

Abstract
Structure-specific characterization and quantitation is often required for effective functional studies of oligosaccharides. Inside the gut, HMOs are preferentially bound and catabolized by the beneficial bacteria. HMO utility by these bacteria employs structure-specific catabolism based on a number glycosidases. Determining the activity of these enzymes requires accurate quantitation of a large number of structures. In this study, we describe a method for the quantitation of human milk oligosaccharide (HMO) structures employing LC/MS and isotopically labeled internal standards. Data analysis was accomplished with a newly developed software tool, LC/MS Searcher, that employs a reference structure library to process LC/MS data yielding structural identification with accurate quantitation. The method was used to obtain a meta-enzyme analysis of bacteria, the simultaneous characterization of all glycosidases employed by bacteria for the catabolism of milk oligosaccharides. Analysis of consumed HMO structures confirmed the utility of a β-1,3-galactosidase in Bifidobacterium longum subsp. infantis ATCC 15697 (B. infantis). In comparison, Bifidobacterium breve ATCC 15700 showed significantly less HMO catabolic activity compared to B. Infantis.
Introduction
Human milk oligosaccharides (HMOs) are an important component of breast milk and provide health benefits that infant formula manufacturers want to mimic; however, the natural composition of human milk is far too complex to synthesize with current technology. Despite this limitation, if a few key structures were selected for their substantial contribution to milk, it would be within our community’s reach to improve infant formula and provide some of the immunological and other advantages that human milk confers. The next step towards the naturalization of infant formula is to identify and quantify specific HMO substrates for their specificity to promote growth of beneficial bacteria over pathogenic bacteria.
Quantitation of HMO catabolism is of general interest because the biological role reveals an evolutionary link between human milk and beneficial bacteria found in an infant’s gut.1-2 Although HMOs constitute one of the most abundant components1 of human milk, it is the intestinal bacteria that are able to catabolize them, not the infant.3 Furthermore, reduced risk for diseases has been demonstrated among breast-fed infants. Structure-specific studies may reveal the health-promoting mechanisms of HMOs and lead to improved infant formulas.4 Bacterial binding to HMOs and subsequent uptake is necessary for providing nutrients to beneficial bacteria and for blocking5 the binding between pathogenic bacteria and gut tissue.6
Bacterial consumption of oligosaccharides has been extensively studied on the basis of composition and for a few select structures.7-9 Methods for relative10 and absolute11 quantitation of oligosaccharides have been demonstrated previously.12-15 Relative quantitation has employed non-identical internal standards such as tetradeuterium-labeled pyridylamino monosaccharides and dextran ladders as well as structure-specific standards that differ from the analyte of interest by only a few heavy isotopes. Quantitation of oligosaccharide mixtures has benefitted substantially from using structurally identical internal standards that account for matrix changes at different elution times and provide the closest possible ionization efficiencies in regard to the analyte. Automated structure-specific analysis has been achieved with tandem MS; however, such methods are generally not able to distinguish isomeric oligosaccharides and may require derivatization.16-17
Oligosaccharide mixtures are typically characterized according to monosaccharide composition by mass spectrometry (MS). In some cases, the carbohydrate composition provides putative structures18; however, unambiguous determination of structures usually requires chromatographic separation of isomers prior to mass spectrometry.19-21,22 Due to the lack of structure libraries for LC/MS, structure-specific analysis is often performed with standard glycan arrays that measure real-time binding of analytes, such as proteins to immobilized carbohydrate standards. The binding events can be detected by a variety of structure-naïve spectrophotometric techniques. The major benefit of the traditional binding arrays is that the structure-specific binding is detected quantitatively with high certainty and sensitivity.23 The drawbacks are the requirements to obtain sufficient amounts of pure standards and to measure binding outside biological conditions. Our approach simulates an array that addresses both issues and presents structure-specific quantitation of non-immobilized (unbound) structures in a similar format as typically used for bound arrays.
Investigation of enzyme activity on glycans is much less common than binding studies. Structural changes of immobilized glycans cannot be easily monitored by spectrophotometric techniques. In addition, glycan reactions may involve enzymes that require the oligosaccharide substrate to be in free form. For example, catabolism requires glycans to be transported24 inside the bacterial cells for enzymatic digestion.25 We have previously employed MS to monitor the mass-specific changes of residual (unbound) glycans during in vitro catabolism.7 However, the method employed only mass spectrometry and therefore did not determine isomeric possibilities.15, 26 Any significant differences in catabolism that correlate with structure during in vitro catabolism may inform us of structure-specific cellular binding and hydrolysis as well as potential targets for improving prebiotic components in infant formula.
In this research, we describe a method for monitoring the catabolism of HMOs using liquid chromatography MS (LC/MS). By identifying and quantifying each component, we can monitor the activity of bacterial enzymes towards specific structures. We present the data in a glycan array format. The array’s structures were identified from a previously published library of over 75 neutral and acidic HMOs.27-28 The library contains the accurate mass and reproducible retention time of each structure, determined by glycosidase digestion and tandem mass spectrometry. In combination with highly reproducible nano-HPLC CHIP/TOF MS and a standard gradient program, the library enables rapid identification of linkage-isomers by LC/MS.
An in-house written software (LC/MS Searcher) has been developed to rapidly identify and quantify oligosaccharides analyzed with LC/MS according to the stable-isotope labeling method.15, 29-30 Many software applications are already freely available for general LC/MS applications such as MZMime31-32 and msInspect33. Many more software tools can be found on the web.34 In contrast to these tools, LC/MS Searcher uniquely automates the library-based identification and quantitation of HMOs with monodeuterated internal standards. LC/MS Searcher is further distinguished from other software tools by its use of a single spectrum at each peak apex to quantify the ratio of co-eluting ions. The combined experimental and analytical methods were used for the first time to quantify the structure-specific catabolism of HMOs from the supernatant of in vitro monoclonal bacteria cultures, specifically, the strains Bifidobacterium longum subsp. infantis ATCC 15697 and Bifidobacterium breve ATCC 15700. Previous work has demonstrated structure specific preferences for a set of standards, revealing some of the specificity of enzymes expressed by B. infantis.35 This application illustrates the utility in determining species and substrate-specific catabolism of oligosaccharides that may have future import in the design of infant formulas.
Methods
Milk oligosaccharide isolation and purification: human milk samples were obtained from the milk banks of San Jose, California and Austin, Texas. Each milk sample (5 mL) was extracted with four volumes (2:1) of the chloroform–methanol solution (v/v). The emulsion was centrifuged at 3500 RPM for 30 min and the lower chloroform layer containing the denatured proteins was discarded. The upper layer was collected and the fraction was freeze-dried. The resultant powder (freeze-dried oligosaccharide-rich fraction) was used for oligosaccharide analysis.
The extracted HMOs were used as bacterial cell growth substrate. Bifidobacterium longum subsp. infantis ATCC 15697 and Bifidobacterium breve ATCC 15700 were anaerobically cultivated (separately) on M17 media with a 2% HMO solution. The cell culture medium supernatant was removed near the end of the exponential growth phase. 50 μl of each batch of supernatant HMOs were reduced by 50 μl of 2 M NaBH4 for one hour at 65° C. The internal standard was generated for each supernatant sample: 50 μl of the original 2% HMO solution was reacted with 50 μl of 2 M NaBD4 for one hour at 65° C. Just prior to removing the borate salts with solid-phase extraction, the non-deuterated and deuterated HMOs were mixed. The non-catabolized samples (the zero time-point) were spiked at a ratio of 1:1 (non-deuterated:deuterated) and the catabolized samples were spiked up to a ratio of 100:1. The different spiking ratios were used to generate H/D ratios in the range of 0.05-20 for linear relative quantitation.
Spiked HMO samples were fractionated by solid-phase extraction using the non-porous graphitized carbon cartridge (GCC). Prior to use, the GCC was washed with 80% acetonitrile in 0.1% trifluoroacetic acetic acid (TFA) (v/v) followed by deionized water. After loading of the oligosaccharide mixture onto a cartridge, salts were removed by washing with several cartridge volumes of deionized water. The oligosaccharides were fractionated using 20% acetonitrile in water (v/v) and 40% acetonitrile in 0.05% TFA (v/v) as the eluting solvents. Each fraction (~9 mL) was combined and evaporated in vacuo prior to MS analysis.
HMOs were chromatographically separated and detected with the Agilent 6210 HPLC-Chip/TOF-MS (Santa Clara, CA). Time-of-flight (TOF) mass spectra were collected every 1.6 seconds with 16,000 transient scans per spectrum. The Chip LC contained a pre-concentration column (40 nL, 4 mm) and an analytical column (43 × 0.075 mm), both made from porous graphitized carbon (PGC). For the gradient, solvent A was 0.1% formic acid in 3% acetonitrile and solvent B was 0.1% formic acid in 90% acetonitrile. The sample (1 μl) was loaded onto the pre-concentration column for two minutes. The gradient program (in reference to solvent B) was 0% between 0-2.5 min, 0 to 16% between 2.5-20 min, 16-44% between 20-30 min, 44-100% between 30-35 min, isocratic at 100% between 35-45 min, and isocratic at 0% between 45 and 65 min. The pre-concentration column was run on 100% A at 4 μL/min, except when the column was in-line with the analytical column, which flowed on the described gradient program at 0.3 μL/min. HMOs were ionized under 1.7 kV spray voltage and the resulting data transients were combined into spectra at rates of either 5 spectra/sec or 0.63 spectra/sec (the former rate permitted more precise evaluation of the shift between the closely spaced deuterated and non-deuterated peak apices). The range of detection was m/z 100-3000. The data was collected in the positive mode and calibrated by a dual nebulizer electrospray source with a wide range of internal calibrant ions: m/z 118.086, 322.048, 622.029, 922.010, 1221.991, 1521.972, 1821.952, 2121.933, 2421.914 and 2721.895.
LC/MS results were exported into mzDATA36 format with Agilent’s MassHunter software and then analyzed with an in-house software tool written in Java called LC/MS Searcher. HMOs were identified by comparing with structures contained in the library that includes accurate precursor masses and retention times.
Results and Discussion
The workflow shown in Figure 1 describes the experimental procedure for quantitation of the relative change in amount of individual HMO isomers by a bacterial culture. The relative change in amount of each HMO structure was monitored for the purpose of finding the structure-specific preferences of enzymes that are expressed by the bacteria for binding and hydrolyzing the HMOs. 15, 25, 37-39
Figure 1.
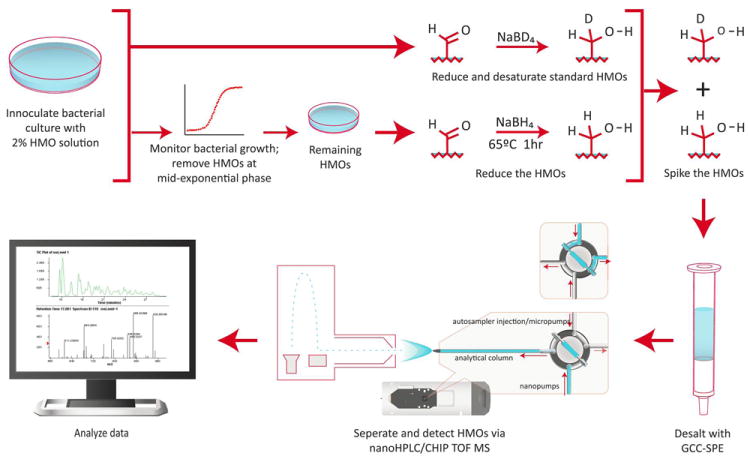
Experimental workflow including sample preparation and analysis.
Identification and quantitation of oligosaccharides
Isomer-specific analysis generally40 requires chromatographic separation prior to MS.41 Identification of oligosaccharides in this research was aided by a structure library that has been published previously.27-28 Identification was performed by comparing samples’ accurate masses and characteristic retention times to those belonging to known structures of the published libraries. Supplemental Figure 1 shows an extracted ion chromatogram (EIC) for three library compounds with the composition corresponding to 4Hex:2HexNAc. The three major components LNH, LNnH, and p-LNH were identified based on the library, which currently contains nearly 100 fully elucidated HMO structures.27-28 The linkage-isomers19 were distinguished experimentally by employing nanoflow porous graphitized carbon HPLC, which has been shown to be effective at separating isomers, while co-eluting glycans were composed of HMOs with different compositions and were readily distinguished by mass. The method has been shown to have high retention time reproducibility (Supplemental Figure 2).
Each structure was quantified relative to a co-eluting internal standard of the same glycan structure that contained a deuterium label. Relative quantitation was based on the ratio of the deuterated and non-deuterated isotopomer for each glycan. Figure 2 shows the implementation of Supplemental Eq. 1 for calculating the H/D ratio and the relative catabolism of each HMO structure. The corresponding isotopic envelopes for representative data are shown in Supplemental Figure 3. The first step in the determination is to calculate the H/D ratio for the original (zero time-point from growth curve) and catabolized samples. The H/D ratio for each structure is calculated separately with Supplemental Eq. 1. The next step is to generate an adjusted H/D ratio for the samples (excluding the zero time-point) for which the amount of spiked HMO was not the same for both the zero time-point and catabolized samples. While the amount of spiked HMO need not be the same for all samples, the differences in the amounts spiked into catabolized samples relative to the amount spiked into the zero time-point must be known. For instance, if the original sample were spiked with 50 μl of 2 mg/mL deuterated HMO, we may choose to spike the catabolized sample with 5 μl of 2 mg/mL deuterated HMO, as long as the relative amount of standard added to the catabolized versus zero time-point samples is included in Supplemental Eq. 2. Once the H/D ratio is adjusted according to the relative amount of spiked HMO, the percent catabolism is calculated with Supplemental Eq. 3.15, 29
Figure 2.
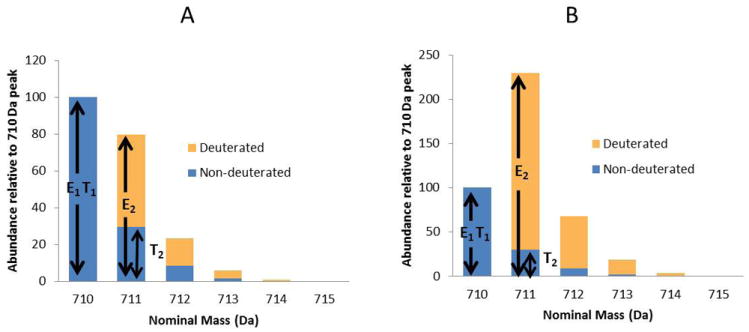
A. Theoretical isotopic distribution of LNT spiked 1:1 (nondeuterated:deuterated). B. Theoretical isotopic distribution of a spiked sample in which the non-deuterated LNT is 75% catabolized. The data in A and B must be applied separately to Eq. 1. E1 and T1 are adjusted to be the same value through Supplemental Eq. 1.
The H/D ratio method has been demonstrated previously29, 42; however, for the quantitation of HMOs it was based on composition15 as opposed to structure. While Supplemental Figure 4A shows a successful standard addition calibration for LNT within a complex matrix, Supplemental Figure 4B demonstrates one of the challenges of HMO quantitation with an external HMO standard by showing that the abundance of the HMO in the complex mixture is significantly attenuated by the matrix and not comparable to the detection of the standard in a simpler matrix. The matrix problem is expected to be specific to each elution time and to each sample, particularly for samples collected at different times of bacterial growth. Furthermore, Supplemental Figure 5 shows that relative quantitation for two HMO samples of similar complexity and composition can be less precise than relative quantitation with an internal (deuterated) standard.
We analyzed a series of nine standard mixtures with known H/D ratios to verify accuracy (slope of 1.0) and linearity (R2 of 0.99) between H/D 0.05-20 (Supplemental Figure 6). Excellent accuracy and linearity of H/D ratio measurement was only realized by first determining the linear response range of the absolute peak intensities of HMOs with TOF MS and diluting samples appropriately as well as spiking later time-points with less standard to obtain H/D ratios between 0.5-20. We have also shown that the ionization efficiencies of the non-deuterated and deuterated HMOs are equivalent (Supplemental Figure 7) and that the experimental first and second isotopes are within 5% of the theoretical relative abundances and are reproducible between runs within 2.0 ±0.4%.
Automated Quantitation with H/D Ratios
Automated structure-specific quantitation of HMOs should address at least five non-ideal chromatographic and mass-spectrometric behaviors: 1) unresolved chromatographic peaks, 2) unpredictable charge-state preferences, 3) suppression of oligosaccharide ionization, 4) retention time shifts, and 5) reduced confidence in identification of each compound after significant catabolism.
Supplemental Figure 8 shows the user interface for the in-house written software, LC/MS Searcher, that displays both the raw data and processed results. LC/MS Searcher in combination with nanoCHIP HPLC/TOF MS provides all the necessary corrections for these challenges within a single algorithm (Supplemental Figure 9). Previously published work43-45 has dealt with most of these issues; however, the authors are not aware of software that automates the structure-specific analysis of HMOs spiked with a pool of monodeuterated standards. A discussion of how we solved each of the non-idealities is presented here, followed by an application of the method toward identifying candidate structures to add to infant formulas.
Unresolved chromatographic peaks
The most common solution for quantifying partially resolved chromatographic peaks is to forgo deconvolution46-49 and assign peak boundaries that partition a portion of the unresolved peak to one compound. Such algorithms establish the peak boundaries with the first and second derivatives along the chromatogram and look for inflection points, local minima, and changes in the acceleration of slopes to identify the front and tail ends of peaks.46, 50 The peak area is then calculated within the artificial boundaries regardless of the underlying peak overlap.51 This solution is straight forward for peaks of equal area and peak shape; however, the expectation of equally sized and shaped peaks is rare. An example of the wide range in peak quality is shown with an EIC for a family of isomers in Supplemental Figure 10.
Our approach is simpler for the sake of introducing less ambiguity in assigning peak abundances. We use the ratio of two ions in a single mass spectrum at the apex of each chromatographic peak to quantify each structure. Quantitation at the peak apex is often affected less from peak overlap, which is generally greater at peak boundaries for the data shown here (Supplemental Figure 11). HPLC employing the more traditional stationary phases generally does not produce sufficient resolving power to separate carbohydrate isomers. However, several recent publications (see the work of Altman and others) have demonstrated the ability of graphitized carbon to effectively separate closely-related carbohydrate structures.52-54 Fortunately, co-elution of non-isobaric glycans is not a problem because they are simultaneously detected at different m/z values.
The use of a single mass spectrum to calculate the H/D ratio versus the use of peak area is demonstrated here. The H/D ratio for a particular sample was calculated with four unique methods for extracting peak abundance. The three standard methods for extracting peak abundance are illustrated in Supplemental Figure 12 for the quantitation of LNT and LNnT. Manual chromatographic deconvolution (Supplemental Eq. 5) was used to obtain H/D ratios of 0.50 and 0.54, respectively (Supplemental Figure 12A). An automated peak boundary method using a dynamically shifting baseline generated H/D ratios of 0.53 and 0.41 (Supplemental Figure 12B). An automated peak boundary method using a static baseline generated values of 0.52 and 0.58 (Supplemental Figure 12C). Finally, the automated method using a single mass spectrum near the peak apex generated H/D ratios of 0.52 and 0.49. These results show that the dynamically shifted baseline deviated the most from the accurate results (i.e. deconvolution). While this analysis does not distinguish the static baseline and single spectrum methods from each other, it does show non-inferiority of the single spectrum method relative to two other broadly applied techniques.
Chromatographic isotope shift
Deuterium-labeled HMOs are more hydrophobic than their non-labeled counterparts and generally elute one to two seconds earlier than the non-deuterated analogs. This systematic chromatographic shift, also explained as a kinetic isotope effect, has been observed previously and is referred to here as the d-shift.12
The d-shift complicates the use of individual mass spectra for calculating the H/D ratios because the two chromatographic peaks are slightly out of phase and produce artificially decreasing H/D ratios across the chromatographic peaks (Figure 3A). The d-shift is particularly problematic when a selected mass spectrum is near the peak boundaries. The artifact H/D ratios can be algorithmically corrected by shifting the deuterated peak to be in-phase with the corresponding non-deuterated peak. The result is a constant H/D ratio from all the mass spectra across the peak (Figure 3B). Constant H/D ratios allow quantitation from any point along the chromatographic peak.
Figure 3.
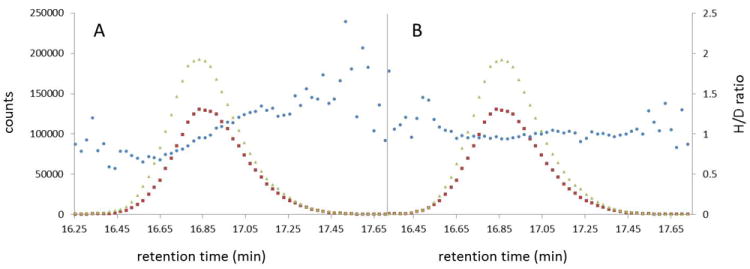
EIC’s for the deuterated (triangle) and nondeuterated (square) peaks of LNT with the H/D ratio (circle) calculated from each point along the peak. A. The H/D ratio increases across the peak because the deuterated peak elutes one second before the nondeuterated peak. B. The H/D ratio is constant across the peak after shifting the deuterated peak intensities to artificially co-elute with the nondeuterated peak.
Retention time shifts
The characteristic retention times had negligible shifts between runs (1 ±0.6 seconds) because the plumbing tolerances are tight and have an effective zero dead volume. With proper care of the HPLC system and careful preparation of the solvents, we have observed nearly identical retention times across months to years (within 6 seconds), approaching the reproducibility of gas chromatography.55 Furthermore, a sufficient amount of time (20 minutes) for column equilibration at the end of each run as well as two warm-up runs (with or without sample) prior to starting a sample set proved to be essential for ensuring reproducibility. Although not a problem in these experiments, for experimental outcomes where the retention times are not sufficiently reproducible, the data should be aligned algorithmically56 or a retention time library of standards should be prepared at the beginning of each series of runs.
Reduced confidence in identification of each compound with increasing catabolism
The deuterated peak profiles are always similar to the original HMO pool, regardless of the change in peak profiles of the catabolized sample. The overlay of the deuterated HMOs on top of the catabolized HMOs is beneficial when the EIC profiles of the catabolized HMOs are no longer comparable to the profile of the known HMO pool. The problem is exacerbated by the presence of several isomers that are detected as proximal chromatographic peaks that must be distinguished from the isomer of interest. The ambiguity is solved by the software feature that identifies peaks based on the deuterated EIC profiles. In addition to the algorithm, the user interface (UI) shows the deuterated EIC profile overlaid on top of the non-deuterated EIC, providing visual verification that the correct peaks were selected (Supplemental Figure 11 and 13).
In-house Software
We developed a software application to standardize and accelerate data analysis. Typical analysis time for one data file is 45 minutes by hand but mere seconds with LC/MS Searcher. Peak selection and quantitation is applied consistently for data sets with multiple files. The user interface includes features for displaying and searching all types and levels of LC/MSn data (Supplemental Figure 8). The chromatographic data can be visualized either two-dimensionally as intensity vs. retention time or three-dimensionally as a heat map (Supplemental Figure 14A). The heat map is unique in that it efficiently reveals impurities such as polymers (Supplemental Figure 14B) as well as isomer families (Supplemental Figure 14C) and could potentially be used as a sample “fingerprint.” The HMO isomer families can be seen as sequentially eluting spots or peaks of the same mass-to-charge ratio. Although ion suppression is significantly reduced for chromatographically separated compounds, a few abundant compounds appear to suppress co-eluting species as shown by the apparent “shadows” cast across the heat map by LNT, 2’FL, and the polymer impurities in Supplemental Figure 14D. In this way, the heat map can provide valuable information for method development regarding ion suppression. The LC/MS Searcher employs a simple algorithm (Supplemental Figure 9) that smooths the raw data for each EIC, identifies all local maxima (presumably the peak apices), and finds the local maxima closest to the library retention times for the structures of interest (illustrated in Supplemental Figure 15).
Structure-specific catabolism study
The ability to precisely discriminate isomers from complex HMO mixtures enables an unprecedented view of bacterial consumption of these important glycans. To illustrate this we examined the HMO isomer consumption pattern of two model bifidobacteria (B. infantis ATCC 15697 and B. breve ATCC 15700)—strains for which we previously characterized the HMO consumption, however only at the compositional level.7 At the time of the analysis, 75 library structures and 41 non-elucidated compounds were simultaneously monitored, providing precise results for 39 structures and 33 non-elucidated compounds with an average relative standard deviation of 11% ± 10 for the percent catabolism (Figure 4). All results are based on library structures except those beginning with the letter ’u’, indicating an abundant compound detected with accurate mass and retention time but where the structure has yet to be elucidated. The results show that certain structures were preferentially catabolized by B. infantis (representative chromatographic separation and identification data is included in Supplemental Figure 16). Catabolism of linkage-isomer HMOs varied significantly within isomer families. The heptamer family containing 4Hex:2HexNAc:1dHex included two prominent isomers (MFLNH I and MFLNH III) that eluted within 30 seconds of each other and yet showed significantly different percent catabolism: 20% ±3 and 83% ±0.5. The only structural difference between these isomers was the location of the fucose residue and its linkage, α(1-2) vs. α(1-3), (Supplemental Figure 16). The α(1-2) linkage is suspect for the decreased catabolism because of a virtually identical correlation in the 11-mer family 5Hex:3HexNAc:2dHex, where two isomers eluted within 30 seconds of each other and differed only in the location and linkage of a fucose, where the less catabolized isomer contained an α(1-2) linkage: 5230a and DFLNnO II of the 11-mer family were consumed 5% ±3 and 79% ±1. A similar structural trend was also observed within the heptamer family, 5Hex:2HexNAc, where the two structures (LNH and p-LNH), both containing one galactose-β(1-3) linkage and one galactose-β(1-4) linkage, were catabolized significantly more than another structure (LNnH) containing only galactose-β(1-4) linkages (56% ±1 and 48% ±3 compared to 6% ±0.5) (Figure 4 and Supplemental Figure 5 and 16).
Figure 4.

Percent consumption of 72 HMO structures by B. infantis ATCC 15697 at mid-exponential growth, as calculated by Supplemental Eq. 3. Orange circles are sized proportionally to the percent consumption.
The bacteria’s preference for α(1-3) fucose linkages over α(1-2) linkages can also be shown by comparing LNH (a non-fucosylated structure) to two isomeric structures that differ from LNH only by the presence of fucose: MFLNH I, containing α(1-2) fucose, and MFLNH III, containing α(1-3) fucose. While LNH showed intermediate percent catabolism, MFLNH I showed less catabolism and MFLNH III showed more (Figure 4 and Supplemental Figure 16). Such trends show structure-specific preferences of bifidobacteria and will inform future research toward selecting key HMOs to be synthesized and tested for bifidogenic benefits to humans.
Although most HMOs decreased in concentration during the B. infantis growth, a few HMOs increased (Figure 4 and Supplemental Figure 17). While this may be due to partial decomposition of larger HMOs, they may also be produced by the bacteria. These results require further studies.
Structure-specific catabolism of HMOs was also quantified from the exponential growth phase of B. breve ATCC 15700 as shown in Figure 5. The 67 HMOs shown in Figure 5 were on average significantly less catabolized than for B. infantis ATCC 15697. Percent catabolism for B. breve clustered around 21% ±12, indicating only minor catabolism and few structure-specific preferences as compared to the broader and higher range for B. infantis, 51% ±22. A representative example of the difference in structure-specificity between the two strains can be shown with the family of isomers with the composition 4Hex:2HexNAc. The catabolism by B. breve of LNH, LNnH and p-LNH is 17% ±4 for all three, whereas for B. infantis the catabolism is different for each: 56% ±1, 6% ±0.5, 48% ±3, respectively. These findings correlate with the total HMO catabolism measurements that were previously published for these strains. Furthermore, the previously published percent catabolism of LNnT by B. breve at 64.5% agrees with our measured value at 59% ±12.38
Figure 5.

Percent consumption of 67 HMO structures by B. breve ATCC 15700 at mid-exponential growth, as calculated by Supplemental Eq. 3. Orange circles are sized proportionally to the percent consumption.
Conclusion
Although the data looks intriguing with clear differences in the consumption pattern between the two organisms, further examination involving multiple replicates with more points along the timeline would be appropriate to make any biological conclusions and guide a collaboration to synthesize key structures and conduct clinical trials with modified infant formula. The method for quantifying structure specific consumption by MS in combination with liquid chromatography appears robust while allowing the study of large groups of oligosaccharides yielding enzymatic information that can be placed in an array format (Figure 4 and 5). Were these oligosaccharides bound to an array, it would be difficult to monitor their specific consumptions as they would not be present in their native unbound form. Furthermore, the quantitation of HMOs in this manner is expected to be more precise than label-free methods, such as those that take ion abundances into consideration. This application allows the characterization of global enzyme activity of organisms that employ a complicated suite of enzymes for oligosaccharide catabolism. While the application illustrated here is for single cultures in vitro, the method can be applied to large microbiomes in vivo.
Supplementary Material
Acknowledgments
Supported in part by grants from the Eunice K. Shriver National Institute of Child Health and Human Development Grant (HD059127), the National Center for Research Resources, a component of the National Institutes of Health, (UL1 RR024146). The infants of mothers delivering at term were enrolled in observational studies funded by the US Department of Agriculture (NRI-CSREES 2008-35200-18776) and supported by the UC Davis Foods for Health Institute. Special thanks to Rachel Strum for illustrating Figure 1 and to Babraham Bioinformatics for use of the mzViewer code available to the public at http://www.bioinformatics.bbsrc.ac.uk/projects/mzviewer
References
- 1.Petherick A. Nature. 2010;468:S5–7. doi: 10.1038/468S5a. [DOI] [PubMed] [Google Scholar]
- 2.Rockova S, Nevoral J, Rada V, Marsik P, Sklenar J, Hinkova A, Vlkova E, Marounek M. Int Dairy J. 2011;21:504–508. [Google Scholar]
- 3.Gnoth MJ, Kunz C, Kinne-Saffran E, Rudloff S. J Nutr. 2000;130:3014–20. doi: 10.1093/jn/130.12.3014. [DOI] [PubMed] [Google Scholar]
- 4.Zivkovic AM, German JB, Lebrilla CB, Mills DA. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4653–8. doi: 10.1073/pnas.1000083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoaf K, Mulvey GL, Armstrong GD, Hutkins RW. Infect Immun. 2006;74:6920–8. doi: 10.1128/IAI.01030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donovan SM. Br J Nutr. 2009;101:1267–9. doi: 10.1017/S0007114508091241. [DOI] [PubMed] [Google Scholar]
- 7.LoCascio RG, Ninonuevo MR, Freeman SL, Sela DA, Grimm R, Lebrilla CB, Mills DA, German JB. J Agric Food Chem. 2007;55:8914–9. doi: 10.1021/jf0710480. [DOI] [PubMed] [Google Scholar]
- 8.Stepans MB, Wilhelm SL, Hertzog M, Rodehorst TK, Blaney S, Clemens B, Polak JJ, Newburg DS. Breastfeed Med. 2006;1:207–15. doi: 10.1089/bfm.2006.1.207. [DOI] [PubMed] [Google Scholar]
- 9.Asakuma S, Hatakeyama E, Urashima T, Yoshida E, Katayama T, Yamamoto K, Kumagai H, Ashida H, Hirose J, Kitaoka M. J Biol Chem. 2011;286:34583–92. doi: 10.1074/jbc.M111.248138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J, Hashii N, Kawasaki N, Itoh S, Kawanishi T, Hayakawa T. J Chromatogr A. 2005;1067:145–52. doi: 10.1016/j.chroma.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 11.Fong B, Ma K, McJarrow P. J Agric Food Chem. 2011;59:9788–95. doi: 10.1021/jf202035m. [DOI] [PubMed] [Google Scholar]
- 12.Jang KS, Kim YG, Gil GC, Park SH, Kim BG. Anal Biochem. 2009;386:228–36. doi: 10.1016/j.ab.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 13.Gil GC, Kim YG, Kim BG. Anal Biochem. 2008;379:45–59. doi: 10.1016/j.ab.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 14.Ridlova G, Mortimer JC, Maslen SL, Dupree P, Stephens E. Rapid Commun Mass Spectrom. 2008;22:2723–30. doi: 10.1002/rcm.3665. [DOI] [PubMed] [Google Scholar]
- 15.Ninonuevo MR, Ward RE, LoCascio RG, German JB, Freeman SL, Barboza M, Mills DA, Lebrilla CB. Anal Biochem. 2007;361:15–23. doi: 10.1016/j.ab.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Tang H, Mechref Y, Novotny MV. Bioinformatics. 2005;21(Suppl 1):i431–9. doi: 10.1093/bioinformatics/bti1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ethier M, Saba JA, Ens W, Standing KG, Perreault H. Rapid Commun Mass Spectrom. 2002;16:1743–54. doi: 10.1002/rcm.779. [DOI] [PubMed] [Google Scholar]
- 18.Stahl B, Thurl S, Zeng J, Karas M, Hillenkamp F, Steup M, Sawatzki G. Anal Biochem. 1994;223:218–26. doi: 10.1006/abio.1994.1577. [DOI] [PubMed] [Google Scholar]
- 19.Yamagaki T, Nakanishi H. Anal Sci. 2001;17:83–7. doi: 10.2116/analsci.17.83. [DOI] [PubMed] [Google Scholar]
- 20.Pfenninger A, Karas M, Finke B, Stahl B. J Am Soc Mass Spectrom. 2002;13:1331–40. doi: 10.1016/S1044-0305(02)00645-1. [DOI] [PubMed] [Google Scholar]
- 21.Pfenninger A, Karas M, Finke B, Stahl B. J Am Soc Mass Spectrom. 2002;13:1341–8. doi: 10.1016/S1044-0305(02)00646-3. [DOI] [PubMed] [Google Scholar]
- 22.Ninonuevo M, An H, Yin H, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Electrophoresis. 2005;26:3641–9. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 23.Laurent N, Voglmeir J, Flitsch SL. Chem Commun (Camb) 2008:4400–12. doi: 10.1039/b806983m. [DOI] [PubMed] [Google Scholar]
- 24.Gnoth MJ, Rudloff S, Kunz C, Kinne RK. J Biol Chem. 2001;276:34363–70. doi: 10.1074/jbc.M104805200. [DOI] [PubMed] [Google Scholar]
- 25.Ward RE, Ninonuevo M, Mills DA, Lebrilla CB, German JB. Appl Environ Microbiol. 2006;72:4497–9. doi: 10.1128/AEM.02515-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ninonuevo MR, Park Y, Yin H, Zhang J, Ward RE, Clowers BH, German JB, Freeman SL, Killeen K, Grimm R, Lebrilla CB. J Agric Food Chem. 2006;54:7471–80. doi: 10.1021/jf0615810. [DOI] [PubMed] [Google Scholar]
- 27.Wu S, Tao N, German JB, Grimm R, Lebrilla CB. J Proteome Res. 2010;9:4138–51. doi: 10.1021/pr100362f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu S, Grimm R, German JB, Lebrilla CB. J Proteome Res. 2011;10:856–68. doi: 10.1021/pr101006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu JL, Huang SY, Chow NH, Chen SH. Anal Chem. 2003;75:6843–52. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 30.Guo K, Li L. Anal Chem. 2010 [Google Scholar]
- 31.Katajamaa M, Miettinen J, Oresic M. Bioinformatics. 2006;22:634–6. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 32.Pluskal T, Castillo S, Villar-Briones A, Oresic M. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.May D, Law W, Fitzgibbon M, Fang Q, McIntosh M. J Proteome Res. 2009;8:3212–7. doi: 10.1021/pr900169w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. [14 July 2012]; http://www.ms-utils.org/wiki/pmwiki.php/Main/SoftwareList.
- 35.Yoshida E, Sakurama H, Kiyohara M, Nakajima M, Kitaoka M, Ashida H, Hirose J, Katayama T, Yamamoto K, Kumagai H. Glycobiology. 2012;22:361–8. doi: 10.1093/glycob/cwr116. [DOI] [PubMed] [Google Scholar]
- 36.Orchard S, Taylor CF, Hermjakob H, Weimin Z, Julian RK, Jr, Apweiler R. Proteomics. 2004;4:2363–5. doi: 10.1002/pmic.200400884. [DOI] [PubMed] [Google Scholar]
- 37.Ward RE, Ninonuevo M, Mills DA, Lebrilla CB, German JB. Mol Nutr Food Res. 2007;51:1398–405. doi: 10.1002/mnfr.200700150. [DOI] [PubMed] [Google Scholar]
- 38.Locascio RG, Ninonuevo MR, Kronewitter SR, Freeman SL, German JB, Lebrilla CB, Mills DA. Microb Biotechnol. 2009;2:333–42. doi: 10.1111/j.1751-7915.2008.00072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marcobal A, Barboza M, Froehlich JW, Block DE, German JB, Lebrilla CB, Mills DA. J Agric Food Chem. 2010;58:5334–40. doi: 10.1021/jf9044205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takegawa Y, Deguchi K, Ito S, Yoshioka S, Sano A, Yoshinari K, Kobayashi K, Nakagawa H, Monde K, Nishimura S. Anal Chem. 2004;76:7294–303. doi: 10.1021/ac0493166. [DOI] [PubMed] [Google Scholar]
- 41.Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Anal Chem. 2007;79:5051–7. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 42.Guo K, Li L. Anal Chem. 2009;81:3919–32. doi: 10.1021/ac900166a. [DOI] [PubMed] [Google Scholar]
- 43.Katajamaa M, Oresic M. BMC Bioinformatics. 2005;6:179. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange E. Analysis of mass spectrometric data peak picking and map alignment. Freie Universtaet Berlin; Berlin: 2008. [Google Scholar]
- 45.Yao W, Yin X, Hu Y. J Chromatogr A. 2007;1160:254–62. doi: 10.1016/j.chroma.2007.05.061. [DOI] [PubMed] [Google Scholar]
- 46.Vivo-Truyols G, Torres-Lapasio JR, van Nederkassel AM, Vander Heyden Y, Massart DL. J Chromatogr A. 2005;1096:133–45. doi: 10.1016/j.chroma.2005.03.092. [DOI] [PubMed] [Google Scholar]
- 47.Vivo-Truyols G, Torres-Lapasio JR, van Nederkassel AM, Vander Heyden Y, Massart DL. J Chromatogr A. 2005;1096:146–55. doi: 10.1016/j.chroma.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 48.Nikitas P, Pappa-Louisi A, Papageorgiou A. J Chromatogr A. 2001;912:13–29. doi: 10.1016/s0021-9673(01)00524-6. [DOI] [PubMed] [Google Scholar]
- 49.Lacey R. Anal Chem. 1986;58:1404–1410. [Google Scholar]
- 50.Stevenson PG, Gritti F, Guiochon G. J Chromatogr A. 2011;1218:8255–63. doi: 10.1016/j.chroma.2011.08.088. [DOI] [PubMed] [Google Scholar]
- 51.Qiu R, Regnier FE. Anal Chem. 2005;77:7225–31. doi: 10.1021/ac050554q. [DOI] [PubMed] [Google Scholar]
- 52.Davies MJ, Smith KD, Carruthers RA, Chai W, Lawson AM, Hounsell EF. J Chromatogr. 1993;646:317–326. doi: 10.1016/0021-9673(93)83344-r. [DOI] [PubMed] [Google Scholar]
- 53.Forgacs E. Journal of Chromatography A. 2002;975:229–43. doi: 10.1016/s0021-9673(99)01250-9. [DOI] [PubMed] [Google Scholar]
- 54.Itoh S, Kawasaki N, Ohta M, Hyuga M, Hyuga S, Hayakawa T. J Chromatogr A. 2002;968:89–100. doi: 10.1016/s0021-9673(02)00951-2. [DOI] [PubMed] [Google Scholar]
- 55.Hua S, Lebrilla C, An HJ. Bioanalysis. 2011;3:2573–85. doi: 10.4155/bio.11.263. [DOI] [PubMed] [Google Scholar]
- 56.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78:779–87. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.