyl ester. The culture dish was placed on the stage of an inverted microscope (TMD-300; Nikon, Tokyo, Japan), which was equipped with a chamber controlled at 37°C. The fura-2 absorption shift that occurs upon binding was determined by scanning the excitation spectra at between 340 and 380 nm while monitoring emission at 510 nm. The resultant fluorescence images were analyzed every 30 s from the individual cells with a fluorescence analyzer (Aqua-cosmos; Hamamatsu Photonics, Hamamatsu, Japan) using an ultra-high-sensitivity television camera (charge-coupled device [CCD]). The imaging system was standardized with a fura-2 calcium-imaging calibration kit (Invitrogen) as described previously (5). Heat-inactivated Ib was prepared by heating at 95°C for 10 min.
RESULTS
Endocytosis of Ib into MDCK cells.
We have shown that Ib can be internalized by MDCK cells independently of Ia (18, 19). Thus, we monitored the entry and intracellular trafficking of Ib using immunofluorescence. Incubation of MDCK cells with Ib at 4°C resulted in plasma membrane staining consistent with the binding of toxin components to a cell surface receptor (Fig. 1A). Upon the transfer of cells from 4°C to 37°C, Ib was internalized. After 5 to 30 min at 37°C, Ib moved from the plasma membranes into intracellular vesicles. After 60 min, Ib was no longer associated with the cell membranes and localized into vesicles in a perinuclear area. From 120 min onwards, immunofluorescent signals were observed in plasma membranes. To investigate the activity of Ib that was transported from cytoplasmic vesicles to plasma membranes, MDCK cells were preincubated with Ib at 4°C for 60 min, washed, and incubated with the medium alone at 37°C to allow internalization. Ia was then added to the cells at 0 to 240 min after the incubation at 37°C. As shown in Fig. 1B, 100% of the cells were round after 4 h when Ia was added immediately after washing (0 min). Most cells were round after 4 h when Ia was applied after 30 min of incubation. On the other hand, the addition of Ia after 60 min decreased the cell rounding activity, and the addition of Ia after over 120 min did not cause cell rounding. Next, to test the binding and oligomerization of Ib on MDCK cells during the incubation period, the cells were preincubated with Ib in DMEM-10% FCS at 4°C for 60 min, washed, and incubated in the same medium at 37°C for various periods. The treated cells were dissolved in Tricine-SDS sample buffer (Invitrogen) and analyzed by Tricine-SDS-PAGE. Ib was detected by Western blotting with anti-Ib antibody (Fig. 1C). When the MDCK cells were incubated with Ib at 4°C for 60 min, only the Ib monomer (76 kDa) was detected. On the other hand, when the cells were incubated with Ib at 37°C, the level of the Ib monomer decreased and that of the Ib oligomer increased in a time-dependent manner during the first 60 min. Later, both bands decreased, and they disappeared at 240 min. Under these experimental conditions, degraded Ib was not detected using the anti-Ib antibody. These results suggest that after internalization, Ib is degraded in the vesicles and transported to plasma membranes.
Fig 1.

Intracellular trafficking and oligomerization of Ib in MDCK cells. (A) MDCK cells were incubated with Ib (1 μg/ml) at 4°C for 1 h, washed, and incubated at 37°C for the period indicated. Cells were fixed, permeabilized, and stained with anti-Ib antibody and DAPI. The Ib (red) and nucleus (blue) were viewed with a confocal microscope. The experiments were repeated three times, and a representative result is shown. Bar, 5 μm. (B) MDCK cells were incubated with Ib (500 ng/ml) at 4°C for 1 h. The washed cells were incubated at 37°C for the period indicated. The cells were treated with Ia (100 ng/ml) and incubated at 37°C for 4 h. Pictures were taken. The total number of cells and number of round cells were counted from the pictures, and the percentage of round cells was calculated. Values are given as the mean ± standard deviation (SD) (n = 3). (C) Cells were incubated with Ib (1 μg/ml) at 4°C for 1 h. The cells were rinsed and incubated at 37°C for the period indicated. They were lysed in Tricine-SDS sample buffer (Invitrogen). The cell lysates were subjected to Tricine-SDS-PAGE and Western blot analysis of Ib and β-actin as a control. A typical result from three experiments is shown.
Endocytic trafficking of Ib.
To investigate the endocytic trafficking of Ib, we studied whether endocytic markers were associated with intracellular compartments containing Ib (Fig. 2A and C). Cells incubated with Ib for specific periods of time at 37°C were fixed, permeabilized, and immunostained with antibodies against Ib and endocytic markers. Ib colocalized with early endosome antigen 1 (EEA1), a marker of early endosomes, after 15 min but not after 30 min. In cells that had been transfected with a plasmid encoding GFP-Rab11, a marker of recycling endosomes, Ib colocalized with GFP-Rab11 for 15 and 30 min but not 60 min. Next, we examined the trafficking of Ib to the late endosomes and lysosomes. Ib did not colocalize with lysosome-associated membrane protein 2 (Lamp2), a marker of late endosomes and lysosomes, after 15 min but did so after 30 and 60 min. Similarly, Ib colocalized with lysosome-GFP, a lysosome marker, after 30 and 60 min. On the other hand, Ib did not colocalize with Golgi 58K, a Golgi marker, and ER-GFP, an endoplasmic reticulum marker. In contrast, negative-control preparations with the additions of Alexa Fluor 568-conjugated anti-rabbit IgG, FITC-conjugated anti-mouse IgG, and DAPI failed to detect the fluorescence signals without DAPI (Fig. 2B). We compared the internalization pathways of Ib and Alexa Fluor 488-labeled cholera toxin subunit B (CTB). CTB is transported from the early endosomes to the endoplasmic reticulum (27). After incubation at 37°C for 15 to 30 min, Ib partially colocalized with CTB (data not shown). These results indicate that internalized Ib is delivered to early endosomes, where sorting occurs. Some Ib is transported to the plasma membrane in Rab11-positive endosomes. The majority of Ib goes to late endosomes, which deliver Ib to lysosomes for degradation.
Fig 2.

Colocalization of Ib and endosome markers in MDCK cells. (A) MDCK cells were incubated with Ib (1 μg/ml) at 4°C for 1 h, washed, and incubated at 37°C for the period indicated. Cells were fixed, permeabilized, and stained with DAPI and antibodies to EEA1, Lamp2, Golgi 58K, and Ib. MDCK cells were transiently transfected with pGFP-Rab11, pGFP-lysosome, or pER-GFP. After 24 h of transfection, the transfected cells were incubated with Ib as described above. Cells were fixed, permeabilized, and stained with anti-Ib antibody and DAPI. Ib (red), endosome markers (green), and the nucleus (blue) were viewed with a confocal microscope. The experiments were repeated three times, and a representative result is shown. Bar, 5 μm. (B) Negative-control preparation. Cells were fixed, permeabilized, and stained with Alexa Fluor 568-conjugated anti-rabbit IgG, FITC-conjugated anti-mouse IgG, and DAPI. (C) Quantification of colocalizations. The percentage of Ib/endocytic marker colocalization represents the ratio of the number of endosomal structures stained for Ib and for endocytic marker to the total number of endosomal structures stained for endocytic marker. The percentage of colocalization was determined for each cell, and the results represent the average ± standard error of the mean (SEM) for several cells (n > 10) obtained from at least three independent experiments.
Effect of endocytosis inhibitors on intracellular trafficking of Ib.
Inhibitors that interfere with known intracellular trafficking pathways were utilized to investigate the route of Ib. We reported that treatment of Vero cells with iota-toxin in the presence of bafilomycin A1 (BAF), which inhibits vacuole-type H+-ATPase and prevents acidification of endosomes, resulted in decreased translocation of iota-toxin from endosomes into the cytosol (18). Treatment of MDCK cells with Ib in the presence of BAF had no effect on the binding of Ib to plasma membranes at 4°C (Fig. 3A). After incubation at 37°C, BAF clearly increased the accumulation of Ib in the cytoplasmic vesicles compared with that in the absence of an inhibitor. Disruption of microtubules with nocodazole and colchicine inhibited the transport from early to late endosomes (7, 20). As shown in Fig. 3A, nocodazole and colchicine disrupted the perinuclear localization of Ib, and Ib-containing vesicles were scattered in the cytosol. Disruption of the F-actin cytoskeleton by cytochalasin D blocked actin-mediated endocytosis (6). A nontoxic concentration of cytochalasin D prevented the internalization of Ib into cytosol (Fig. 3A). On the other hand, BAF inhibited rounding of the cells induced by Ia plus Ib (Fig. 3B). In contrast, nocodazole and colchicine did not inhibit iota-toxin-induced cell rounding. These inhibitors themselves did not have any morphological effects under our experimental conditions. These results indicated that Ia was translocated from acidic endosomes to the cytosol and that iota-toxin was trafficked by microtubule-based transport.
Fig 3.

Effect of inhibitors on intracellular trafficking of Ib. (A) MDCK cells were incubated with bafilomycin A1 (50 nM), nocodazole (20 μg/ml), colchicine (50 μM), or cytochalasin D (0.5 μM) at 37°C for 1 h and then rinsed. Ib (1 μg/ml) was added and incubated at 4°C for 1 h. Cells were rinsed and incubated at 37°C for 0 and 30 min. Cells were fixed, permeabilized, and stained with anti-Ib antibody and DAPI. The Ib (red) and nucleus (blue) were viewed with a confocal microscope. The experiments were repeated three times, and a representative result is shown. Bar, 7.5 μm. (B) MDCK cells were pretreated with dimethyl sulfoxide (DMSO), bafilomycin A1 (50 nM), nocodazole (20 μg/ml), or colchicine (50 μM) at 37°C for 1 h. The cells were incubated with Ia (100 ng/ml) and Ib (100 ng/ml) at 37°C for 4 h. Pictures were taken. The total number of cells and number of round cells were counted from the pictures, and the percentage of round cells was calculated. Values are given as the mean ± SD (n = 3).
Ib-induced Ca2+ influx into MDCK cells.
As shown in Fig. 1A, degraded Ib was transported to plasma membranes. We investigated the intracellular trafficking of degraded Ib. The fusion of lysosomes with the plasma membranes might occur in the cells upon rises in intracellular free-Ca2+ concentrations. To examine whether the Ib-induced elevation in the intracellular Ca2+ concentration [Ca2+]i correlated with the internalization of Ib, the effect of Ib on the increase was examined using MDCK cells (Fig. 4). Intracellular Ca2+ levels were measured using fura-2, a fluorescent Ca2+ indicator. MDCK cells were loaded with fura-2 AM and treated with various concentrations of Ib. [Ca2+]i was then estimated from the fluorescence intensity. The concentration increased after the addition of Ib (within 15 min) in a dose-dependent manner (Fig. 4). On the other hand, in Ca2+-free buffer, Ib did not induce an elevation in the intracellular Ca2+ level (data not shown). We reported that extracellular Ca2+ had no effect on the binding of Ib to cells (16). No increase in [Ca2+]i was evoked by heat-inactivated Ib, and the elevation in [Ca2+]i caused by Ib was completely neutralized by an anti-Ib antibody (data not shown). This result indicates that Ib promotes Ca2+ influx from the extracellular buffer during internalization by endocytosis.
Fig 4.
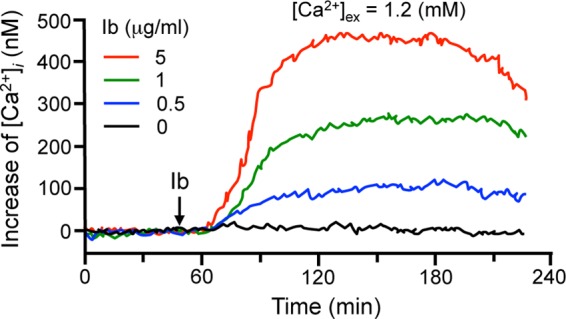
Ib-induced increase in [Ca2+]i resulting from an influx of extracellular Ca2+. MDCK cells were loaded with the intracellular Ca2+ indicator fura-2 AM. [Ca2+]i was calculated as described in Materials and Methods. Changes in [Ca2+]i induced by Ib were measured in cells in extracellular buffer containing 1.2 mM CaCl2. Ib was added at the time indicated by the arrow. The data represent three independent experiments.
DISCUSSION
In the present study, we first demonstrated the dynamics of the intracellular trafficking of Ib. After the internalization of iota-toxin into the cells, Ia is released from acidic early endosomes to the cytosol, Ib is trafficked to lysosomes through an endocytic pathway, and degraded Ib is transported to plasma membranes.
C. perfringens iota-toxin enters host cells and induces toxicity by exploiting the endocytic trafficking (3, 26). Gibert et al. (9) reported that an interleukin-2 (IL-2) receptor endocytic pathway is used by iota-toxin. Ib recognizes specific cell surface receptors, oligomerizes into heptameric ring structures on lipid rafts of plasma membranes, forms transmembrane channels, and docks with Ia (3, 11, 19). Recently, Papatheodorou et al. (22) reported that LSR is a receptor for iota-toxin. After endocytosis through a clathrin-independent and Rho-dependent pathway, iota-toxin migrates until it reaches endocytic carrier vesicles (8, 9). Ia probably passes through the channel formed by oligomeric Ib following a pH gradient, and in addition, a membrane potential gradient is necessary for translocating Ia (8). Furthermore, it has been reported that heat shock protein 90 (Hsp-90) and cyclophilin A are crucial for translocation of Ia across the endosomal membrane (14). In the cytosol, Ia ADP-ribosylates G-actin, resulting in actin depolymerization and cell rounding. However, the intracellular route of Ib after the early endosome has not been described in detail. In the present study, we investigated the internalization of Ib by endocytosis. When Ib was incubated with MDCK cells at 37°C, it colocalized with EEA1 after 15 min, indicating that it reaches the early endosomes. Ib no longer localized with EEA1 at 30 min. After 15 to 30 min, it colocalized with Rab11 (10), indicating that some Ib is delivered to the recycling endosomes. After 30 to 60 min, Ib colocalized with Lamp2 and lysosome-GFP, indicating that Ib moves to the late endosomes and lysosomes. The oligomeric band of Ib was detected after 15 to 60 min, and the addition of Ia to cells preincubated with Ib at 37°C for 30 to 60 min caused cell rounding. Therefore, some Ib is recycled back to the plasma membranes through Rab11-positive recycling endosomes. Recycling may be important for Ib to extend the entry of Ia. These results demonstrate that Ib is endocytosed and sorted from early endosomes to recycling endosomes or late endosomes and lysosomes. After 120 min, immunofluorescence signals of Ib were observed in plasma membranes, and the Ib oligomer decreased and disappeared at 240 min. It was reported that the acidification of the endosomes triggers the conversion of the Ib oligomer into pores and the insertion of Ib pores in endosomal membranes (8). Lysosomes are in general responsible for the degradation of membrane and extracellular proteins that enter cells by endocytosis (2). Therefore, we proposed that, by fusing between lysosomes and endosomes containing Ib, the luminal domain of Ib is degraded in the lysosomal environment but not the membrane-inserted domain of Ib. It was reported that lysosomes move to plasma membranes via a Ca2+-dependent step and fuse with the membranes, indicating that lysosomes are the Ca2+-regulated exocytic vesicles (2, 13). We showed that Ib induces an increase in Ca2+ influx from extracellular compartments during endocytosis. These findings indicated that a rise in the intracellular Ca2+ concentration caused by Ib triggers the fusion with plasma membranes of lysosomes, and degraded Ib is transported to plasma membranes. Further studies are needed to understand the role of degraded Ib. We demonstrated that among bacterial toxins, Ib caused the fusion of lysosomes with the plasma membranes in the cells upon rises in the intracellular Ca2+ concentration. The Ca2+-regulated fusion of lysosomes with plasma membranes induced by Ib might lead to membrane repair after pore formation of Ib. Ib is useful for understanding this fusion process.
Bafilomycin A1, an inhibitor of vacuole-type ATPases, inhibited the cell rounding in response to iota-toxin and caused the accumulation of Ib in endosomes. This result was attributed to its inhibition of intracellular acidification. After acidification of early endosomes, Ib forms channels which mediate the passage of Ia into the cytosol, as previously reported (8). It has been reported that bafilomycin A1 also blocks endocytic transport (12). Thus, since bafilomycin A1-induced endosome alkalinization does not perturb endocytic trafficking, endosomal acidification is required for the translocation of Ia and endocytic transport of Ib. Cytochalasin D, an actin cytoskeleton-disrupting drug, affects the actin-mediated endocytosis (6). We found that cytochalasin D blocked the internalization of Ib, suggesting that actin filaments are required for the initial internalization of Ib. Nocodazole and colchicine prevent the polymerization of microtubules and depress membrane trafficking from early to late endosomes (12). In this experiment, cell rounding by iota-toxin was not inhibited by nocodazole and colchicine. As the disruption of microtubules by nocodazole and colchicine does not inhibit the initial acidification of endosomes (12), Ia is translocated from endosomes to the cytosol. However, treatment of the cells with nocodazole and colchicine restricted the transport of Ib toward the perinuclear region, where the majority of lysosomes are localized, and caused the dispersal of Ib-containing vesicles, indicating that intracellular movement of Ib was impaired. The data support that after endocytosis, Ia is translocated from acidic endosomes to the cytosol and Ib is trafficked from early endosomes to late endosomes and lysosomes by microtubule-based transport.
In conclusion, we demonstrated the intracellular route of Ib. Ib is internalized, transported to early endosomes, and sorted into recycling endosomes and late endosomes. From late endosomes, Ib is delivered to lysosomes for degradation. Due to an increase in the intracellular Ca2+ concentration caused by Ib, the fusion of lysosomes with the plasma membranes occurs and degraded Ib is exposed at the cell surface.
ACKNOWLEDGMENTS
We thank A. Kanbayashi for technical assistance.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, MEXT.SENRYAKU, 2010.
Footnotes
Published ahead of print 23 July 2012
REFERENCES
- 1. Aktories K, Wegner A. 1989. ADP-ribosylation of actin by clostridial toxins. J. Cell Biol. 109:1385–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrews NW. 2000. Regulated secretion of conventional lysosomes. Trends Cell Biol. 10:316–321 [DOI] [PubMed] [Google Scholar]
- 3. Barth H, Aktories K, Popoff MR, Stiles BG. 2004. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 68:373–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blöcker D, Behlke J, Aktories K, Barth H. 2001. Cellular uptake of the Clostridium perfringens binary iota-toxin. Infect. Immun. 69:2980–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buttigieg J, Brown ST, Lowe M, Zhang M, Nurse CA. 2008. Functional mitochondria are required for O2 but not CO2 sensing in immortalized adrenomedullary chromaffin cells. Am. J. Physiol. Cell Physiol. 294:C945–C956 [DOI] [PubMed] [Google Scholar]
- 6. Chu JJH, Ng ML. 2004. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 78:10543–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng Q, Zhang Y, Barbieri JT. 2007. Intracellular trafficking of Pseudomonas ExoS, a type III cytotoxin. Traffic 8:1331–1345 [DOI] [PubMed] [Google Scholar]
- 8. Gibert M, et al. 2007. Differential requirement for the translocation of clostridial binary toxins: iota toxin requires a membrane potential gradient. FEBS Lett. 581:1287–1296 [DOI] [PubMed] [Google Scholar]
- 9. Gibert M, et al. 2011. Endocytosis and toxicity of clostridial binary toxins depend on a clathrin-independent pathway regulated by Rho-GDI. Cell. Microbiol. 13:154–170 [DOI] [PubMed] [Google Scholar]
- 10. Gruenberg J. 2001. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2:721–730 [DOI] [PubMed] [Google Scholar]
- 11. Hale ML, Marvaud JC, Popoff MR, Stiles BG. 2004. Detergent-resistant membrane microdomains facilitate Ib oligomer formation and biological activity of Clostridium perfringens iota-toxin. Infect. Immun. 72:2186–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huotari J, Helenius A. 2011. Endosome maturation. EMBO J. 30:3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Idone V, et al. 2008. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaiser E, et al. 2011. Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect. Immun. 79:3913–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knapp O, Benz R, Gibert M, Marvaud JC, Popoff MR. 2002. Interaction of Clostridium perfringens iota-toxin with lipid bilayer membranes. Demonstration of channel formation by the activated binding component Ib and channel block by the enzyme component Ia. J. Biol. Chem. 277:6143–6152 [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi K, et al. 2008. Role of Ca2+-binding motif in cytotoxicity induced by Clostridium perfringens iota-toxin. Microb. Pathog. 44:265–270 [DOI] [PubMed] [Google Scholar]
- 17. Nagahama M, Sakaguchi Y, Kobayashi K, Ochi S, Sakurai J. 2000. Characterization of the enzymatic component of Clostridium perfringens iota-toxin. J. Bacteriol. 182:2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagahama M, Nagayasu K, Kobayashi K, Sakurai J. 2002. Binding component of Clostridium perfringens iota-toxin induces endocytosis in Vero cells. Infect. Immun. 70:1909–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagahama M, et al. 2004. Binding and internalization of Clostridium perfringens iota-toxin in lipid rafts. Infect. Immun. 72:3267–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagahama M, et al. 2011. Cellular vacuolation induced by Clostridium perfringens epsilon-toxin. FEBS J. 278:3395–3407 [DOI] [PubMed] [Google Scholar]
- 21. Nagahama M, et al. 2011. Clostridium perfringens iota-toxin b induces rapid cell necrosis. Infect. Immun. 79:4353–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Papatheodorou P, et al. 2011. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. U. S. A. 108:16422–16427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakurai J, Kobayashi K. 1995. Lethal and dermonecrotic activities of Clostridium perfringens iota toxin: biological activities induced by cooperation of two nonlinked components. Microbiol. Immunol. 39:249–253 [DOI] [PubMed] [Google Scholar]
- 24. Sakurai J, Nagahama M, Ochi S. 1997. Major toxins of Clostridium perfringens. J. Toxicol. Toxin Rev. 16:195–214 [Google Scholar]
- 25. Sakurai J, Nagahama M, Hisatsune J, Katunuma N, Tsuge H. 2003. Clostridium perfringens iota-toxin, ADP-ribosyltransferase: structure and mechanism of action. Adv. Enzyme Regul. 43:361–377 [DOI] [PubMed] [Google Scholar]
- 26. Sakurai J, Nagahama M, Oda M, Tsuge H, Kobayashi K. 2009. Clostridium perfringens iota-toxins: structure and function. Toxins 1:208–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sandvig K, van Deurs B. 2002. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 529:49–53 [DOI] [PubMed] [Google Scholar]
- 28. Songer JG. 1996. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9:216–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stiles BG, Hale ML, Marvaud JC, Popoff MR. 2000. Clostridium perfringens iota toxin: binding studies and characterization of cell surface receptor by fluorescence-activated cytometry. Infect. Immun. 68:3475–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stiles BG, Hale ML, Marvaud JC, Popoff MR. 2002. Clostridium perfringens iota toxin: characterization of the cell-associated iota b complex. Biochem. J. 367:801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takahashi M, et al. 2007. Cholesterol controls lipid endocytosis through Rab11. Mol. Biol. Cell 18:2667–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsuge H, et al. 2003. Crystal structure and site-directed mutagenesis of enzymatic components from Clostridium perfringens iota-toxin. J. Mol. Biol. 325:471–483 [DOI] [PubMed] [Google Scholar]
- 33. Tsuge H, et al. 2008. Structural basis of actin recognition and arginine ADP-ribosylation by Clostridium perfringens iota-toxin. Proc. Natl. Acad. Sci. U. S. A. 105:7399–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]