

Abstract
Studies of innate immunity in metazoans have largely focused on detection of microbial molecules by host pattern recognition receptors (PRRs). A complementary mode of innate immune recognition, based on detection of pathogen-encoded activities, has long been recognized in plants, where it is termed effector-triggered immunity; however, little is known about the possibility of effector-triggered immunity in metazoans. Legionella pneumophila is an intracellular bacterial pathogen that causes Legionnaires' disease, an inflammatory pneumonia. We recently demonstrated that macrophages infected with L. pneumophila exhibit mitogen-activated protein (MAP) kinase (MAPK) activation that is independent of known PRRs but dependent on a functional bacterial secretion system. Here, we show that five secreted L. pneumophila effectors are responsible for the activation of host MAP kinases. These five effectors inhibit host translation, and their activity is required for host MAPK activation. We demonstrate that MAPK activation by these effectors shapes the host transcriptional response to L. pneumophila. Furthermore, we find that uninfected macrophages treated with two different translation inhibitors exhibit activation of MAP kinases and upregulation of target genes, indicating that translation inhibition alone is sufficient to elicit this response in macrophages. MAP kinase pathways are crucial in many aspects of the immune response, including inflammation and cell motility. Our results demonstrate that this important host pathway can be activated in response to a pathogen-encoded activity, adding to an emerging body of evidence in support of this novel mode of innate immune detection in metazoans.
INTRODUCTION
Historically, studies of innate immune surveillance have centered on pattern recognition receptors (PRRs), which are germ-line-encoded host proteins that recognize molecules that are highly conserved among microbes but absent in the host (29). These microbial molecules—called pathogen-associated molecular patterns (PAMPs)—may be unique to a certain class of microbe, as in the case of viral double-stranded RNA or bacterial lipopolysaccharide (LPS), allowing the generation of an immune response that is tailored to the pathogen at hand. While the PRR-PAMP model is fundamental to our understanding of innate immune surveillance, it does not provide a straightforward mechanism for immune discrimination between harmless and harmful microbes, since PAMPs are found on pathogens and nonpathogens alike. Therefore, it has been suggested that in addition to detection of microbial molecules, the host might also screen for patterns of pathogenesis (49), the activities that pathogens utilize to invade, replicate, and spread within the host (16, 38, 49). Indeed, several reports of host responses to such pathogen-encoded activities have recently emerged in the literature. Examples in mammals include activation of the Nlrp3 inflammasome by a viral ion channel (26) and activation of host mitogen-activated protein (MAP) kinases (MAPKs) by Salmonella (9, 42) or Escherichia coli (8, 41) effectors that target host Rho family GTPases. Additionally, disruptions of conserved host pathways have recently been reported to elicit immune responses in the nematode Caenorhabditis elegans (13, 37, 39) and in the fruit fly Drosophila melanogaster (8). Together, these host responses appear to represent a novel mode of immunosurveillance that relies not on ligand-receptor interaction but instead on the host's detection of the disruption of a crucial cellular process.
The intracellular pathogen Legionella pneumophila has proven an effective tool for probing novel mechanisms of innate immune sensing (16). L. pneumophila is a Gram-negative, motile bacterium that evolved as a parasite of freshwater amoebae. It can also infect macrophages in the mammalian lung, causing a severe inflammatory pneumonia known as Legionnaires' disease (14). Because L. pneumophila does not appear to be transmitted between mammals (31), it has not evolved significant immune evasion mechanisms. Thus, it can be used to reveal immunosurveillance pathways in the absence of the manipulation or evasion that is common with other, better-adapted pathogens.
Upon phagocytosis by the host amoeba or macrophage, L. pneumophila employs a type IV secretion system, called the Dot/Icm system, to translocate over 200 effector proteins into the host cytosol (51). These effectors manipulate host cell processes to remodel the Legionella-containing vacuole, preventing fusion with lysosomes and resulting in creation of a specialized compartment in which the bacteria can replicate (25, 28). The Dot/Icm apparatus is essential for bacterial replication and virulence, but its deployment also exposes the bacteria to cytosolic host surveillance pathways (discussed below).
Various PRRs are known to recognize L. pneumophila, leading to activation of multiple downstream pathways. Several Toll-like receptors (TLRs), especially the acylated lipoprotein sensor TLR2, recognize both Dot/Icm-positive (Dot/Icm+) and Dot/Icm-negative (Dot/Icm−) L. pneumophila isolates in the extracellular or endosomal compartment (1–3, 12, 22, 23). In addition, multiple cytosolic PRRs respond specifically to Dot/Icm+ L. pneumophila isolates that can access the cytosol through type IV secretion. These include the peptidoglycan sensors Nod1 and Nod2 (6, 15, 17, 45), the inflammasome proteins Naip5 and Nlrc4 (20, 32, 35, 43, 50), and an additional pathway that utilizes the adaptor protein Mavs (11, 40). The outputs of these various sensing pathways are distinct: Naip5 and Nlrc4 initiate cytokine processing and caspase-1-dependent macrophage death in response to bacterial flagellin (32, 35), while the Mavs-dependent pathway results in transcription of type I interferons and coregulated genes (40). The TLRs and Nod1/Nod2 both induce overlapping transcriptional responses resulting from activation of host MAP kinases and the proinflammatory transcription factor NF-κB (15, 45).
Interestingly, we recently reported that macrophages deficient in both TLR and Nod signaling still exhibit MAP kinase activation when infected with Dot/Icm+ L. pneumophila bacteria (45). This activation appeared to involve secreted bacterial effectors, since host MAPK activation was abrogated during infection with a bacterial mutant lacking IcmS (45), a chaperone protein required for secretion of many Dot/Icm effectors (10). The MAPK activation could not be explained by the Mavs-dependent cytosolic pathway, as the ΔicmS mutant robustly induces interferon (45, 46). Thus, a previously unknown, IcmS-dependent signal led to activation of host MAP kinases in macrophages infected with L. pneumophila.
In a separate publication, we also demonstrated a specific innate immune response to five secreted L. pneumophila effector enzymes (Lgt1, Lgt2, Lgt3, SidI, SidL) that inhibit host translation through inactivation of the host elongation factor eEF1a (4, 5, 44). Induction of the immune response required the activity of the effectors, prompting us to call it the effector-triggered response (ETR) (15). We characterized several downstream consequences of the ETR, including NF-κB activation; robust transcription of a subset of genes, including stress response genes and proinflammatory cytokines; and production of cytokine protein. Mutant L. pneumophila lacking the five effectors was defective in induction of the ETR, as was the ΔicmS mutant (15).
The immune response to translation inhibition resembled the previously observed TLR/Nod-independent MAP kinase activation, in that both responses required type IV secretion and the chaperone protein IcmS (15, 45). Furthermore, we noted that some transcriptional targets in the effector-triggered response to translation inhibition have been reported to be induced downstream of MAPK signaling (7, 30, 45). Therefore, we hypothesized that the five effectors might also be responsible for the activation of host MAP kinases in TLR/Nod-deficient macrophages. However, a connection between these two findings—the PRR-independent activation of host MAP kinases and the response to the five bacterial effectors that inhibit protein translation—had not been established.
Here we find that activation of host MAP kinases in macrophages lacking TLR and Nod signaling is indeed due to the activity of the five L. pneumophila effectors that inhibit host translation. We demonstrate that these effectors shape the transcriptional response to L. pneumophila via MAP kinase activation. Finally, we show that other diverse translation inhibitors induce similar MAPK and transcriptional responses in macrophages, suggesting that translation inhibition itself—rather than direct detection of bacterial molecules—can elicit this response. These results provide an important link between two previously published reports, giving us a clearer picture of how the immune system may recognize and respond to pathogen-associated activities independently of PRR-PAMP interactions.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (41a). The protocol was approved by the Animal Care and Use Committee at the University of California, Berkeley (protocol number R301-0311BRC).
Mice and cell culture.
Macrophages were derived from the bone marrow of C57BL/6J mice (Jackson Laboratory) or Myd88−/− Nod1−/− Nod2−/− mice on the B6 background (generated from crosses at the University of California, Berkeley). Macrophages were derived by 8 days of culture in RPMI supplemented with 10% serum, 100 μM streptomycin, 100 U/ml penicillin, 2 mM l-glutamine, and 10% supernatant from 3T3–macrophage colony-stimulating factor cells, with feeding on day 5.
Bacterial strains.
LP02 is a streptomycin-resistant thymidine auxotroph derived from L. pneumophila LP01. The ΔflaA, Δ5, and Δ5 ΔflaA strains were generated on the LP02 background and have been described previously (15, 43). Mutants were complemented with the wild-type effector Lgt3 or a catalytically inactive mutant with a point mutation (15) expressed from the L. pneumophila sidF promoter in the plasmid pJB908, which encodes thymidine synthetase as a selectable marker.
Infection and stimulation.
For harvesting of RNA, macrophages were plated in 6-well dishes at a density of 1.5 × 106 cells per well and infected at a multiplicity of infection (MOI) of 1. For Western blot analyses, macrophages were plated in 6-well dishes at a density of 2 × 106 cells per well and infected at an MOI of 2. For enzyme-linked immunosorbent assay (ELISA), macrophages were plated in 24-well dishes at a density of 5 × 105 cells per well and infected at an MOI of 1. After infection, plates were centrifuged for 10 min at 400 × g. Where indicated, wells were treated with cycloheximide (10 μg/ml; Sigma), exotoxin A (500 ng/ml; List Biological Labs), or the p38 inhibitor SB203580 or the JNK inhibitor II (both at 1 μM; Calbiochem).
Quantitative RT-PCR.
Macrophage RNA was isolated at 4 h postinfection using an RNeasy kit (Qiagen) according to the manufacturer's protocol. RNA samples were treated with RQ1 DNase (Promega) prior to reverse transcription (RT) with Superscript III (Invitrogen). cDNA reactions were primed with poly(dT). Quantitative PCR was performed as described previously (40) using a Step One Plus RT-PCR system (Applied Biosystems) with Platinum Taq DNA polymerase (Invitrogen) and EvaGreen (Biotium). Transcript levels were normalized to those of Rps17. The following primer sequences were used: for Il1a, 5′-ATGACCTGCAACAGGAAGTAAAA and 3′-TGTGATGAGTTTTGGTGTTTCTG; for Dusp1, 5′-ACGGGGCTCAGCCTCCC and 3′-GTCAAGCATATCCTTCCGAGAA; for Fos, 5′-GAAGGGGCAAAGTAGAGCAG and 3′-CAACGCAGACTTCTCATCTTCA; and for Rps17, 5′-CGCCATTATCCCCAGCAAG and 3′-TGTCGGGATCCACCTCAATG.
Western blot analyses.
At the indicated time points postinfection or poststimulation, macrophages were lysed in radioimmunoprecipitation assay buffer supplemented with 2 mM NaVO3, 50 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, and 1× complete protease inhibitor cocktail (Roche). Protein levels were normalized using a micro-bicinchoninic acid kit (Pierce), and then lysates were separated on 10% NuPAGE bis-Tris gels (Invitrogen). Proteins were transferred to polyvinylidene difluoride membranes and immunoblotted with antibodies to phospho-p38, total p38, phospho-stress-activated protein kinase (SAPK)/Jun N-terminal protein kinase (JNK), or total SAPK/JNK (all from Cell Signaling). For phosphoprotein-specific blots, 2 mM NaVO3 and 20 mM NaF were added to blocking and antibody solutions.
ELISA.
After 24 h of infection, supernatants were collected, cleared by centrifugation, and analyzed by ELISA using paired interleukin-1α (IL-1α) antibodies (BD Biosciences). Recombinant IL-1α (eBioscience) was used as a standard.
RESULTS
Effector-triggered activation of host MAP kinases in L. pneumophila infection.
Previously, we showed that Dot/Icm+ L. pneumophila bacteria (but not Dot/Icm− bacteria) induced activation of both p38 and SAPK/JNK MAP kinases in Myd88−/− Rip2−/− macrophages, which lack TLR and Nod signaling in response to L. pneumophila (45). (While Toll/interleukin-1 receptor domain-containing adaptor inducing beta interferon [Trif]-dependent signaling downstream of TLR4 is intact in these macrophages, L. pneumophila LPS is a very poor substrate for TLR4; most TLR-dependent recognition of L. pneumophila occurs via TLR2 [18].) We confirmed this finding in Myd88−/− Nod1−/− Nod2−/− (triple-knockout [TKO]) macrophages (Fig. 1A), using flagellin-deficient bacteria to avoid Naip5/Nlrc4-dependent host cell death. To determine whether the recently described bacterial effectors that inhibit host protein translation (15) were responsible for MAPK activation, we infected the TKO macrophages with a mutant strain of L. pneumophila, called Δ5, that lacks these five secreted effectors (Lgt1, Lgt2, Lgt3, SidI, SidL [4, 5, 44]). MAP kinase activation was abrogated during infection with the Δ5 strain, as well as with the Dot/Icm− mutant, the ΔdotA strain (Fig. 1A). MAPK activation could be restored by complementing the Δ5 strain with one of the secreted effectors expressed constitutively from a plasmid (Fig. 1B). Importantly, however, complementation with a catalytically inactive effector did not rescue MAPK activation, indicating that the activity of the effector is required for MAPK activation (Fig. 1B).
Fig 1.

L. pneumophila effectors activate host MAP kinases in Myd88−/− Nod1−/− Nod2−/− (TKO) macrophages. (A) TKO macrophages were infected with the indicated L. pneumophila strains for the designated times. Cell lysates were blotted for phosphorylated and total p38 and SAPK/JNK. All panels in each row are from the same gel and exposure; intervening irrelevant lanes have been spliced out. (B) TKO macrophages were infected and blotted as described for panel A. Indicated strains were complemented with a plasmid expressing either a functional effector (plgt3) or a mutant with a point mutation lacking catalytic activity (plgt3*). wt, wild type; un, uninfected. Results shown are representative of at least three experiments.
Effector-triggered induction of MAPK-dependent host genes.
It has been demonstrated that MAP kinase activation by L. pneumophila leads to upregulation of specific transcriptional targets, including Il1a, which encodes the proinflammatory cytokine IL-1α (45). We wondered whether induction of these targets was also dependent on the five effectors. Consistent with a role for these effectors in MAPK activation, we observed defective induction of Il1a in TKO macrophages infected with L. pneumophila Δ5 (Fig. 2A). We also examined several other transcriptional targets of MAPK signaling, namely, Dusp1, which encodes a regulator of MAPK signaling (7), and the canonical MAPK target transcript Fos (30). As expected, a pharmacological p38 inhibitor abrogated induction of each of these genes (Fig. 2A to C). Furthermore, consistent with their role in MAP kinase activation (Fig. 1A and B), the five effectors were required for induction of these genes (Fig. 2B and C). Thus, in the absence of TLR and Nod signaling, MAP kinase activation by L. pneumophila-secreted effectors shapes the host transcriptional response to this pathogen.
Fig 2.
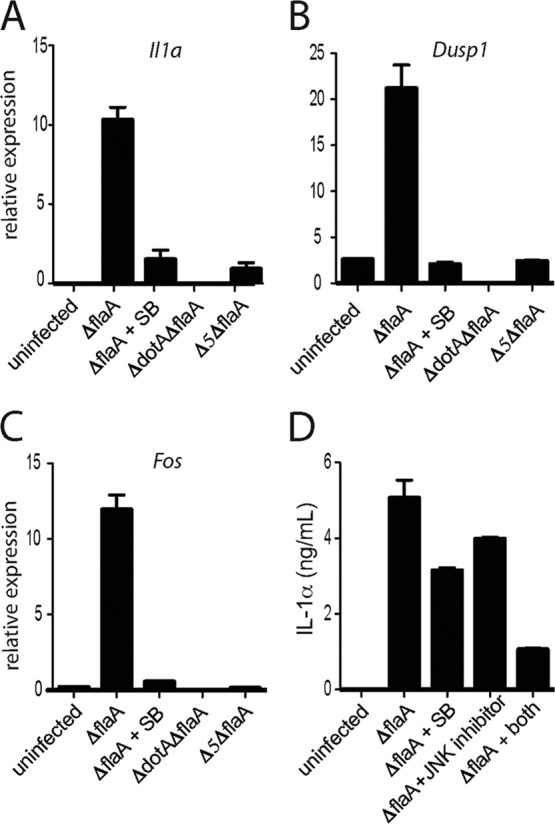
The five L. pneumophila effectors cause upregulation of MAPK-dependent host genes. (A to C) TKO macrophages were infected for 4 h with the indicated strains of L. pneumophila. A p38 inhibitor (SB203580 [SB]; 1 μM) was added 30 min before infection, where specified. Levels of Il1a (A), Dusp1 (B), and Fos (C) mRNA were measured by quantitative RT-PCR. (D) B6 macrophages were infected with L. pneumophila ΔflaA for 24 h alone or in the presence of p38 inhibitor (1 μM), JNK II inhibitor (1 μM), or both (1 μM each). IL-1α protein in the supernatant was measured by ELISA. Data shown are representative of three experiments (mean ± SD for panels A to C).
Since L. pneumophila inhibits host translation during infection, it was important to examine whether the transcriptional response downstream of MAP kinase activation could actually result in production of protein. To that end, we measured IL-1α protein in the supernatant of infected wild-type macrophages. Consistent with previous reports of cytokine induction by wild-type L. pneumophila (15), we found that infected macrophages did indeed secrete substantial levels of IL-1α (Fig. 2D), despite a greatly reduced translational capacity (15). This protein production could be partially inhibited by either a p38 or an SAPK/JNK inhibitor and almost completely inhibited by the combination of both inhibitors (Fig. 2D). We note that transcription of Il1a, measured at 4 h postinfection, can be abrogated by a p38 inhibitor alone (Fig. 2A), while full suppression of IL-1α protein production, measured at 24 h postinfection, requires inhibition of both p38 and SAPK/JNK (Fig. 2D). Taken together, these results suggest that p38 and SAPK/JNK can both mediate upregulation of IL-1α, but with different kinetics. Thus, host MAPK activation can result in production and secretion of protein and may therefore impact the course of infection and the development of the immune response.
Recapitulation of MAPK activation by noninfectious inhibitors of translation.
We wished to further investigate whether translation inhibition itself was the precise signal that led to activation of host MAP kinases. To do this, we treated uninfected macrophages with other inhibitors of translation: the pharmacological agent cycloheximide and the Pseudomonas aeruginosa-secreted toxin exotoxin A (ExoA). Interestingly, we found that treatment with each translation inhibitor resulted in activation of p38 and SAPK/JNK in TKO macrophages (Fig. 3A and B). We also observed robust upregulation of MAPK-dependent transcriptional targets in uninfected macrophages treated with cycloheximide or ExoA (Fig. 4A to C; note the log scale), indicating that translation inhibition alone is sufficient to induce these genes. Importantly, each agent that we tested—the L. pneumophila-secreted effectors, cycloheximide, and ExoA—has a unique structure and inhibits protein translation by a distinct mechanism. Thus, direct molecular recognition or nonspecific effects are unlikely to account for the common activation of MAP kinases. Instead, these data suggest that the block in translation itself leads to MAPK activation and transcription of downstream target genes.
Fig 3.

Other inhibitors of translation activate MAP kinases in macrophages. TKO macrophages were infected with L. pneumophila or treated with cycloheximide (CHX; 10 μg/ml) (A) or exotoxin A (ExoA; 500 ng/ml) (B) for the indicated time points. Cell lysates were blotted for phosphorylated and total p38 and SAPK/JNK. Results shown are representative of three experiments. un, untreated.
Fig 4.
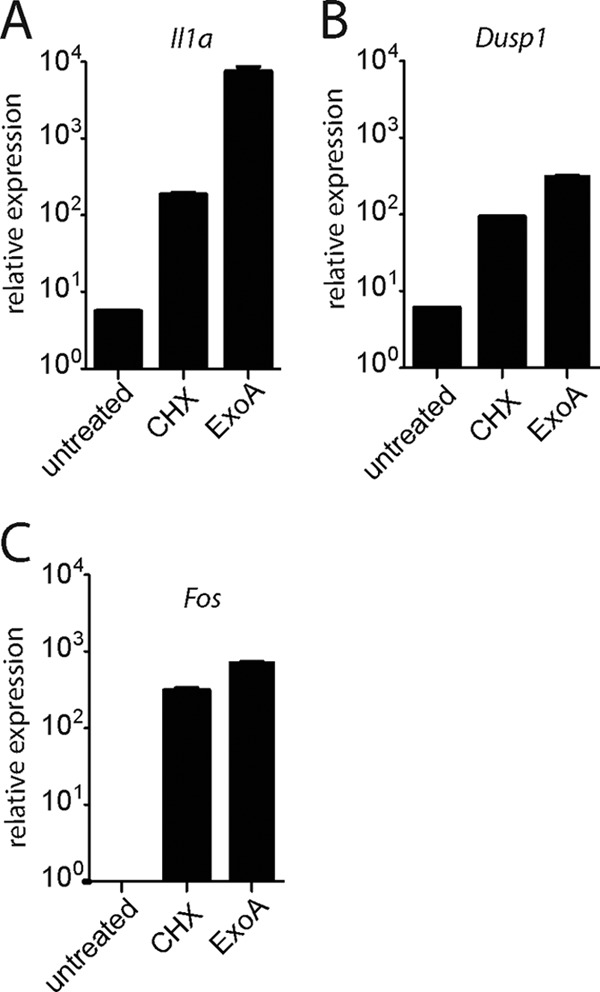
Translation inhibition is sufficient to induce transcription of target genes. Uninfected TKO macrophages were treated with cycloheximide (CHX; 10 μg/ml) or exotoxin A (ExoA; 500 ng/ml) for 4 h. Levels of Il1a (A), Dusp1 (B), and Fos (C) mRNA were measured by quantitative RT-PCR. Data shown are representative of 3 experiments (mean ± SD). Note that in contrast to the graphs in Fig. 2, data are expressed in log scale to accommodate the wide range of values (e.g., >1-log-unit difference between cycloheximide and ExoA in panel A).
Possible mechanisms.
We attempted to determine the mechanism of MAPK activation by translation inhibition in macrophages. Studies in other cell types have implicated the RNA-dependent protein kinase PKR and the ribotoxic stress response in activation of MAP kinases by certain translation inhibitors, for example, Shiga toxin (19, 27, 34). However, we were unable to block MAP kinase activation with a PKR inhibitor (CAS 608512-97-6; Calbiochem; data not shown), suggesting that a ribotoxic stress response is not involved in this cell type or with this stimulus. In addition, we examined whether translation inhibition might prevent resynthesis of some unidentified short-lived inhibitor of MAPK signaling, resulting in activation of MAP kinases. We reasoned that this hypothetical mechanism would resemble the previously reported activation of NF-κB by L. pneumophila, in which translation inhibition results in loss of the labile inhibitor of NF-κB, IκB, allowing NF-κB activation and transcription of target genes (15). However, contrary to our hypothesis, we observed intact MAP kinase activation in macrophages infected with L. pneumophila in the presence of two different proteasome inhibitors (data not shown). This suggests that MAPK activation does not require degradation of an inhibitor, although we have not definitely ruled out this possibility; proteasome inhibition may be incomplete, or a putative MAPK inhibitor could be degraded or inactivated in a proteasome-independent manner.
DISCUSSION
In previous papers, we demonstrated (i) the PRR-independent activation of host MAP kinases by L. pneumophila (45) and (ii) a specific immune response resulting from inhibition of host protein synthesis by L. pneumophila effectors (15). However, it was not clear whether the observed MAP kinase activation was related to inhibition of host protein synthesis; the latter report focused instead on the mechanism of NF-κB activation in response to translation inhibition. Here we have investigated a possible link between these two published reports and found that, indeed, they describe distinct components of a single immunosurveillance pathway—namely, host monitoring of the integrity of its translational machinery. We have shown that host MAP kinases are activated by a specific pathogen-encoded activity, resulting in alteration of host transcriptional and translational responses. Our results therefore provide an important supplement to the literature and are crucial for understanding the multiple downstream signaling events that occur upon inhibition of translation by a pathogen. In macrophages, MAP kinases activate both stress-related and immune-related genes, including proinflammatory cytokines; thus, it is logical that they should be activated in response to pathogen-induced stress.
Several toxins and small molecules that inhibit translation have previously been reported to induce MAP kinase activation in diverse cell types, including intestinal epithelial, monocyte, and fibroblast cell lines (19, 24, 27, 48). However, in these cases, the precise disruption that leads to MAPK activation is thought to be not translation inhibition itself but a ribotoxic stress response resulting from damage to the 28S rRNA and transmitted by the RNA-dependent protein kinase PKR (27, 47). Evidence for this model stemmed from the observation that agents that targeted the 28S rRNA peptidyl transferase center, such as anisomycin and Shiga toxins, activated MAP kinases in a PKR-dependent manner in a fibroblast cell line, while other translation inhibitors such as ExoA did not activate MAP kinases (27).
In contrast to these reports, all translation inhibitors that we tested—including ExoA—were capable of activating MAP kinases in macrophages. Furthermore, the MAPK activation that we observed could not be abrogated by inhibition of PKR. These results imply that there are cell-type-specific differences in the mechanisms of MAP kinase activation in macrophages compared with those in other cell types. Furthermore, given that each of the translation inhibitors that we tested has a distinct structure and mode of action, we favor the hypothesis that the block in translation itself—rather than either direct molecular recognition or more indirect effects, such as ribosomal damage—gives rise to MAP kinase activation in macrophages. As with other cell types (19, 24, 27, 34, 48), further research is needed to elucidate the molecular interactions that lead to MAP kinase activation downstream of translation inhibition in macrophages. Nevertheless, this report provides a novel example of immune activation by an intracellular pathogen and identifies both the bacterial factors that elicit the response and the host pathway that is activated.
Because L. pneumophila's natural hosts, amoebae, do possess MAP kinase pathways, it is possible that L. pneumophila has evolved to intentionally activate host MAP kinases to further its own survival (21). However, in the mammalian host, MAP kinase activation results in an immune response that does not exist in the amoebae and that may help restrict growth and spread of the bacteria (33). Translation inhibition by L. pneumophila also results in activation of the proinflammatory NF-κB pathway (15, 36), which is not present in amoebae and is therefore unlikely to be an intentional target of L. pneumophila survival strategies (21). In consideration of all these factors, we favor the hypothesis that MAP kinase activation in response to L. pneumophila represents a host mechanism that has evolved to detect disruptions in the vital process of protein synthesis. Indeed, such a mechanism may be conserved in metazoans: several recent papers report that translation inhibition also elicits an immune response in C. elegans and that this response is partially dependent on the p38 MAP kinase PMK-1 (13, 37, 39).
Conceptually, host detection of pathogen-associated activities bears resemblance to the well-established mode of PAMP-PRR recognition in several important ways. Since PRRs are relatively few in number and are hardwired in the germ line, they must target a relatively low number of slow-to-evolve microbial features. Unlike many microbial molecules, pathogen-associated activities cannot easily be mutated without negative consequences for the pathogen. Furthermore, many diverse pathogens—including both bacteria and viruses—perturb a relatively small set of host physiological processes, such as plasma membrane integrity, vesicle trafficking, host translation, and cytoskeletal dynamics (49). Thus, pathogen-associated activities are equivalent to a set of highly conserved, difficult-to-modify features that make attractive potential targets for innate immune recognition. While recognition of pathogenic activities may not provide information about the class of pathogen present, this information may be obtained by simultaneous detection of microbial molecules, chosen in turn for their high conservation and slow evolvability. Recognition of pathogenic activities therefore provides an important complement to the long-established recognition of microbial molecules that serves as the cornerstone of innate immunity.
ACKNOWLEDGMENTS
We thank J. von Moltke for technical assistance. We also thank Zhao-Qing Luo and Simran Banga for bacterial strains.
Work in R.E.V.'s laboratory is supported by NIH grants AI063302, AI075039, and AI080749. Work in S.S.'s laboratory is supported by NIH grant AI087963.
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Akamine M, et al. 2005. Differential roles of Toll-like receptors 2 and 4 in in vitro responses of macrophages to Legionella pneumophila. Infect. Immun. 73:352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Archer KA, Alexopoulou L, Flavell RA, Roy CR. 2009. Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell. Microbiol. 11:21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Archer KA, Roy CR. 2006. MyD88-dependent responses involving Toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires' disease. Infect. Immun. 74:3325–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belyi Y, et al. 2006. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U. S. A. 103:16953–16958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belyi Y, Tabakova I, Stahl M, Aktories K. 2008. Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190:3026–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berrington WR, et al. 2010. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur. J. Immunol. 40:3519–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boutros T, Chevet E, Metrakos P. 2008. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol. Rev. 60:261–310 [DOI] [PubMed] [Google Scholar]
- 8. Boyer L, et al. 2011. Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway. Immunity 35:536–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruno VM, et al. 2009. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 5:e1000538 doi:10.1371/journal.ppat.1000538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cambronne ED, Roy CR. 2007. The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188 doi:10.1371/journal.ppat.0030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiu YH, Macmillan JB, Chen ZJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coers J, Vance RE, Fontana MF, Dietrich WF. 2007. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9:2344–2357 [DOI] [PubMed] [Google Scholar]
- 13. Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. 2012. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fields BS, Benson RF, Besser RE. 2002. Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15:506–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fontana MF, et al. 2011. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7:e1001289 doi:10.1371/journal.ppat.1001289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fontana MF, Vance RE. 2011. Two signal models in innate immunity. Immunol. Rev. 243:26–39 [DOI] [PubMed] [Google Scholar]
- 17. Frutuoso MS, et al. 2010. The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect. 12:819–827 [DOI] [PubMed] [Google Scholar]
- 18. Girard R, et al. 2003. Lipopolysaccharides from Legionella and Rhizobium stimulate mouse bone marrow granulocytes via Toll-like receptor 2. J. Cell Sci. 116:293–302 [DOI] [PubMed] [Google Scholar]
- 19. Gray JS, Bae HK, Li JC, Lau AS, Pestka JJ. 2008. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 105:322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Growney JD, Dietrich WF. 2000. High-resolution genetic and physical map of the Lgn1 interval in C57BL/6J implicates Naip2 or Naip5 in Legionella pneumophila pathogenesis. Genome Res. 10:1158–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haenssler E, Isberg RR. 2011. Control of host cell phosphorylation by Legionella pneumophila. Front. Microbiol. 2:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hawn TR, et al. 2007. Altered inflammatory responses in TLR5-deficient mice infected with Legionella pneumophila. J. Immunol. 179:6981–6987 [DOI] [PubMed] [Google Scholar]
- 23. Hawn TR, Smith KD, Aderem A, Skerrett SJ. 2006. Myeloid differentiation primary response gene (88)- and Toll-like receptor 2-deficient mice are susceptible to infection with aerosolized Legionella pneumophila. J. Infect. Dis. 193:1693–1702 [DOI] [PubMed] [Google Scholar]
- 24. Hershko DD, Robb BW, Wray CJ, Luo GJ, Hasselgren PO. 2004. Superinduction of IL-6 by cycloheximide is associated with mRNA stabilization and sustained activation of p38 map kinase and NF-kappaB in cultured Caco-2 cells. J. Cell. Biochem. 91:951–961 [DOI] [PubMed] [Google Scholar]
- 25. Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283 [DOI] [PubMed] [Google Scholar]
- 26. Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11:404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iordanov MS, et al. 1997. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 17:3373–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janeway CA, Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216 [DOI] [PubMed] [Google Scholar]
- 30. Karin M. 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270:16483–16486 [DOI] [PubMed] [Google Scholar]
- 31. Katz SM, Habib WA, Hammel JM, Nash P. 1982. Lack of airborne spread of infection by Legionella pneumophila among guinea pigs. Infect. Immun. 38:620–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. LeibundGut-Landmann S, Weidner K, Hilbi H, Oxenius A. 2011. Nonhematopoietic cells are key players in innate control of bacterial airway infection. J. Immunol. 186:3130–3137 [DOI] [PubMed] [Google Scholar]
- 34. Leyva-Illades D, Cherla RP, Lee MS, Tesh VL. 2012. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by Shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 80:2109–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lightfield KL, et al. 2008. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9:1171–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Losick VP, Isberg RR. 2006. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203:2177–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McEwan DL, Kirienko NV, Ausubel FM. 2012. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11:364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Medzhitov R. 2010. Innate immunity: quo vadis? Nat. Immunol. 11:551–553 [DOI] [PubMed] [Google Scholar]
- 39. Melo JA, Ruvkun G. 2012. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149:452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monroe KM, McWhirter SM, Vance RE. 2009. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5:e1000665 doi:10.1371/journal.ppat.1000665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munro P, et al. 2004. Activation and proteasomal degradation of rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J. Biol. Chem. 279:35849–35857 [DOI] [PubMed] [Google Scholar]
- 41a. National Institutes of Health 2011. Guide for the care and use of laboratory animals, 8th ed National Institutes of Health, Bethesda, MD [Google Scholar]
- 42. Patel JC, Galan JE. 2006. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J. Cell Biol. 175:453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2:e18 doi:10.1371/journal.ppat.0020018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shen X, et al. 2009. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell. Microbiol. 11:911–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shin S, et al. 2008. Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4:e1000220 doi:10.1371/journal.ppat.1000220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stetson DB, Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103 [DOI] [PubMed] [Google Scholar]
- 47. Tesh VL. 2012. Activation of cell stress response pathways by Shiga toxins. Cell. Microbiol. 14:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thorpe CM, Smith WE, Hurley BP, Acheson DW. 2001. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 69:6140–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vance RE, Isberg RR, Portnoy DA. 2009. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zamboni DS, et al. 2006. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7:318–325 [DOI] [PubMed] [Google Scholar]
- 51. Zhu W, et al. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638 doi:10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]