

Abstract
Evidence suggests that aldehydic molecules generated during lipid peroxidation (LPO) are causally involved in most pathophysiological processes associated with oxidative stress. 4-hydroxy-2-nonenal (4-HNE), the LPO-derived product, is believed to be responsible for much of the cytotoxicity. To counteract the adverse effects of this aldehyde, many tissues have evolved cellular defense mechanisms which include the aldehyde dehydrogenases (ALDHs). Our laboratory has previously characterized the tissue distribution and metabolic functions of ALDHs, including ALDH3A1, and demonstrated that these enzymes may play a significant role in protecting cells against 4-HNE. To further characterize the role of ALDH3A1 in the oxidative stress response, rabbit corneal keratocyte cell lines (TRK43) were stably transfected to over-express human ALDH3A1. These cells were studied following treatment with 4-HNE to determine their abilities to: a) maintain cell viability, b) metabolize 4-HNE and its glutathione conjugate, c) prevent 4-HNE-protein adduct formation, d) prevent apoptosis, e) maintain glutathione homeostasis, and f) preserve proteasome function. The results demonstrated a protective role for ALDH3A1 against 4-HNE. Cell viability assays, morphological evaluations and Western blot analyses of 4-HNE-adducted proteins revealed that ALDH3A1 expression protected cells from the adverse effects of 4-HNE. Based on the present results, it is apparent that ALDH3A1 provides exceptional protection from the adverse effects of pathophysiological concentrations of 4-HNE such as may occur during periods of oxidative stress.
Keywords: aldehyde dehydrogenase, ALDH3A1, metabolism, lipid peroxidation, 4-hydroxy-2-nonenal, cornea
Introduction
Solar ultraviolet radiation (UV-R) is a well-documented environmental insult to biological systems. Absorption of UV-R by organic molecules, including nucleic acids, proteins, fatty acids and others within living cells, can result in cellular damage through the formation of reactive oxygen species (ROS). ROS can initiate lipid peroxidation (LPO) by reacting with polyunsaturated fatty acids (PUFAs) in the cell membrane bilayer and thereby lead to the formation of: a) lipid radicals that propagate the reaction and b) lipid hydroperoxide reaction products. It is the degradation of the lipid hydroperoxides that yields a variety of breakdown products including more than 200 species of aldehydes (1). When compared with the ROS that initiated LPO, the aldehydes are relatively, allowing them to diffuse intra- and inter-cellularly from the site of their generation. Several of these aldehydic breakdown products are electrophilic in nature, including acrolein, malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE). These aldehydes readily react with cellular nucleophiles (including nucleic acids, proteins, and phospholipids) to elicit their cytotoxic and genotoxic actions (1).
4-HNE has received significant attention over the last 30 years after it was identified as the most cytotoxic breakdown product generated from LPO (2). It is also one of the most abundant α,β-unsaturated aldehydes generated from LPO through beta-cleavage of hydroperoxides originating from the ω-4 and ω-6 PUFAs, arachidonic and linoleic acid, respectively (2, 3). The high reactivity of 4-HNE is attributed to its three main functional groups (the carbon-carbon double bond, the carbonyl and the hydroxyl groups), which appear to react synergistically with biomolecules containing amino and thiol groups (4, 5). The cellular consequences of 4-HNE synergistic reactivities include growth inhibition, decreases in glutathione (GSH) levels, decreases in sulfhydryl- and thiol-containing proteins, inhibition of enzyme activities, inhibition of calcium sequestration by microsomes, inhibition of protein synthesis and degradation, as well as alterations in signal transduction and gene expression profiles (3, 6-12). Intracellular regulation of 4-HNE is controlled primarily through several biotransformation pathways (13-17). Excessive production of 4-HNE during periods of oxidative stress may lead to the saturation, inhibition or degradation of these normal metabolic pathways responsible for maintaining its intracellular homeostasis (18) and thereby result in its accumulation and consequent reactivity with other cellular constituents. As a direct result of the ability of 4-HNE to affect cellular functions, a great deal of research has been conducted over the last 15 years to understand the enzyme systems responsible for its metabolism and disposition (19).
The cornea, an avascular tissue at the anterior surface of the eye, is structurally and functionally organized to serve as a protective barrier between the external environment and the internal ocular elements. Like other surface tissues of the body, it is exposed to a variety of environmental assaults, including UV-R. The cornea comprises three distinct layers: a self-renewing outermost squamous epithelial layer, a collagenous stromal matrix and a thin endothelial layer. It possesses an armamentarium of non-enzymatic and enzymatic defense systems to combat oxidative stress, including high levels of albumin, ascorbate, ferritin, superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione-S-transferases (GST), aldose reductase (AR), alcohol dehydrogenases (ADH), and aldehyde dehydrogenases (ALDH), which enable this tissue to maintain its structural integrity and functional role in light transparency (19-21).
ALDH3A1 is an enzyme present in the corneal tissues that efficiently metabolizes 4-HNE (13, 14, 22, 23). This cytosolic, NADP+-dependent protein catalyzes the oxidation of a wide variety of endogenous and exogenous aldehydes to their corresponding carboxylic acids. [ALDH3A1 is also considered to be a mammalian corneal crystalline, attaining high concentrations in this ocular tissue (24). Its presence has been identified by immunofluorescence in both the epithelial and stromal matrix layers of the normal human cornea, but not in the endothelial layer (13, 25). It was originally identified as a highly expressed protein in the bovine cornea (26) and has subsequently been shown to constitute 10 to 40% of the total water-soluble protein in the corneas of mammalian species (25, 27, 28). The precise role of this enzyme in corneal function remains to be elucidated. However, data suggest ALDH3A1 plays a significant role as an enzymatic antioxidant in the protection of cells from 4-HNE-induced cytotoxicity (22, 29). Such a protective function of ALDH3A1 has also been shown in corneal stromal keratocytes exposed to a variety of oxidative stress-inducing agents, including H2O2, the chemotherapeutic agents mitomycin-C and the etoposide VP-16 (30). The capacity of ALDH3A1 to metabolize 4-HNE by direct conversion to 4-hydroxy-2-nonenoic acid (HNA) (13, 14) supports the notion that this enzyme is an integral part of the cellular defense mechanisms protecting corneal tissues from UVR-induced damage (13, 14, 22). In addition, conjugation of 4-HNE to glutathione via GST isozymes to form the 4-HNE glutathione conjugate, GS-HNE, has been reported in a number of tissues as a predominant metabolic pathway for detoxification of this aldehyde (15-17). Given that GSH conjugates may themselves adversely affect cellular function, elimination of GS-HNE may be an important step in driving this detoxification pathway. Accordingly, ALDH enzymes could contribute to this pathway by converting GS-HNE to GS-HNA. To date, no studies have reported a role for ALDH-mediated metabolism in the elimination of GS-HNE, although it has been alluded to in a review (31). Hence, the demonstration of a metabolic role of ALDH3A1 in eliminating the GS-HNE conjugate amplifies its significance in cellular protection from 4-HNE-induced toxicity.
In spite of the high abundance of ALDH3A1 in corneal tissues, primary culture and established cell lines of mammalian corneal cells lose constitutive ALDH3A1 expression (32). Therefore, to study the role of ALDH3A1 in the disposition of 4-HNE in the corneal stromal matrix, we utilized the SV-40-immortalized rabbit corneal stromal keratocyte cell line (TRK43), which were stably-transfected with human ALDH3A1 (30). This paper presents the results of a series of studies in which this transfected cell model was exposed to pathophysiological levels of 4-HNE. The objectives were to determine the effects of ALDH3A1 expression on: (i) 4-HNE-induced cytotoxicity, (ii) metabolism of 4-HNE and GS-HNE, (iii) 4-HNE-protein adduct formation, and (iv) cellular glutathione levels and proteasome activity. The results of these experiments demonstrated a role for ALDH3A1 in protecting cells from the deleterious effects of 4-HNE.
Materials & Methods
Reagents
All tissue culture media, supplements, growth factors, assay reagents, protein inhibitors and buffers were purchased from Gibco BRL (Gaithersburg, MD, USA) or Sigma-Aldrich Co (St. Louis, MO, USA) unless otherwise specified. Lipofectamine Plus and hygromycin reagents were purchased from Invitrogen (Carlsbad, CA, USA). The bicinchoninic acid (BCA) kit was purchased from Pierce Chemical Co (Rockford, IL, USA). The poly-vinylidene difluoride (PVDF) membranes were obtained from Immobilon-P Millipore (Bedford, MA, USA). 4-HNE was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). The monoclonal anti-4-HNE antibody was purchased from Oxis International Inc (Portland, OR, USA). Polyclonal anti-4-HNE antibody was provided as a generous gift from Dr. D. Peterson (University of Colorado Denver, Aurora, CO). The monoclonal anti-ALDH3A1 antibody was developed and described by our laboratory (13). Horseradish peroxidase-conjugated secondary antibody was obtained from The Jackson Laboratory (West Grove, PA, USA). The chemiluminescence assay kit was obtained from NEN Life Science Products (Boston, MA, USA). The Oxyblot detection kit was purchased from Chemicon International (Temecula, CA, USA). 4-HNE stock concentrations were prepared and confirmed by measurement of UV absorbance at 224 nm (extinction coefficient (ε) = 13750 M-1 cm-1; A224 = ε × concentration × 1 cm). GS-HNE was prepared and measured per the methods described by Tjalkens et al. (33).
ALDH3A1 Expressing Corneal Stromal Keratocyte cell line (TRK43)
SV-40-immortalized TRK43 cells were derived from rabbit corneal stromal keratocytes and were provided as a generous gift from Dr. James Jester (University of California at Irvine, Irvine, USA) (34). Cells were maintained at 37°C in a humidified 5% CO2 incubator and grown on 100 mm culture plates in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin solution. Parental TRK43 cells were transfected with the ΔpCEP4Δ mammalian expression vector containing human ALDH3A1 cDNA (ΔpCEP4Δ-ALDH3A1) or the empty vector (ΔpCEP4Δ) using Lipofectamine Plus reagent as described elsewhere (30). Stably transfected cells were selected in culture medium containing 0.4 mg/mL hygromycin for 4-6 wk before individual colonies were obtained. Hygromycin-resistant clones were isolated from individual colonies, further expanded and characterized for ALDH3A1 expression by enzymatic assay, silver staining and Western immunoblot analyse. The TRK43-ALDH3A1 Clone 10 showed high ALDH3A1 expression and therefore was used for all experiments described herein between passages 10 and 20. The mock cells (TRK43-vector) behaved the same as the parental TRK43 cells (data not shown).
Preparation of Whole Cell Extracts
Cell cultures were washed twice with ice-cold PBS, collected by scraping and resuspended in cell lysis buffer containing 25 mM Tris, 0.25 M sucrose (pH 7.4), protease inhibitors (0.5 μg/mL leupeptin, 0.5 μg/mL aprotinin, 1 μg/mL pepstatin, 100 μg/mL phenylmethylsulfonyl fluoride) and 0.1% Triton X-100. Cell lysates were subjected to three cycles of sonication for 5 s on ice using a Branson Sonifier 250 (VWR Scientific, Willard, OH) and cooled on ice for 30 s. The resultant cell lysates were centrifuged at 18,000 × g for 30 min at 4°C and the supernatants collected for Western blotting, ALDH enzyme activity and proteasome activity assays. Protein concentrations of supernatant samples were determined using the bicinchoninic acid (BCA) method according to the manufacturer’s protocol.
Cell Viability Assay and Calculation of LC50 for 4-HNE
Cell viability of TRK43 transfectants was measured using the MTT (3-[4,5-dimethylthiazol-2-yl]2,5-diphenyl tetrazolium bromide) assay (35). Briefly, cells (3 × 104) were seeded in 96-well tissue culture plates containing complete medium and incubated overnight at 37°C. The medium was removed and replaced with serum-free treatment medium containing 4-HNE (0 - 1 mM) for 1 hr. Cells were then allowed to recover by replacing the treatment medium with complete medium and incubated a further 2 hr. Following the recovery, cells were incubated in serum-free medium containing MTT (1 mg/mL) for 2 hr, followed by the optical density reading at a wavelength of 550 nm using a microplate reader (Thermomax, Molecular Devices Corp., Sunnyvale, CA, USA). The acute LC50 values for 4-HNE were calculated by regression analysis of cell viability data using the equation of a four-parameter logistic curve (SigmaPlot software, version 7.0, 2001).
Detection of 4-HNE Protein Adducts
Cells were treated with 4-HNE (0 - 100 μM) for 1 hr and whole cell lysate was prepared. The presence of 4-HNE-adducted proteins in these lysates was determined by Western immunoblotting using polyclonal rabbit anti-4-HNE-KLH antibodies (1:2000). Densitometry data were normalized to β-actin and analyzed using the Scion Image software (http://www.scioncorp.com). To confirm these results, Western immunoblots were repeated using the Oxyblot® protein oxidation detection kit per the manufacturer’s protocol.
Metabolism of 4-HNE and GS-HNE
The capacity of TRK43 cell lysate and human recombinant ALDH3A1 to metabolize 4-HNE and GS-HNE was measured by monitoring the production of NADPH at 340 nm over a 3 min interval using a Beckman DU-640 spectrophotometer (Fullerton, CA, USA) as previously described (22). The reaction solution consisted of 100 mM sodium pyrophosphate buffer (pH 8.0), 2.5 mM NADP+, 1 mM pyrazole (to inhibit alcohol dehydrogenase activity) and the enzyme (200 μg lysate or 10-50 μg recombinant ALDH3A1). Reactions were initiated by adding 5.0 mM benzaldehyde. Purification of human ALDH3A1 protein was accomplished using affinity chromatography as described previously (36).
DNA Fragmentation as a Measure for Apoptosis
Cells were seeded (5 × 105) in 100 mm tissue culture plates containing complete medium and incubated overnight at 37°C. The medium was removed and replaced with serum-free treatment medium containing 4-HNE (0 - 100 μM) for 1 hr. Cells were then allowed to recover for 9 hr by replacing the treatment medium with complete medium. At the end of recovery period, medium was removed and the cells were gently washed, collected by scraping, re-suspended in lysis buffer (10 mM Tris-HCl, pH 7.4 containing 10 mM EDTA, 10 mM NaCl, 0.5% SDS and 0.1 mg/mL proteinase K) and incubated overnight at 50°C. DNA was extracted using phenol-chloroform and subsequently incubated with RNase A (0.1 mg/mL) for 1 hr at 37°C. DNA samples were analyzed using conventional electrophoresis in a 1.5% (w/v) agarose gel and visualized under UV illumination using a Fluor-S MultiImager (Bio-Rad Laboratories, Richmond, CA, USA).
Measurement of Total Glutathione (GSH + GSSG) Levels
Cells were seeded (5 × 105) in 100 mm tissue culture plates containing complete medium and incubated overnight at 37°C. The medium was removed and replaced with serum-free treatment medium containing 4-HNE (0 - 50 μM) for 1 hr. Treatment medium was gently removed and replaced with complete medium and the cells further incubated for 0, 4 and 8 hr recovery. Following the recovery, cells were gently washed with ice-cold PBS and collected by scraping. An aliquot from each cell suspension was taken for cell counting using a hemocytometer and trypan blue staining in order to normalize total glutathione results. The remaining cells were collected by centrifugation and re-suspended in 10 mM HCl to inactivate γ-glutamyl-transpeptidase. Cells were lysed via three freeze/thaw cycles involving -80°C for 30 min and 37°C for 15 min. Protein within these cell lysates was precipitated by addition of 5% 5-sulfosalicylic acid and centrifugation at 15,000 × g for 10 min. Supernatants from these samples were then collected for total glutathione (GSH + GSSG) analysis according to the methods of Anderson (37). Briefly, 25 μL of sample supernatant was suspended in 875 μL of a sodium phosphate (143 mM) / Na4-EDTA (6.3 mM) buffer (pH 7.5) containing NADPH (0.248 mg/mL), 5,5’-dithiobis 2-nitrobenzoic acid substrate (DTNB; 0.6 mM) and glutathione reductase (250 U/mL). Each sample was monitored for the formation of 2-nitro-5-thiobenzoic acid (TNB) at 412 nm over a 10 min interval using a Beckman-Coulter DU-640 spectrophotometer. Total glutathione content was calculated from a GSH standard curve, normalized to total cell counts and the values were expressed as a percentage of the experimental control groups.
20S Proteasome Activity Assay
Cells were seeded (5 × 105) in 100 mm tissue culture plates containing complete medium and incubated overnight at 37°C. The medium was removed and replaced with serum-free treatment medium containing 4-HNE (0 - 20 μM) for 1 hr. Treatment medium was gently removed and replaced with complete medium and the cells further incubated for 0, 2, 16 or 24 hr. Following this recovery, cells were harvested and whole cell lysate were prepared as described above. Cell extracts were diluted to 0.1 mg/mL protein in PBS and assayed for chymotrypsin-like activity using the fluorogenic peptide substrate, succinyl-Leu-Leu-Val-Tyr-amidomethylcoumarin, in black 96-well plates (Costar, Corning Inc.) as described previously (38). This activity (as reflected by the increase of fluorescence associated with the release of 7-amino-4-methylcoumarin from the peptide substrate) was monitored at an excitation and emission wavelength of 360 and 430 nm (respectively) over a 2 hr interval at 37°C using a fluorescence plate reader (Spectra MAX Gemini EM, Sunnyvale, CA). Parallel samples were prepared in the presence of the proteasome inhibitor N-benzyloxycarbonyl-Leu-Leu-leucinal (MG132; 20 μM) in order to allow determination of the specific activity for the 20S proteasome. The rates of chymotrypsin-like enzymatic activity were then calculated by linear regression of the kinetic plots recorded.
Statistical Analysis
All experiments performed and all data points taken were developed in triplicate. Data are presented as means ± standard deviation (SD). All comparisons of mean values between groups were performed using one-way analysis of variance (ANOVA). P < 0.05 was considered to be statistically significant.
Results
Generation of high-expression ALDH3A1 corneal stromal fibroblast cell model
To study whether ALDH3A1 has a protective role against 4-HNE-induced toxicity in the corneal stromal keratocytes, a cell model was generated from parental TRK43 corneal stromal fibroblasts by stable transfection with an expression vector containing human ALDH3A1 cDNA. Silver stain (data not shown) and Western immunoblots using specific antibodies against human ALDH3A1 verified the expression of a protein band at 50 kDa in cells transfected with ALDH3A1 expression construct (TRK43-ALDH3A1), but not in mock cells (TRK43-vector) (Figure 1).
Figure 1.
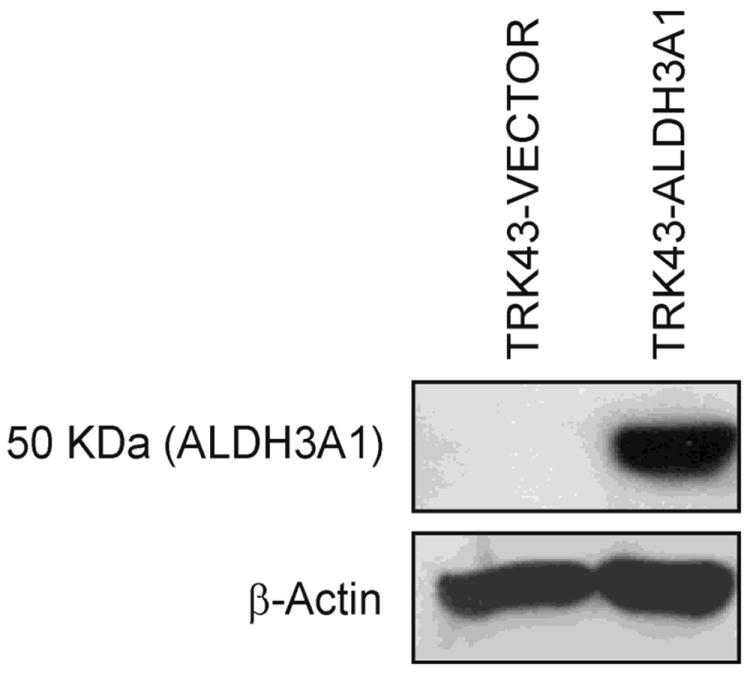
Western immunoblot of human ALDH3A1. Rabbit corneal keratocytes (TRK43) were stably transfected with empty vector (TRK43-VECTOR) or human ALDH3A1 (Clone 10) (TRK43-ALDH3A1). Expression of ALDH3A1 was only detected in TRK43-ALDH3A1 cells by Western immunoblotting. Expression of β-actin was examined as the loading control.
Expression of ALDH3A1 in corneal stromal fibroblasts protects cells from 4-HNE-induced cytotoxicity
Following 1 hr of 4-HNE treatment, cells were examined under light microscopy for gross morphology. At all tested 4-HNE concentrations, the control (TRK43-vector) cells revealed an increased stress response, characterized by a decrease in normal fibroblast morphology, cellular rounding and detachment from culture plates, (Figure 2). In contrast, the morphology of TRK43-ALDH3A1 cells appeared to be relatively preserved when 4-HNE concentrations were less than 50 μM. Such a protective effect of ALDH3A1 was also observed in the MTT cell viability study wherein 4-HNE-induced cell death occurred at a 3-fold higher LC50 in TRK43-ALDH3A1 cells (75 μM) than in TRK43-vector cells (25 μM) (Figure 2).
Figure 2.
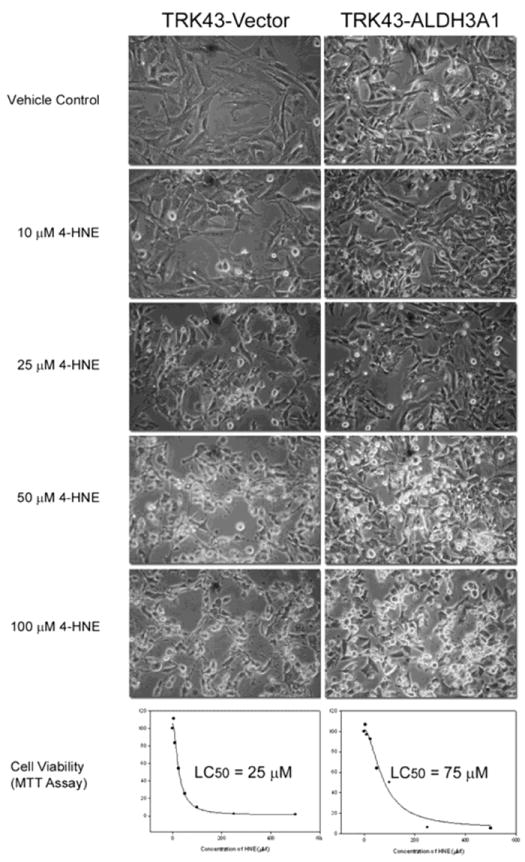
Gross morphology and cell viability of rabbit corneal keratocytes stably transfected with human ALDH3A1 (TRK43-ALDH3A1) or empty vector (TRK43-vector) following 1 hr of treatment with 4-HNE (10 - 100 μM) or vehicle (Vehicle Control).
ALDH3A1 prevents 4-HNE protein adduction in corneal stromal fibroblasts
As one of the most reactive products of LPO, 4-HNE interacts with numerous cellular proteins causing the formation of protein adducts and alterations in cellular function (5). Treatment with 4-HNE resulted in a concentration-dependent increase in 4-HNE-protein adduct formation in the TRK43 cells (Figure 3). The threshold for manifestation of protein adducts varied between the transfectants such that elevated levels of adducts were detectable in TRK43-vector (control) cells at 4-HNE exposure concentrations ≥ 10 μM. In TRK43-ALDH3A1cells, however, only at the highest 4-HNE exposure concentration (100 μM) were protein adducts observed. To confirm these results, Western immunoblots were repeated using the Oxyblot® protein oxidation detection kit. In these studies, protein oxidation patterns correlated with those produced by the polyclonal antibodies for 4-HNE-protein adducts (data not shown).
Figure 3.
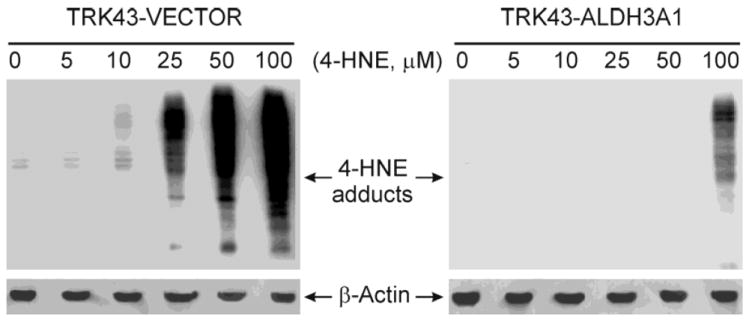
Western immunoblot of 4-HNE protein adducts in rabbit corneal keratocytes stably transfected with human ALDH3A1 (TRK43-ALDH3A1) or empty vector (TRK43-vector) following treatment with 4-HNE (5 – 100 μM) or vehicle (0 μM). Expression of β-actin was examined as the loading control.
4-HNE is effectively metabolized by ALDH3A1
The capacity of ALDH3A1 to metabolize 4-HNE or its glutathione conjugate, GS-HNE, was determined in TRK43 transfectant cell lysate and in purified recombinant human ALDH3A1 protein. In TRK43 cells, 4-HNE was metabolized in a concentration-dependent manner (Figure 4, upper panel). The extent of metabolism varied greatly between the transfectants such that ALDH activity at the highest 4-HNE concentration tested (400μM) was 0.48 nmol NADPH/min.mg protein in TRK43-vector cells and 264 nmol NADPH/min.mg protein in TRK43-ALDH3A1 cells. In contrast, when GS-HNE was used as the substrate (Figure 4, lower panel), both cell lines showed low ALDH activity with the greatest activity of 1.1 and 2.1 nmol NADPH/min.mg protein for control and ALDH3A1 extracts, respectively. Metabolism of 4-HNE and GS-HNE by purified human ALDH3A1 also occurred in a concentration-dependent manner, with peak ALDH activity corresponding to the highest concentration of substrate tested, viz. 600 μM. Under these circumstances, ALDH3A1 exhibited a much greater capacity to metabolize 4-HNE (1680 nmol NADPH/min.mg protein) than GS-HNE (170 nmol NADPH/min.mg protein) (Figure 5).
Figure 4.
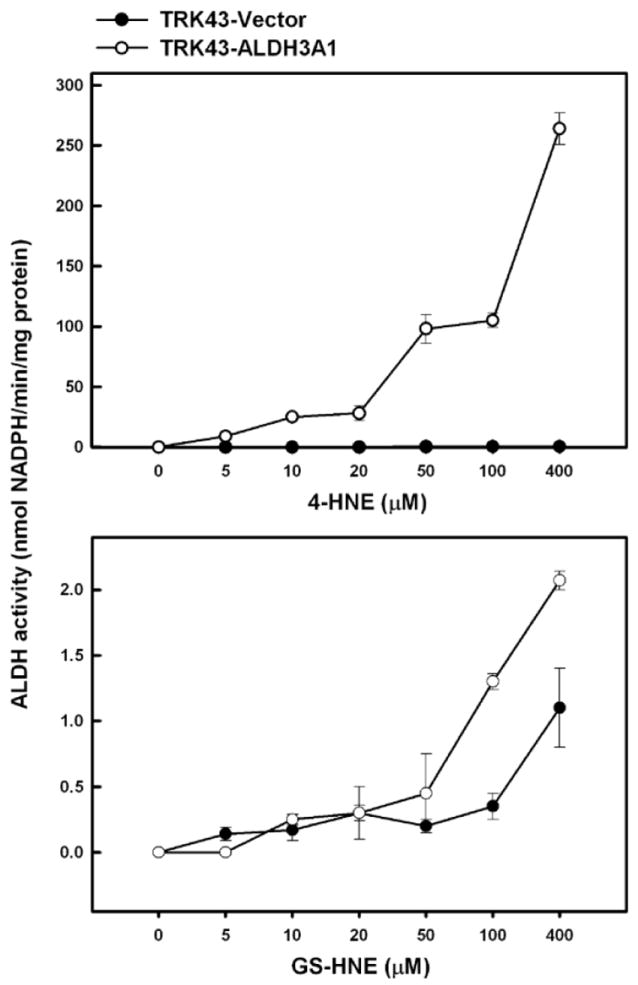
ALDH activity on 4-HNE and the GS-HNE in lysates of rabbit corneal keratocytes stably transfected with human ALDH3A1 (TRK43-ALDH3A1) or empty vector (TRK43-vector). ALDH activity was measured by monitoring the production of NADPH at 340 nm using various concentrations of 4-HNE or GS-HNE to initiate the reaction.
Figure 5.
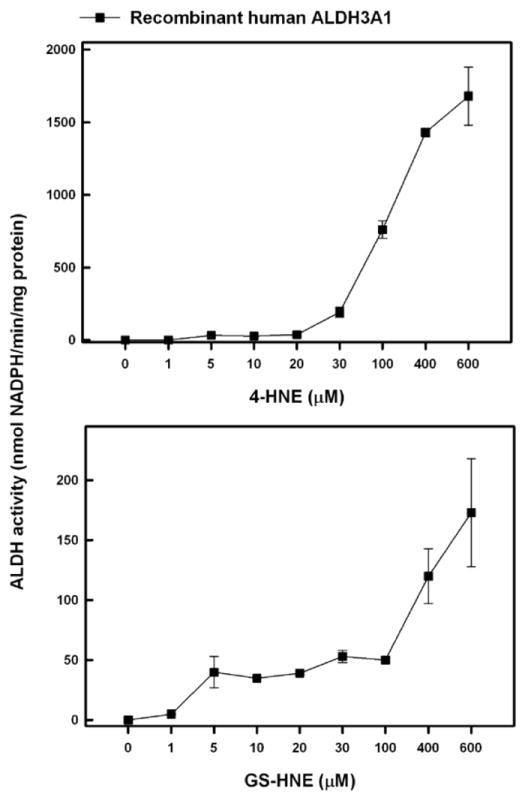
ALDH activity on 4-HNE and GS-HNE by purified human ALDH3A1. ALDH activity was measured by monitoring the production of NADPH at 340 nm using various concentrations of 4-HNE or GS-HNE to initiate the reaction.
ALDH3A1 protects cells from 4-HNE-induced apoptotic DNA fragmentation
The ability of ALDH3A1 to prevent 4-HNE-induced apoptosis was examined by monitoring DNA fragmentation, a hallmark of apoptosis. In TRK43-vector (control) cells, 4-HNE induced fragmentation at concentrations exceeding 10 μM (Figure 6). No such fragmentation was observed in TRK43-ALDH3A1 cells over the 4-HNE concentration range tested, viz. up to 40 μM.
Figure 6.
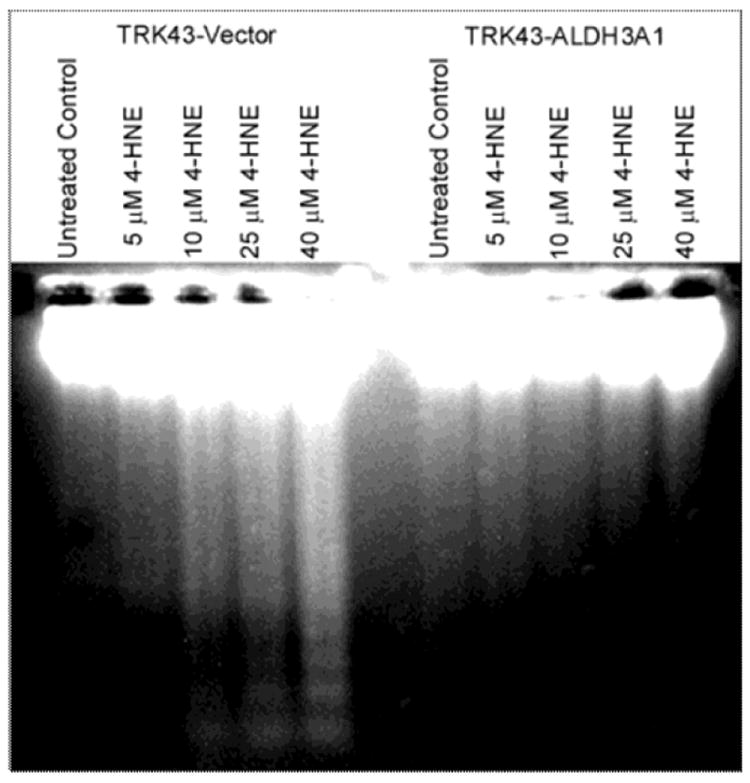
DNA fragmentation in rabbit corneal keratocytes stably transfected with human ALDH3A1 (TRK43-ALDH3A1) or empty vector (TRK43-vector) following 1 hr treatment with 4-HNE (5-40 μM) and 8 hrs recovery.
ALDH3A1 limits 4-HNE-induced depletion of total glutathione levels
Total glutathione (GSH + GSSG) was measured at various times following a 1 hr exposure to 4-HNE. Independent of the treatment concentration, 4-HNE led to acute and transient depletion of glutathione in TRK43-vector (control) cells, followed by an increase to basal or higher levels over the ensuing 8 hr (Table 1). In TRK43-ALDH3A1 cells, total glutathione levels remained stable over the 4 hr following 4-HNE exposure, and increased further by 30-80% in cells exposed to 25 or 50 μM 4-HNE.
Table 1.
Total Cellular Glutathione Levels after 4-HNE Exposure
| % Vehicle a | |||
|---|---|---|---|
| 0 Hr | 4 Hr | 8 Hr | |
| TRK43-VECTOR | |||
| Vehicle | 100 | 100 | 100 |
| 10 μM HNE | 43 ± 10 † | 199 ± 1 † | 154 ± 11 † |
| 25 μM HNE | 37 ± 7 † | 130 ± 11 † | 91 ± 13 |
| 50 μM HNE | 52 ± 4 † | 93 ± 3 | 71 ± 11 † |
| TRK43-ALDH3A1 | |||
| Vehicle | 100 | 100 | 100 |
| 10 μM HNE | 89 ± 9 | 101 ± 13 | 89 ± 13 |
| 25 μM HNE | 101 ± 14 | 79 ± 16 | 181 ± 6 † |
| 50 μM HNE | 83 ± 19 | 73 ± 11 † | 132 ± 5 † |
Total glutathione levels were measured in lysates of TRK43 cells stably transfected with human ALDH3A1 or empty vector following 1 hr treatment with 4-HNE (10 – 50 μM) and 0, 4 or 8 hrs recovery. Data are expressed as a percentage of the levels in vehicle-treated (vehicle) cells. Values are expressed as mean ± SD from triplicate experiments.
P < 0.05 one-way ANOVA compared with the Control cells in the same transfectant population at the same time point.
ALDH3A1 limits 4-HNE-induced inhibitory effects on 20S proteasome activity
Following 1-hr exposure to 4-HNE, a concentration-independent inhibition (≈50% of control) of the chymotrypsin-like activity of the 20S proteasome was observed in TRK43-vector (control) cells within 16 hr of treatment; this activity then climbed up to 140-225% of control 24 hr after the end of the 4-HNE exposure (Table 2). In contrast, TRK43-ALDH3A1 cells demonstrated little change in chymotrypsin-like proteasome activity immediately following the 4-HNE exposure; increases in this activity occurred 2 and 16 hr thereafter. By 24 hr, the activity had returned to levels comparable to those found in vehicle-treated cells (Table 2).
Table 2.
20S Proteasome Chymotrypsin-like Activity
| % Vehicle a | ||||
|---|---|---|---|---|
| 0 Hr | 2 Hr | 16 Hr | 24 Hr | |
| TRK43-VECTOR | ||||
| Vehicle | 100 | 100 | 100 | 100 |
| 10 μM HNE | 96 ± 8 | 57 ± 6 † | 54 ± 10 † | 137 ± 9 † |
| 20 μM HNE | 65 ± 5 † | 49 ± 7 † | 53 ± 7 † | 225 ± 7 † |
| TRK43-ALDH3A1 | ||||
| Vehicle | 100 | 100 | 100 | 100 |
| 10 μM HNE | 98 ± 3 | 123 ± 8 † | 157 ± 5 † | 106 ± 10 |
| 20 μM HNE | 91 ± 5 | 139 ± 7 † | 164 ± 4 † | 116 ± 14 |
20S proteasome chymotrypsin activity was measured in lysates of TRK43 cells stably transfected with human ALDH3A1 or empty vector following 1 hr treatment with 4-HNE (10 or 20 μM) or vehicle and 0, 2, 16 or 24 hrs recovery. Data are expressed as a percentage of the levels in vehicle treated (vehicle) cells. Values are expressed as mean ± SD from triplicate experiments.
P < 0.05 one-way ANOVA compared with the Control cells in the same transfectant population at the same time point.
Discussion
Aldehyde dehydrogenases catalyze the oxidation of a wide spectrum of endogenous and exogenous aldehydes to their corresponding carboxylic acids (39). As a corneal crystallin, ALDH3A1 is thought to play an integral non-enzymatic role in maintaining the corneal functions of light transparency and refraction (40). In addition, it has been implicated in the oxidative metabolism of 4-HNE, displaying apparent Km values of 44 μM - 110 μM (13, 22, 41). 4-HNE, a highly reactive LPO-generated aldehyde, significantly affects normal cell functions, as well as inducing cell death through necrotic and apoptotic processes through its ability to interact with thiol and amino groups. The enzyme activity of ALDH3A1 is sufficient to detoxify levels of 4-HNE encountered under pathophysiologic (>10 μM) conditions (3, 4). The capacity of ALDH3A1 to metabolize 4-HNE may serve to abrogate its cellular accumulation during periods of UVR-induced oxidative stress and thereby protect the viability and gross functions of corneal tissues. In support of this notion, human corneas with decreased ALDH3A1 activity were associated with a higher incidence of corneal pathologies (42). In addition, mice sensitive to corneal injury from UVB exposure have been shown to lack ALDH3A1 expression due to mutations in the structural gene of ALDH3A1 (23, 35).
In the present report, we sought to further characterize the protective role of ALDH3A1 against 4-HNE-induced cellular damage in corneal stromal keratocytes. An established rabbit corneal stromal fibroblastic cell line (TRK43) that does not constitutively express ALDH3A1 was used in these experiments. Human ALDH3A1 expression was induced in these cells through the establishment of stably-transfected clones. The ability of 4-HNE to elicit adverse cellular effects in the presence of elevated expression of ALDH3A1 in TRK cells was then assessed. Our results demonstrate that ALDH3A1 is effective in protecting the corneal stromal fibroblasts from 4-HNE-induced cytotoxicity through efficient metabolism of 4-HNE. ALDH3A1 expression reduced the toxic potency of 4-HNE (LC50 increased from 25 to 75 μM), decreased 4-HNE-protein adduct formation and resulted in DNA fragmentation occurring at higher 4-HNE exposure concentrations. Such protective effects of ALDH3A1 is in agreement with considerable 4-HNE-metabolizing ALDH activity observed in ALDH3A1-expressing cells and in purified human ALDH3A1 protein.
The possibility that ALDH3A1 could metabolize the 4-HNE conjugate, GS-HNE, was examined in the present study, with the idea that this may represent an alternative pathway by which 4-HNE could be detoxified. GS-HNE-initiated ALDH activity was observed in cell lysates derived from both control and ALDH3A1-expressing TRK cells at similar levels. However, such activity was minor when compared to the capacity of the ALDH3A1-expressing extracts to metabolize unconjugated 4-HNE, i.e., peak activity ≈ 2 versus 264 nmol NADPH/min.mg protein. The same trend was observed when GS-HNE-initiated ALDH activity was examined for purified human ALDH3A1 enzyme, with the peak activity against GS-HNE (170 nmol NADPH/min.mg protein) being considerably less than that against 4-HNE (>1600 nmol NADPH/min.mg protein). These data suggest that ALDH3A1 has the capacity to metabolize glutathione-conjugated 4-HNE, albeit to a relatively small extent. The significance of this activity remains to be established. Further, the interpretation of the result may be complicated by the possibility that some of the activity measured may have been related to unconjugated 4-HNE, i.e., 4-HNE that may not have conjugated to GSH in the GS-HNE preparation process. Whatever the case, the results of the present study would suggest that it is minor.
Glutathione (GSH), the most abundant thiol compound in animal tissues, is a 3-amino acid molecule. GSH plays a vital role in maintaining cellular redox balance viz. it’s nucleophilic and reducing properties that protect against reactive oxygen species, maintain protein sulfhydryl groups and enable conjugation to reactive intermediates (43). Its conjugation to 4-HNE is believed to be the prevalent pathway by which 4-HNE is metabolized in a number of ocular tissues (42, 44). The utilization of GSH by such conjugation may serve to deplete cellular GSH reserves that might otherwise function to protect the cell from other redox imbalances and thus exacerbate oxidative stress-related pathologies. Metabolism of 4-HNE to HNA by ALDH would, therefore, be predicted to preserve cellular GSH by directing 4-HNE away from the GSH-conjugation pathway. Our current study supports this notion, in part, by showing that total glutathione levels in ALDH3A1-expressing cells were not nearly as impacted by 4-HNE exposure as the control cells, particularly at lower concentrations. However, depletion of total GSH was observed in ALDH3A1-expressing cells at higher concentrations of 4-HNE (>10 μM) within 4 hours of exposure, suggesting GSH conjugation is not shifted to the ALDH3A1 oxidation pathway regardless of this enzyme’s level of expression and that GSH conjugation to 4-HNE likely functions in conjunction with ALDH3A1 oxidation of this aldehyde. It would have been interesting to delineate the contribution of reduced (GSH) and oxidized (GSSG) to the total glutathione that we measured. This would have allowed us to determine the GSH/GSSG ratio, a useful index of cellular redox status.
The 20S proteasome is essential for protein degradation in mammalian cells. It has been reported to be vulnerable to 4-HNE-induced inhibition, the consequences of which would result in an accumulation of oxidized proteins and overt cytotoxicity (45-47). The ALDH3A1-expressing TRK cells appear to be protected from 4-HNE induced inhibition of the chymotrypsin-like activity of the 20S proteasome. This is consistent with ALDH3A1 efficiently metabolizing 4-HNE, thereby preventing this reactive aldehyde from inhibiting the 20S proteasome.
The results presented herein demonstrate that expression of ALDH3A1 protects corneal stromal keratinocytes from pathophysiological concentrations of 4-HNE that could arise from UV-induced lipid peroxidation. The capacity of ALDH3A1 to oxidize 4-HNE appears to contribute to the cellular protection observed. This is the first study reporting that purified ALDH3A1 is capable of oxidizing GS-HNE as a substrate. While limited in extent (relative to 4-HNE), such an action may assist in the removal of this conjugate from the cell. Based on the present results and previous reports showing a low Km for human (45 μM; (13)), we speculate that the oxidative capacity of ALDH3A1 becomes increasingly important during periods of oxidative stress and helps preserve the integrity of corneal stromal keratocytes.
Highlights.
Expression of ALDH3A1 in corneal stromal fibroblasts protects cells from 4-HNE-induced cytotoxicity.
4-HNE is effectively metabolized by ALDH3A1.
ALDH3A1 prevents 4-HNE protein adduction.
ALDH3A1 limits 4-HNE-induced glutathione depletion and inhibitory effects on proteasome activity.
Acknowledgments
We thank our colleagues for valuable discussions and critical reading of the manuscript. This work was supported by NIH grants EY11490 (V.V.) and EY17963 (V.V.).
Abbreviations
- ADH
alcohol dehydrogenases
- ALDH
aldehyde dehydrogenases
- AR
aldose reductase
- CAT
catalase
- GSH
glutathione
- GPx
glutathione peroxidase
- GST
glutathione-S-transferases
- 4-HNE
4-hydroxy-2-nonenal
- 4-HNA
4-hydroxy-2-nonenoic acid
- LPO
lipid peroxidation
- MDA
malondialdehyde
- PUFAs
polyunsaturated fatty acids
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TRK
rabbit corneal keratocyte cell line
- UV-R
ultraviolet radiation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. The American journal of clinical nutrition. 1993;57:779S–785S. doi: 10.1093/ajcn/57.5.779S. discussion 785S-786S. [DOI] [PubMed] [Google Scholar]
- 2.Benedetti A, Comporti M, Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim Biophys Acta. 1980;620:281–296. doi: 10.1016/0005-2760(80)90209-x. [DOI] [PubMed] [Google Scholar]
- 3.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 4.Poli G, Schaur RJ. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life. 2000;50:315–321. doi: 10.1080/713803726. [DOI] [PubMed] [Google Scholar]
- 5.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 6.Dianzani MU. 4-Hydroxynonenal and cell signalling. Free Radic Res. 1998;28:553–560. doi: 10.3109/10715769809065811. [DOI] [PubMed] [Google Scholar]
- 7.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. HNE interacts directly with JNK isoforms in human hepatic stellate cells. The Journal of clinical investigation. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonarduzzi G, Arkan MC, Basaga H, Chiarpotto E, Sevanian A, Poli G. Lipid oxidation products in cell signaling. Free Radic Biol Med. 2000;28:1370–1378. doi: 10.1016/s0891-5849(00)00216-1. [DOI] [PubMed] [Google Scholar]
- 9.Leonarduzzi G, Robbesyn F, Poli G. Signaling kinases modulated by 4-hydroxynonenal. Free Radic Biol Med. 2004;37:1694–1702. doi: 10.1016/j.freeradbiomed.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Weigel AL, Handa JT, Hjelmeland LM. Microarray analysis of H2O2-, HNE-, or tBH-treated ARPE-19 cells. Free Radic Biol Med. 2002;33:1419–1432. doi: 10.1016/s0891-5849(02)01082-1. [DOI] [PubMed] [Google Scholar]
- 11.Kumagai T, Kawamoto Y, Nakamura Y, Hatayama I, Satoh K, Osawa T, Uchida K. 4-hydroxy-2-nonenal, the end product of lipid peroxidation, is a specific inducer of cyclooxygenase-2 gene expression. Biochem Biophys Res Commun. 2000;273:437–441. doi: 10.1006/bbrc.2000.2967. [DOI] [PubMed] [Google Scholar]
- 12.Feng Z, Hu W, Tang MS. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc Natl Acad Sci U S A. 2004;101:8598–8602. doi: 10.1073/pnas.0402794101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pappa A, Estey T, Manzer R, Brown D, Vasiliou V. Human aldehyde dehydrogenase 3A1 (ALDH3A1): biochemical characterization and immunohistochemical localization in the cornea. Biochem J. 2003;376:615–623. doi: 10.1042/BJ20030810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manzer R, Qamar L, Estey T, Pappa A, Petersen DR, Vasiliou V. Molecular cloning and baculovirus expression of the rabbit corneal aldehyde dehydrogenase (ALDH1A1) cDNA. DNA Cell Biol. 2003;22:329–338. doi: 10.1089/104454903322216671. [DOI] [PubMed] [Google Scholar]
- 15.Sharma R, Yang Y, Sharma A, Dwivedi S, Popov VL, Boor PJ, Singhal SS, Awasthi S, Awasthi YC. Mechanisms and physiological significance of the transport of the glutathione conjugate of 4-hydroxynonenal in human lens epithelial cells. Invest Ophthalmol Vis Sci. 2003;44:3438–3449. doi: 10.1167/iovs.03-0051. [DOI] [PubMed] [Google Scholar]
- 16.Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 17.Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Del Corso A, Dal Monte M, Vilardo PG, Cecconi I, Moschini R, Banditelli S, Cappiello M, Tsai L, Mura U. Site-specific inactivation of aldose reductase by 4-hydroxynonenal. Arch Biochem Biophys. 1998;350:245–248. doi: 10.1006/abbi.1997.0488. [DOI] [PubMed] [Google Scholar]
- 19.Lassen N, Black WJ, Estey T, Vasiliou V. The role of corneal crystallins in the cellular defense mechanisms against oxidative stress. Semin Cell Dev Biol. 2008;19:100–112. doi: 10.1016/j.semcdb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Rao NA, Romero JL, Fernandez MA, Sevanian A, Marak GE., Jr Role of free radicals in uveitis. Surv Ophthalmol. 1987;32:209–213. doi: 10.1016/0039-6257(87)90096-8. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Mehta G, Vasiliou V. Antioxidant defenses in the ocular surface. Ocul Surf. 2009;7:176–185. doi: 10.1016/s1542-0124(12)70185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pappa A, Chen C, Koutalos Y, Townsend AJ, Vasiliou V. Aldh3a1 protects human corneal epithelial cells from ultraviolet- and 4-hydroxy-2-nonenal-induced oxidative damage. Free Radic Biol Med. 2003;34:1178–1189. doi: 10.1016/s0891-5849(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 23.Choudhary S, Xiao T, Vergara LA, Srivastava S, Nees D, Piatigorsky J, Ansari NH. Role of aldehyde dehydrogenase isozymes in the defense of rat lens and human lens epithelial cells against oxidative stress. Invest Ophthalmol Vis Sci. 2005;46:259–267. doi: 10.1167/iovs.04-0120. [DOI] [PubMed] [Google Scholar]
- 24.Jester JV. Corneal crystallins and the development of cellular transparency. Semin Cell Dev Biol. 2008;19:82–93. doi: 10.1016/j.semcdb.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pappa A, Sophos NA, Vasiliou V. Corneal and stomach expression of aldehyde dehydrogenases: from fish to mammals. Chem Biol Interact. 2001;130-132:181–191. doi: 10.1016/s0009-2797(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 26.Abedinia M, Pain T, Algar EM, Holmes RS. Bovine corneal aldehyde dehydrogenase: the major soluble corneal protein with a possible dual protective role for the eye. Exp Eye Res. 1990;51:419–426. doi: 10.1016/0014-4835(90)90154-m. [DOI] [PubMed] [Google Scholar]
- 27.Piatigorsky J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: the “refracton” hypothesis. Cornea. 2001;20:853–858. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 28.Piatigorsky J. Review: A case for corneal crystallins. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics. 2000;16:173–180. doi: 10.1089/jop.2000.16.173. [DOI] [PubMed] [Google Scholar]
- 29.Townsend AJ, Leone-Kabler S, Haynes RL, Wu Y, Szweda L, Bunting KD. Selective protection by stably transfected human ALDH3A1 (but not human ALDH1A1) against toxicity of aliphatic aldehydes in V79 cells. Chem Biol Interact. 2001;130-132:261–273. doi: 10.1016/s0009-2797(00)00270-2. [DOI] [PubMed] [Google Scholar]
- 30.Lassen N, Pappa A, Black WJ, Jester JV, Day BJ, Min E, Vasiliou V. Antioxidant function of corneal ALDH3A1 in cultured stromal fibroblasts. Free Radic Biol Med. 2006;41:1459–1469. doi: 10.1016/j.freeradbiomed.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Alary J, Fernandez Y, Debrauwer L, Perdu E, Gueraud F. Identification of intermediate pathways of 4-hydroxynonenal metabolism in the rat. Chem Res Toxicol. 2003;16:320–327. doi: 10.1021/tx025671k. [DOI] [PubMed] [Google Scholar]
- 32.Jester JV, Budge A, Fisher S, Huang J. Corneal keratocytes: phenotypic and species differences in abundant protein expression and in vitro light-scattering. Invest Ophthalmol Vis Sci. 2005;46:2369–2378. doi: 10.1167/iovs.04-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tjalkens RB, Cook LW, Petersen DR. Formation and export of the glutathione conjugate of 4-hydroxy-2, 3-E-nonenal (4-HNE) in hepatoma cells. Arch Biochem Biophys. 1999;361:113–119. doi: 10.1006/abbi.1998.0946. [DOI] [PubMed] [Google Scholar]
- 34.Barry-Lane PA, Wilson SE, Cavanagh HD, Petroll WM, Jester JV. Characterization of SV40-transfected cell strains from rabbit keratocytes. Cornea. 1997;16:72–78. [PubMed] [Google Scholar]
- 35.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 36.Lindahl R, Baggett DW, Winters AL. Characterization of aldehyde dehydrogenase from HTC rat hepatoma cells. Biochim Biophys Acta. 1985;843:180–185. doi: 10.1016/0304-4165(85)90137-0. [DOI] [PubMed] [Google Scholar]
- 37.Anderson ME. Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- 38.Bulteau AL, Moreau M, Nizard C, Friguet B. Impairment of proteasome function upon UVA- and UVB-irradiation of human keratinocytes. Free Radic Biol Med. 2002;32:1157–1170. doi: 10.1016/s0891-5849(02)00816-x. [DOI] [PubMed] [Google Scholar]
- 39.Vasiliou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/s0009-2797(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 40.Estey T, Piatigorsky J, Lassen N, Vasiliou V. ALDH3A1: a corneal crystallin with diverse functions. Exp Eye Res. 2007;84:3–12. doi: 10.1016/j.exer.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 41.King G, Holmes RS. Human corneal aldehyde dehydrogenase: purification, kinetic characterisation and phenotypic variation. Biochem Mol Biol Int. 1993;31:49–63. [PubMed] [Google Scholar]
- 42.Choudhary S, Srivastava S, Xiao T, Andley UP, Srivastava SK, Ansari NH. Metabolism of lipid derived aldehyde, 4-hydroxynonenal in human lens epithelial cells and rat lens. Invest Ophthalmol Vis Sci. 2003;44:2675–2682. doi: 10.1167/iovs.02-0965. [DOI] [PubMed] [Google Scholar]
- 43.Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava SK, Singhal SS, Bajpai KK, Chaubey M, Ansari NH, Awasthi YC. A group of novel glutathione S-transferase isozymes showing high activity towards 4-hydroxy-2-nonenal are present in bovine ocular tissues. Exp Eye Res. 1994;59:151–159. doi: 10.1006/exer.1994.1093. [DOI] [PubMed] [Google Scholar]
- 45.Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Friguet B, Szweda LI. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Lett. 1997;405:21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 47.Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. The Journal of biological chemistry. 1999;274:23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]