

Abstract
Molecular dynamics simulations have to be sufficiently long to draw reliable conclusions. However, no method exists to prove that a simulation has converged. We suggest the method of “lagged RMSD-analysis” as a tool to judge if an MD simulation has not yet run long enough. The analysis is based on RMSD values between pairs of configurations separated by variable time intervals Δt. Unless RMSD(Δt) has reached a stationary shape, the simulation has not yet converged.
1. Introduction
Mathematical modeling and computational biology have proved extremely successful in the efforts to understand the mechanisms of immunologic reactions, for example, in modeling the T-cell proliferation dynamics following hepatitisC, HIV infection [1], or the growth of an immunogenic tumor [2]. Methods of statistical mechanics may successfully be applied in order to quantitatively understand the immune system [3, 4], and different modeling approaches are adequate for acquired immunity, as surveyed in a seminal paper by Perelson and Weisbush [5]. Modeling and simulation can be performed on different levels, starting at the top level with agent-based models [6, 7] for the cooperation of numerous large biomolecules in the formation of the immune synapse [8–11] down to more detailed models of antigen binding [12], recognition, and signaling [13–20]. T-cell proliferation modulated by interleukin2 has been simulated [21]. Applications aiming to predict the reaction to epitopes [22] improved vaccine design [23], vaccination against tumors [24], and for improved patient care have been devised [25–27], also in personalized medicine [28]. In particular, the mechanisms how T-cell receptors (TCRs) detect antigen peptides(p) presented by major histocompatibility complex (MHC) molecules can be investigated by molecular dynamics(MD) on a molecular or even atomic level [29–32]. However, TCRpMHC complexes are huge protein complexes, which have to be studied in water, which adds an even larger number of atoms to model and simulate. For these reasons, sufficiently long simulations are mandatory in order to obtain realistic results. This is even more so since molecular recognition phenomena may operate on rather long timescales as compared to the length of usual MD trajectories. Hence, efforts to assess sampling quality are mandatory in such simulations.
Given the trajectory of an actual MD simulation, it is clear from first principles that no formal check whatsoever can prove that complete sampling has been achieved. If parts of the phase space have not been visited yet, there is no possibility that this becomes evident from looking only at data from those other parts that have been visited. Zhang et al. [33] proposed a formal approach to decompose the phase space into Voronoi polyhedra [34, 35].
At any rate, formal checks of sampling quality can only draw on simulation data already produced and at the most detect possible shortcomings of those trajectories. The situation is analogous to tests for the randomness of pseudorandom-number generators [36]. One can never prove randomness as such. It is only possible to detect specific deviations from randomness, such as serial correlations—and any such detection works only with a preselected error rate.
Likewise, MD trajectories may be investigated for specific markers of nonrandomness, the most important type being trends of energy and molecular deformations. This work focuses on the latter, deviations in shape being quantified via the root mean square deviation (RMSD) [37] at time t 2 with respect to a given reference structure at time t 1:
| (1) |
where x i(t) is the position of atom i at time t and N is the total number of atoms in the molecule.
Often, the first frame of a trajectory (t 1) is used as a reference, and values of RMSD(t 1, t 2) are computed for all successive (t 2 > t 1) frames; see the black curve in Figure 1. RMSD monitored this way shows large rapid fluctuations on top of long-term variations and jumps. Generally, it is difficult to identify such long-term variations, which may relate to functional modes. A recent study [38] reflected the insufficiency of visual RMSD inspection alone.
Figure 1.
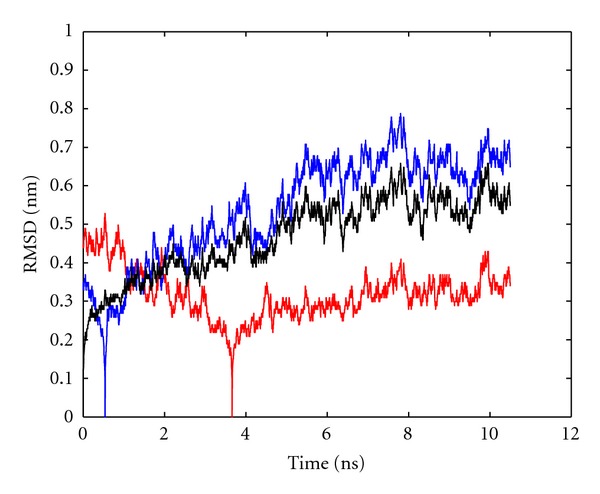
RMSD as a function of time for trajectory1 (10 ns). Black curve: RMSD between first configuration and all successive ones. Blue curve: for a given configuration (t j), RMSD to each of the other configurations of the same trajectory was computed and averaged, yielding . That value of t j for which is maximum is then adopted as a reference (t 1) to plot RMSD values of the whole trajectory. Red curve: as blue curve, but for minimum . Note that the RMSD between the reference configuration and itself is zero by definition.
Extensive work has been published on using the RMSD for the characterization of structural changes, drifts, and trends; see [33] and the references cited therein. Grossfield and Zuckerman [39] gives a seminal conceptual discussion of ergodicity, absolute and relative convergence and proposes checks for overall sampling quality. Block averaging was proposed [40] to reduce short-term fluctuations and to obtain more reliable indicators of long-term trends. However, averaging RMSD over all configurations within a block involves many different time intervals Δt = t j − t i, thus reducing the specificity of the resulting average for one particular time interval.
For this reason we consider the RMSD between pairs of configurations separated by a constant time lag Δt as described in Section 2.2.1. In this way we obtain more stable estimates (averages) which are nevertheless perfectly specific for a given time interval Δt.
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.1.1. Employed Structures
We applied the proposed methodology to a total of 6 MD simulations. For this purpose we used 2 different TCRpMHC complexes: an immunogenic wild-type peptide bound between TCR/MHC and the same TCR/MHC with a less immunogenic mutant peptide. We employed the crystal structure of the LC13 TCR bound to HLA-B*08 : 01 and the Epstein Barr Virus peptide FLRGRAYGL. This structure is available from the Protein Data Bank (PDB) [41] via PDB accession code 1mi5 [42] and is referred to as wild-type. For the mutant complex we substituted the side chain of tyrosine at position 7 of the peptide to alanine (Y7A). This was performed using SCWRL [43] since we could previously show that this tool is most appropriate for mutations in pMHC complexes [44, 45].
The LC13 TCR in complex with the FLRGRAYGL peptide and HLA-B*08 : 01 is an ideal test set for molecular dynamics simulations since this complex was crystallized and described in its parts and as a whole. Initially Kjer-Nielsenetal. crystallized the TCR [46] and the MHC [47] separately while they published a structure of the whole TCRpMHC system [42] afterwards. These available data give substantial insight into the LC13/EBV/HLA-B*08 : 01 system and led to the choice of our wild-type and mutated system for this study. The whole system is illustrated in Figure 2.
Figure 2.
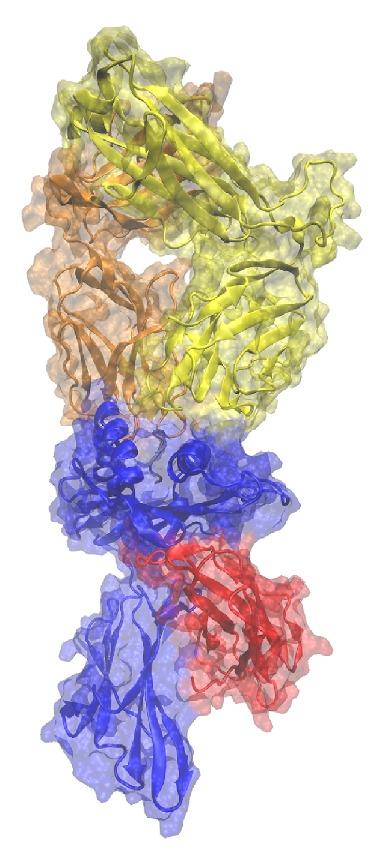
Illustration of the LC13 TCR in complex with HLA-B*08 : 01. Blue: MHC, red: β2-microglobulin, gray: peptide, orange: TCR alpha-chain, yellow: TCR beta-chain.
2.1.2. Molecular Dynamics Simulation Protocol
The wild-type and the mutant complex were simulated in independent runs for 10, 50, and 200 ns yielding a total of 6 simulations (see Table 1). The following protocol for the simulations was employed. First, we minimized the energy of the systems using a steepest descent method. Then we immersed the complexes in explicit SPC [48] artificial water baths allowing for a minimal distance of 2 nm between the box boundary and the protein. Next, we warmed the complex up to 310 K using position restraints. Finally, we carried out the simulations using GROMACS4 [49] and the GROMOS96 force field [50]. All further parameters were set in accordance with [51].
Table 1.
Molecular dynamics simulation runs.
| t max (ns) | Δt config (ps) | Peptide | n | |
|---|---|---|---|---|
| Trajectory1 | 10 | 3 | FLRGRAYGL | 3500 |
| Trajectory2 | 50 | 50 | FLRGRAYGL | 1000 |
| Trajectory3 | 200 | 50 | FLRGRAYGL | 4322 |
| Trajectory4 | 10 | 3 | FLRGRAAGL | 3500 |
| Trajectory5 | 50 | 50 | FLRGRAAGL | 1000 |
| Trajectory6 | 200 | 50 | FLRGRAAGL | 4322 |
2.1.3. RMSD Calculation
After the simulations were finished, we calculated the RMSD values for each configuration in a given trajectory with respect to every other configuration of the same trajectory using the standard g_rms function of GROMACS. This yields an n × n matrix of RMSD values where n is the number of configurations in the trajectory.
2.2. RMSD between Configurations of Trajectories
2.2.1. Averaging and Modeling of Lagged RMSD
Given one configuration x(t i) of an MD simulation as a reference, the RMSD to some other configuration x(t j) may be considered a “distance measure” along the time interval Δt = t j − t i. If Δt is short enough, it may be shifted along the whole trajectory and RMSD values be sampled. The average is characteristic for the difference between configurations separated by Δt in the particular simulation run considered.
Small values of Δt characterize configurations close to each other in time. Increasing Δt means to compare configurations more distant to each other in time, which are—intuitively speaking—“less related” to each other. This fact should be reflected in for increasing values of Δt.
As Δt increases, dependences should diminish and approach the level of “unrelated” or “independent” configurations. In order to quantify such a saturation trend, we applied the Hill equation [52]:
| (2) |
where the parameter a reflects the maximum value to which the function is asymptotic (“plateau value” ), τ is the time lag Δt for which (i.e., the value of Δt for which half-saturation is achieved), and the Hill coefficient γ is a parameter that determines the shape, that is, the level of sigmoidicity, of the model functions. The parameters were estimated by fitting the Hill equation (2) to the measured values of using the nlinfit function as implemented in the Statistics Toolbox of MATLAB(Mathworks, Natick, MA, USA). The maximum time lag Δt was chosen half the total simulation time of the respective trajectory.
2.2.2. Assessing the Influence of Initial Conditions
The initial phase t ≤ t offset of each MD simulation strongly depends on the starting configuration, usually a crystal structure, which can by no means be representative for a configuration obtained from a trajectory. This is even true after energy minimization and warming up. Hence, if the initial phase of an MD trajectory is included in , a bias will result. This is not only true for individual values of but also for the parameters estimated from the fit. In particular, the limiting plateau value will also be biased and depend on t offset: in fact, a = a(t offset). We modeled this dependence as
| (3) |
where a 0 is the limiting value, a 0 = a(t offset = ∞), and β and λ are scaling parameters. Note that a 0 is an extrapolated estimate for and t offset = ∞, that is, the estimate for an RMSD between two totally unrelated configurations of a trajectory, independent of initial phase effects.
Values for t offset were selected in the interval 0 ≤ t offset ≤ t max/2, where t max denotes the total simulation time of the respective trajectory.
3. Results and Discussion
3.1. Convergence of RMSD with Increasing Time Lag
Each panel of Figure 3 shows a representative plot (for reasons of conciseness we display results in the figures only for trajectory 1–3) of mean RMSD values (red circles, y-axis) obtained between configurations of one MD trajectory (see figure caption and Table 1), separated by respective time lags Δt (x-axis). As the time-lag between configurations increases, mean RMSD approaches a plateau.
Figure 3.
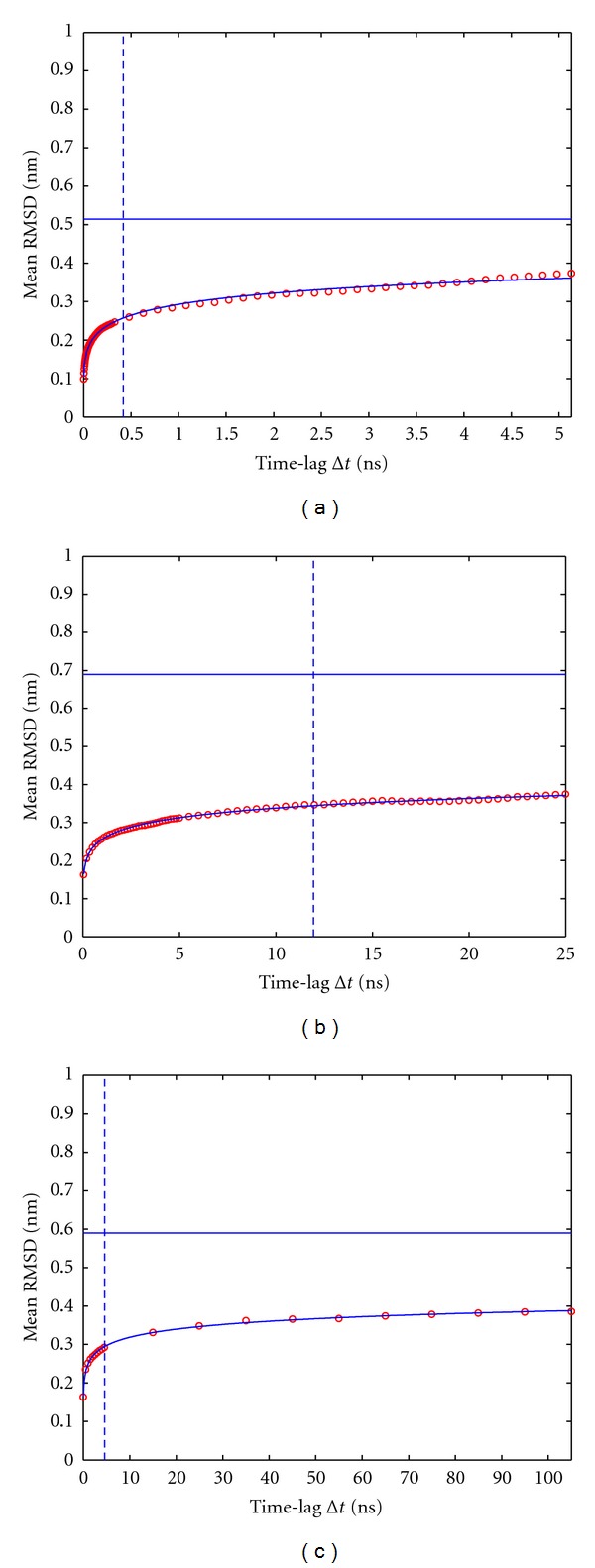
Dependence of mean RMSD on the time lag Δt for t offset = 0. (a) 10ns trajectory1, (b) 50 ns trajectory2, (c) 200 ns trajectory3; see Table 1. Note that due to the different total durations of the simulations, the maximum time lag considered (equal to half the simulation length) varies. Vertical lines indicate the time interval τ, for which half-saturation is achieved; see also (2). The horizontal line indicates the estimated plateau of the mean RMSD, corresponding to the parameter a in (2).
Fits of the model (2) to the values of are displayed as solid lines. Parameters obtained from the fits can readily be interpreted as follows. The estimate for parameter a in (2) represents the limiting value of and is indicated by the horizontal line. The estimate of the parameterτ corresponds to a “characteristic” (“half-saturation”) time interval Δt and is shown as a vertical line in the plots.
Note that the initial phase of each trajectory strongly determines the shape of which should be properly represented by the fitted model. In contrast, the remainder of each trajectory is characteristic for the long-term trend of . Although it shows much smaller changes in RMSD, the remainder contains naturally by far more data points as compared to the initial phase. To achieve an appropriate balance, we increased the lag length in small steps during an initial phase (cf. the initially dense succession of red circles in Figure 3) and in larger steps (more loose succession of circles) later on. This procedure puts increased weight on the data points for the initial phase.
3.2. Influence of the Offset
Applying different temporal offsets t offset before starting to analyze the respective trajectories changes the dependence of as a function of the time lag Δt; see the typical results displayed in Figure 4.
Figure 4.

Dependence of mean RMSD on t offset for trajectory1 (10 ns). (a): t offset = 0 ps, (b): t offset = 200 ps. Vertical lines indicate the time lag τ, for which half-saturation is achieved; see also (2). The horizontal line indicates the estimated plateau of the mean RMSD, corresponding to the parameter a in (2).
The larger the offset t offset from the start configuration, the smaller the time lag Δt necessary for the system to level off to its plateau. The exemplary display of the dependence of fitted parameters on t offset, as shown previously, is systematically analyzed as follows; see Figure 5. With increasing t offset both the plateau value a and the half-saturation parameter t decrease, whereas the shape parameter γ is fairly constant and almost independent of t offset.
Figure 5.
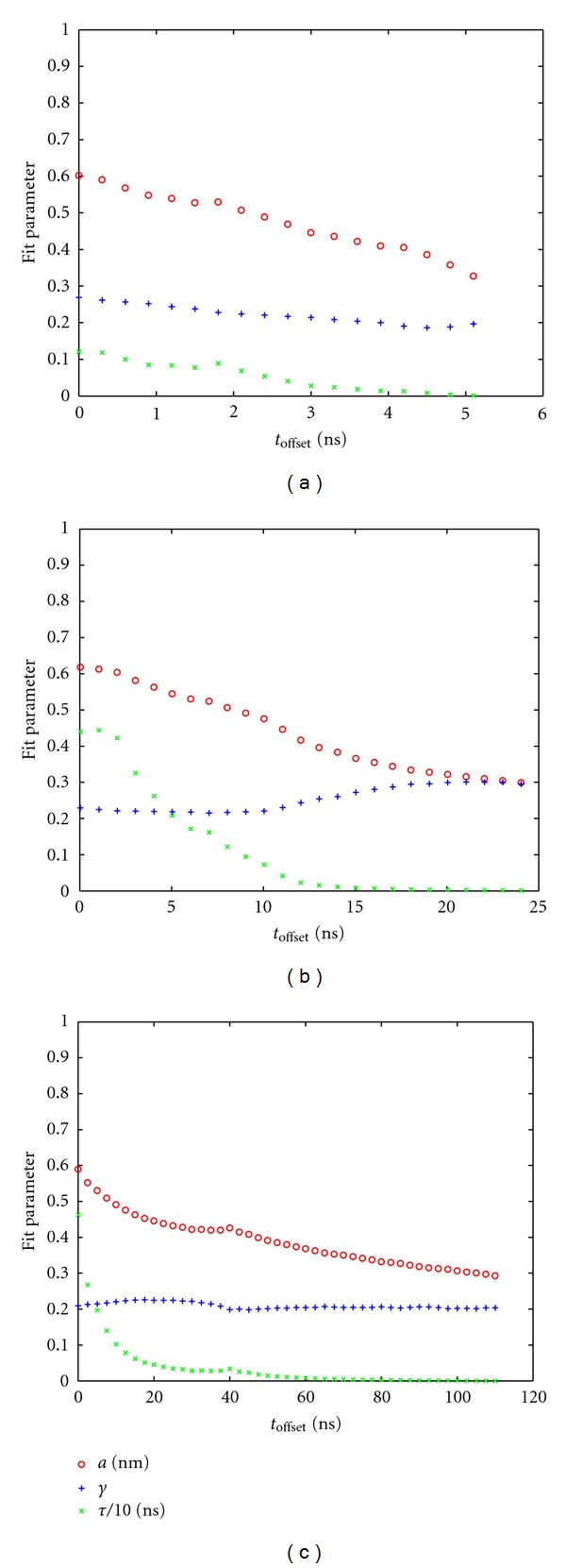
Increasing t offset changes fitted parameters of , (2). (a) 10 ns trajectory, (b) 50 ns trajectory, (c) 200 ns trajectory; see Table 1.
In a next step the systematic dependence of the extrapolated plateau on t offset was fitted via (3); see Figure 6. The monoexponential decay model of (3) represents the most parsimonious choice, given the shape of the simulation results as displayed in Figure 6.
Figure 6.
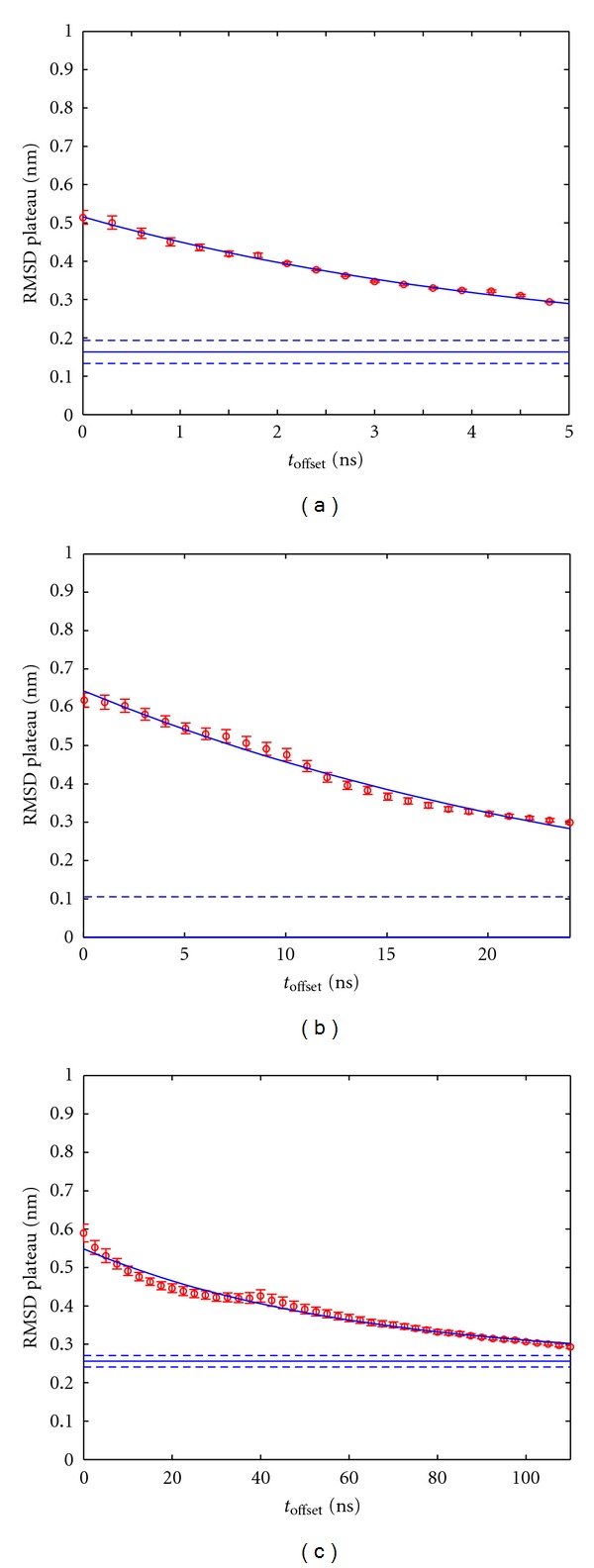
Fit and extrapolation of plateau values for increasing t offset. (a) 10 ns trajectory1, (b) 50 ns trajectory2, (c) 200 ns trajectory3; see Table 1. Red circles represent plateau values a(t offset) the error bars denote their asymptotic standard errors. The solid blue curve shows a nonlinear least-squares fit of (3) to the plateau values a(t offset). From the latter fit the limiting plateau value a 0 = a(t offset = ∞) was extracted and is shown as a solid horizontal line together with its asymptotic standard error as obtained from the fit of (3).
Figures 6(a)–6(c) illustrates the influence of the offset t offset from the start configuration on the respective plateau values as estimated from the parameter a of the model (2). plateau values tend to decrease with increasing t offset.
4. Conclusions
For molecular dynamics simulations of proteins, questions of “convergence” and sampling are important issues if sensible conclusions are to be drawn from such simulations. Since convergence of a particular simulation (in the sense that statistical sampling of the phase space is complete enough with respect to the specific phenomena studied) cannot be judged in advance [39], various techniques have been advised to demonstrate that a simulation has not yet converged [37, 53–56].
Here, we propose a method to assess if a simulation has not yet run long enough, based on RMSD analysis of successive configurations of a given MD trajectory, separated by time lags Δt of varying length (up to half the total simulation time). As long as a simulation has not yet converged, the shape of the function still considerably depends on t offset, that is, the time point along the trajectory, where the analysis interval Δt starts. As t offset is large enough, the shape of becomes stationary; that is, in Figure 4 the shape of the mean RMSD curve is different in the left and right panel, but further increasing t offset would not further change its overall appearance. This is also reflected in the respective model parameters, converging to constant values for large values of t offset; see Figure 5.
To describe this Δt dependence of the mean RMSD values,, for a given t offset, the Hill function was used. This function type is frequently applied in enzyme kinetics to model saturation phenomena together with the number of reactive sites on an enzyme. In the present work, the Hill function proved flexible enough to model the functional form of and to identify the respective parameters (plateau value, shape parameter, and half-saturation time). Contrary to a simple exponential saturation function, the Hill function is able to model different levels of sigmoidicity.
In order to quantify the influence of the initial conditions and the equilibration phase on , we systematically increased the time t offset before starting to analyze each trajectory. With the exception of the shape parameter γ all the other parameters of the Hill function describing exhibit a distinctive dependence on the initial phase of the trajectories and thus indicate that biased estimates would result if the initial phase would be included in the analysis.
As pointed out in the introduction, the method proposed here combines the smoothing effect of averaging, while retaining the specificity of a precise time interval between configurations being compared. For example, the height of the extrapolated plateau a 0 (see Figure 6) may be interpreted as “configurational distance” between two arbitrarily selected, totally unrelated configurations of the particular part of phase space currently visited. If one chooses an arbitrary configuration out of it as the reference, all others visited by the trajectory will have an average distance a 0. In contrast, Figure 1 shows two limiting cases of reference configurations: (i) the configuration with maximum average distance (i.e., RMSD) to all others (blue curve), which represents the “maximum outlier” ever seen in this trajectory and (ii) the configuration with minimum average distance (i.e., RMSD) to all others (red curve), which is “most central” within the trajectory.
Note that extrapolated plateau values are significantly larger than the actually reached in the trajectories (see Figure 3). Only if we consider the extrapolated plateau as a function of t offset, we obtain realistic (i.e., lower) estimates (see Figure 6) to aim at during actual simulations. In this sense, we may attribute the proposed fitting procedure some forecast capability regarding the level of RMSD to be finally expected if the trajectory were carried on. This extrapolated mean RMSD corresponds to pairs of configurations separated by time intervals large enough to consider such configurations approximately uncorrelated, independent representatives of the configuration space of the respective molecule.
Likewise, the “half-saturation time” τ obtained from the fit decreases with increasing t offset. Thus, taking configurations with large enough t offset as a reference, it takes only a short time to get close to the plateau a 0. In each panel of Figure 5, the parameter τ approaches an almost constant level at about half the maximum t offset considered (i.e., 25% of the total simulation time). This might suggest that—regardless of the total simulation length—the fraction of usable (independent) configurations remains the same (final 75% of the trajectory). As the length of the simulation run increases, the criterion derived from the run itself gets increasingly more stringent. From the 200 ns run the first 50 ns should be discarded, which is more than 5times the total length of the 10 ns trajectory. If using the latter as a basis of estimate, however, the last 7.5 ns seem to be trustable.
Although the analysis reported in the present work is specific to TCRpMHC complexes, we expect the method of lagged RMSD analysis to be applicable to similar molecular systems, such as membrane proteins comparable in size and structure. The approach presented here is designed to assess the degree of convergence of MD simulations and hence the statistical quality of conclusions drawn from such simulations.
Acknowledgments
The authors are thankful toL. Litov andP. Petkov for the discussions and constructive remarks. This work was supported in part by the Bulgarian Science Fund under Grant DCoE-02/1/2009 and in part by the Austrian Science Fund (FWF P22258-B12).
References
- 1.Perelson AS. Modelling viral and immune system dynamics. Nature Reviews Immunology. 2002;2(1):28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 2.Kuznetsov VA, Makalkin IA, Taylor MA, Perelson AS. Nonlinear dynamics of immunogenic tumors: parameter estimation and global bifurcation analysis. Bulletin of Mathematical Biology. 1994;56(2):295–321. doi: 10.1007/BF02460644. [DOI] [PubMed] [Google Scholar]
- 3.Barra A, Agliari E. A statistical mechanics approach to autopoietic immune networks. Journal of Statistical Mechanics. 2010;2010(7)P07004 [Google Scholar]
- 4.Barra A, Agliari E. Stochastic dynamics for idiotypic immune networks. Physica A. 2010;389(24):5903–5911. [Google Scholar]
- 5.Perelson AS, Weisbush G. Immunology for physicists. Reviews of Modern Physics. 1997;69(4):1219–1267. [Google Scholar]
- 6.Adam JA. Effects of vascularization on lymphocyte/tumor cell dynamics: qualitative features. Mathematical and Computer Modelling. 1996;23(6):1–10. [Google Scholar]
- 7.Vodovotz Y, Clermont G, Chow C, An G. Mathematical models of the acute inflammatory response. Current Opinion in Critical Care. 2004;10(5):383–390. doi: 10.1097/01.ccx.0000139360.30327.69. [DOI] [PubMed] [Google Scholar]
- 8.Burroughs NJ, Wülfing C. Differential segregation in a cell-cell contact interface: the dynamics of the immunological synapse. Biophysical Journal. 2002;83(4):1784–1796. doi: 10.1016/S0006-3495(02)73944-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dustin ML. Stop and go traffic to tune T cell responses. Immunity. 2004;21(3):305–314. doi: 10.1016/j.immuni.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 10.Favier B, Burroughs NJ, Wedderburn L, Valitutti S. TCR dynamics on the surface of living T cells. International Immunology. 2001;13(12):1525–1532. doi: 10.1093/intimm/13.12.1525. [DOI] [PubMed] [Google Scholar]
- 11.Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nature Immunology. 2001;2(6):487–492. doi: 10.1038/88678. [DOI] [PubMed] [Google Scholar]
- 12.Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunology. 2008;9, article 8 doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295(5559):1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 14.Bell E. T-cell activation—T-cell-APC interactions. Nature Reviews Immunology. 2004;4, article 930 [Google Scholar]
- 15.Borovsky Z, Mishan-Eisenberg G, Yaniv E, Rachmilewitz J. Serial triggering of T cell receptors results in incremental accumulation of signaling intermediates. Journal of Biological Chemistry. 2002;277(24):21529–21536. doi: 10.1074/jbc.M201613200. [DOI] [PubMed] [Google Scholar]
- 16.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436(7050):578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 17.Dustin ML. The cellular context of T cell signaling. Immunity. 2009;30(4):482–492. doi: 10.1016/j.immuni.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gil D, Schrum AG, Alarcón B, Palmer E. T cell receptor engagement by peptide—MHC ligands induces a conformational change in the CD3 complex of thymocytes. Journal of Experimental Medicine. 2005;201(4):517–522. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones LL, Colf LA, Bankovich AJ, et al. Different thermodynamic binding mechanisms and peptide fine specificities associated with a panel of structurally similar high-affinity T cell receptors. Biochemistry. 2008;47(47):12398–12408. doi: 10.1021/bi801349g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanzavecchia A, Iezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96(1):1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 21.Baker CTH, Bocharov GA, Paul CAH. Mathematical modelling of the interleukin-2 T-cell system: a comparative study of approaches based on ordinary and delay direrential equations. Journal of Theoretical Medicine. 1997;2:117–128. [Google Scholar]
- 22.Bianca C. Mathematical modeling for keloid formation triggered by virus: malignant effects and immune system competition. Mathematical Models and Methods in Applied Sciences. 2011;21(2):389–419. [Google Scholar]
- 23.Pappalardo F, Halling-Brown MD, Rapin N, et al. ImmunoGrid, an integrative environment for large-scale simulation of the immune system for vaccine discovery, design and optimization. Briefings in Bioinformatics. 2009;10(3):330–340. doi: 10.1093/bib/bbp014. [DOI] [PubMed] [Google Scholar]
- 24.Bianca C, Pennisi M. The triplex vaccine effects in mammary carcinoma: a nonlinear model in tune with SimTriplex. Nonlinear Analysis: Real World Applications. 2012;13:1913–1940. [Google Scholar]
- 25.Pappalardo F, Lollini PL, Castiglione F, Motta S. Modeling and simulation of cancer immunoprevention vaccine. Bioinformatics. 2005;21(12):2891–2897. doi: 10.1093/bioinformatics/bti426. [DOI] [PubMed] [Google Scholar]
- 26.Pennisi M, Catanuto R, Pappalardo F, Motta S. Optimal vaccination schedules using simulated annealing. Bioinformatics. 2008;24(15):1740–1742. doi: 10.1093/bioinformatics/btn260. [DOI] [PubMed] [Google Scholar]
- 27.Zhang GL, DeLuca DS, Keskin DB, et al. MULTIPRED2: a computational system for large-scale identification of peptides predicted to bind to HLA supertypes and alleles. Journal of Immunological Methods. 2011;374(1–2):53–61. doi: 10.1016/j.jim.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pappalardo F, Zhang P, Halling-Brown M, et al. Computational simulations of the immune system for personalized medicine: state of the art and challenges. Current Pharmacogenomics and Personalized Medicine. 2008;6(4):260–271. [Google Scholar]
- 29.Wan S, Coveney PV, Flower DR. Molecular basis of peptide recognition by the TCR: affinity differences calculated using large scale computing. Journal of Immunology. 2005;175(3):1715–1723. doi: 10.4049/jimmunol.175.3.1715. [DOI] [PubMed] [Google Scholar]
- 30.Zacharias M, Springer S. Conformational flexibility of the MHC Class I α1-α2 domain in peptide bound and free states: a molecular dynamics simulation study. Biophysical Journal. 2004;87(4):2203–2214. doi: 10.1529/biophysj.104.044743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knapp B, Omasits U, Bohle B, et al. 3-Layer-based analysis of peptide-MHC interaction: in silico prediction, peptide binding affinity and T cell activation in a relevant allergen-specific model. Molecular Immunology. 2009;46(8-9):1839–1844. doi: 10.1016/j.molimm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Knapp B, Omasits U, Schreiner W, Epstein MM. A comparative approach linking molecular dynamics of altered peptide ligands and MHC with In Vivo immune responses. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011653.e11653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Bhatt D, Zuckerman DM. Automated sampling assessment for molecular simulations using the effective sample size. Journal of Chemical Theory and Computation. 2010;6(10):3048–3057. doi: 10.1021/ct1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Held M. VRONI: an engineering approach to the reliable and efficient computation of Voronoi diagrams of points and line segments. Computational Geometry. 2001;18(2):95–123. [Google Scholar]
- 35.Okabe A, Boots B, Sugihara K, Chiu SN, Kendall DG. Spatial Tessellations: Concepts and Applications of Voronoi Diagrams. 2nd edition. Chichester John Wiley & Sons; 2000. [Google Scholar]
- 36.Knuth D. The Art of Computer Programming. 2nd edition. Vol. 2. Reading, Mass, USA: Addison-Wesley; 1989. (Seminumerical Algorithms). [Google Scholar]
- 37.García AE. Large-amplitude nonlinear motions in proteins. Physical Review Letters. 1992;68(17):2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 38.Knapp B, Frantal S, Cibena M, Schreiner W, Bauer P. Is an intuitive convergence definition of molecular dynamics simulations solely based on the root mean square deviation possible? Journal of Computational Biology. 2011;18(8):997–1005. doi: 10.1089/cmb.2010.0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grossfield A, Zuckerman DM. Chapter 2 quantifying uncertainty and sampling quality in biomolecular simulations. Annual Reports in Computational Chemistry. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flyvbjerg H, Petersen HG. Error estimates on averages of correlated data. The Journal of Chemical Physics. 1989;91(1):461–466. [Google Scholar]
- 41.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kjer-Nielsen L, Clements CS, Purcell AW, et al. A structural basis for the selection of Dominant αβ T cell receptors in antiviral immunity. Immunity. 2003;18(1):53–64. doi: 10.1016/s1074-7613(02)00513-7. [DOI] [PubMed] [Google Scholar]
- 43.Canutescu AA, Shelenkov AA, Dunbrack RL. A graph-theory algorithm for rapid protein side-chain prediction. Protein Science. 2003;12(9):2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knapp B, Omasits U, Schreiner W. Side chain substitution benchmark for peptide/MHC interaction. Protein Science. 2008;17(6):977–982. doi: 10.1110/ps.073402508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knapp B, Omasits U, Frantal S, Schreiner W. A critical cross-validation of high throughput structural binding prediction methods for pMHC. Journal of Computer-Aided Molecular Design. 2009;23(5):301–307. doi: 10.1007/s10822-009-9259-2. [DOI] [PubMed] [Google Scholar]
- 46.Kjer-Nielsen L, Clements CS, Brooks AG, Purcell AW, McCluskey J, Rossjohn J. The 1.5 Å crystal structure of a highly selected antiviral T cell receptor provides evidence for a structural basis of immunodominance. Structure. 2002;10(11):1521–1532. doi: 10.1016/s0969-2126(02)00878-x. [DOI] [PubMed] [Google Scholar]
- 47.Kjer-Nielsen L, Clements CS, Brooks AG, et al. The structure of HLA-B8 complexed to an immunodominant viral determinant: peptide-induced conformational changes and a mode of MHC class I dimerization. Journal of Immunology. 2002;169(9):5153–5160. doi: 10.4049/jimmunol.169.9.5153. [DOI] [PubMed] [Google Scholar]
- 48.Berendsen HJ, Postma JPM, Van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. Intermolecular Forces. 1981:331–342. [Google Scholar]
- 49.Hess B, Kutzner C, van der Spoel D, Lindahl E. GRGMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 50.Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 51.Omasits U, Knapp B, Neumann M, et al. Analysis of key parameters for molecular dynamics of pMHC molecules. Molecular Simulation. 2008;34(8):781–793. [Google Scholar]
- 52.Hill AV. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. Journal of Physiology. 1910;40, supplement:4–7. [Google Scholar]
- 53.Faraldo-Gómez JD, Forrest LR, Baaden M, et al. Conformational sampling and dynamics of membrane proteins from 10-nanosecond computer simulations. Proteins. 2004;57(4):783–791. doi: 10.1002/prot.20257. [DOI] [PubMed] [Google Scholar]
- 54.Chodera JD, Swope WC, Pitera JW, Seok C, Dill KA. Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. Journal of Chemical Theory and Computation. 2007;3(1):26–41. doi: 10.1021/ct0502864. [DOI] [PubMed] [Google Scholar]
- 55.Smith LJ, Daura X, Van Gunsteren WF. Assessing equilibration and convergence in biomolecular simulations. Proteins. 2002;48(3):487–496. doi: 10.1002/prot.10144. [DOI] [PubMed] [Google Scholar]
- 56.Hess B. Convergence of sampling in protein simulations. Physical Review E. 2002;65(3):10 pages. doi: 10.1103/PhysRevE.65.031910.031910 [DOI] [PubMed] [Google Scholar]