

Abstract
Host responses can contribute to the severity of viral infection, through the failure of innate antiviral mechanisms to recognize and restrict the pathogen, the development of intense systemic inflammation leading to circulatory failure or through tissue injury resulting from overly exuberant cell-mediated immune responses. High-throughput genomics methods are now being used to identify the biochemical pathways underlying ineffective or damaging host responses in a number of acute and chronic viral infections. This article reviews recent gene expression studies of 1918 H1N1 influenza and Ebola hemorrhagic fever in cell culture and animal models, focusing on how genomics experiments can be used to increase our understanding of the mechanisms that permit those viruses to cause rapidly overwhelming infection. Particular attention is paid to how evasion of type I IFN responses in infected cells might contribute to over-activation of inflammatory responses. Reviewing recent research and describing how future studies might be tailored to understand the relationship between the infected cell and its environment, this article discusses how the rapidly growing field of high-throughput genomics can contribute to a more complete understanding of severe, acute viral infections and identify novel targets for therapeutic intervention.
Keywords: 1918 Influenza virus, Influenza, Ebola virus, Filovirus, Interferon, Host response evasion
1. Introduction
The development of expression microarrays and other high-throughput genomics and proteomics techniques has promised to unravel many of the complexities of host–pathogen interactions (Fornek et al., 2007, Geiss et al., 2000, Jenner and Young, 2005, Korth et al., 2005, Liu et al., 2006). Although viruses employ diverse strategies to control host cellular processes at the post-transcriptional level, the regulation of the gene expression is central to the virus–host relationship (Kash et al., 2006a). Messenger RNA expression microarray methods that allow simultaneous measurement of tens of thousands of genes have greatly increased our ability to document and characterize these key interactions.
Regulation of host gene expression is especially important for RNA viruses, which have only a limited number of enzymatic activities encoded in their genomes, and therefore require a facilitative host environment for efficient replication. RNA viruses thus need to regulate their host gene expression environment to ensure an adequate supply of metabolites for RNA, protein and lipid biosynthesis for the production of new virions, while at the same time suppressing antiviral and cellular defense response pathways to prevent their detection and clearance by the immune system. Of primary importance to RNA virus replication is antagonism of the broad spectrum of antiviral activities associated with activation of pathogen-associated molecular pattern (PAMP) receptors and type I interferon (IFN) responses (reviewed in (Garcia-Sastre and Biron, 2006, Loo and Gale, 2007, Randall and Goodbourn, 2008, Sadler and Williams, 2008). However, over-strong viral antagonism and/or evasion of these responses can lead to very high rates of viral replication, significant inflammation and severe disease pathology. This is especially true of highly virulent RNA viruses, such as Ebola and pandemic influenza (Bray, 2001, Garcia-Sastre, 2001, Garcia-Sastre, 2004, Garcia-Sastre, 2006, Harcourt et al., 1999, Villinger et al., 1999).
Conventional antiviral therapies are typically designed to directly inhibit viral replication by targeting the enzymatic activities of viral proteins. However, given the ability of RNA viruses to rapidly mutate, such countermeasures often lead to the development of drug resistance. One approach to help mitigate the impact of such mutations is the additional targeting of host responses to reduce pathogenesis. Multi-target therapies designed to limit viral replication and to mitigate immunopathology promise to greatly increase our capacity to combat viral diseases and improve human health. If highly virulent viruses dysregulate immune responses in the same manner, treatment modalities might be developed that target host response pathways common to many viruses. Moreover, drugs that modulate inflammatory responses could be used early in a critical care setting, before an accurate diagnosis can be made and virus-specific antiviral drugs can be administered.
Although expression microarray experiments have been used to study the host response to many different RNA viruses, including SARS-CoV (Baas et al., 2008, Cameron et al., 2007, de Lang et al., 2007, Reghunathan et al., 2005), dengue (Fink et al., 2007, Ubol et al., 2008, Warke et al., 2003), West Nile (Fredericksen et al., 2004, Venter et al., 2005) and hepatitis C viruses (reviewed in Walters and Katze, 2009), this article will focus on the application of genomics technology to study different aspects of highly pathogenic influenza and Ebola virus infection in cell culture and animal models. Both 1918 influenza and Ebola viruses are examples of RNA viruses that cause rapidly progressive fulminant infections with marked activation of inflammatory responses. Moreover, they also are significant human pathogens with epidemic and pandemic potentials and are important areas of antiviral drug research. The characterization of how these viruses alter gene expression in vitro and in vivo provide complementary perspectives of the RNA virus–host relationship.
Cell culture models are critical to understanding what might be termed the “replication component” of infectious disease because they afford the opportunity to study the primary response of host cells in the absence of immune cells and inflammatory responses. Complementarily, studies in animal models can be used to investigate what might be called the “disease component” of infection because they can reveal the whole infection process including immune cell-mediated effects. Thus, when combining the advantages of greater control of experimental variables in vitro with the biological relevance and complexity of in vivo models, studying the global host response using functional genomics promises to expand development of new antiviral drug targets and host immune responses for therapeutic intervention. This article does not aim to present a comprehensive review of microarray studies of RNA viral infections, but to describe how functional genomics technologies have increased our understanding of severe, acute viral infections in vitro and in vivo, using 1918 influenza and Ebola viruses as examples. How this approach can be further developed to promote the identification of novel and multi-target antiviral therapies will also be discussed.
2. Viral infection and type I interferon responses
A key requirement for successful protection against viral infection is the early detection of viral biomolecules and the activation of host responses to limit replication and activate the immune response. Central to this early warning system are the pathogen associated molecular pattern (PAMP) receptors (Saito and Gale, 2007). These include multiple families of proteins that sample both the extracellular and intracellular environments for a diversity of biomolecules, including lipoproteins, DNA and RNA. A second cornerstone of an effective antiviral response is the rapid triggering of a broad spectrum of antiviral activities by the PAMP receptors, such as the type I IFN response. Type I IFN refers to a large family of proteins composed of seven classes that includes 14 distinct IFNα genes and 1 IFNβ gene (Pestka et al., 2004). Why humans encode so many distinct type I IFNs is unknown, but in general, IFNα and IFNβ are the dominant antiviral IFN that are produced when cells are exposed to viral infection or dsRNA that trigger expression of interferon stimulated genes (ISGs) (Borden et al., 2007). An alternate activation pathway for expression of ISGs is mediated by type III IFN, or IFN-l that signal through heterodimeric IL-28Rα/IL-10Rβ to activate ISG expression (reviewed in Ank et al., 2006). Type III IFN has recently been shown to be an important component of antiviral responses in epithelial cells and to participate in the host response to influenza virus infection (Ank et al., 2008, Kotenko et al., 2003, Mordstein et al., 2008, Onoguchi et al., 2007, Osterlund et al., 2007, Sommereyns et al., 2008). At present the number of ISGs activated by stimulation of the type I IFN receptor is thought to be approximately 150–200 unique genes. Interestingly, the mechanisms of action of only a handful of these key antiviral proteins are currently understood.
Replication of RNA viruses results in the synthesis of dsRNA molecules that can be detected by PAMP receptors, including TLR3, Mda5, RIG-I and PKR (Balachandran and Barber, 2007, Takeuchi and Akira, 2008). Recently, an ER-associated trans-membrane PAMP receptor, called STING (stimulator of interferon genes), has been identified and shown to play an important role in innate antiviral responses (Ishikawa and Barber, 2008). Activation of these PAMP receptors by viral RNAs leads to the rapid activation of transcription factors, including IRF1, IRF3, IRF7 and NFκB, synthesis of type I IFN proteins and subsequent expression of ISGs (Saito and Gale, 2007). Because of the potent antiviral defense properties of the IFN response, RNA viruses have evolved diverse strategies to either prevent activation of PAMP receptors, inhibit the signaling of the IFN receptor, limit ISG expression or evade ISG antiviral activity (Garcia-Sastre and Biron, 2006). For example, the influenza virus protein NS1 has several described activities, including antagonism of RIG-I, MDA5 and PKR, and inhibition of antiviral response pre-mRNA processing and translation (Hale et al., 2008, Kash et al., 2006a). Ebola viruses similarly encode proteins that greatly limit host cell IFN responses, including the inhibition of IFN response activation by the VP35 and VP24 proteins (Basler et al., 2000, Basler et al., 2003, Enterlein et al., 2006, Hartman et al., 2006, Hartman et al., 2008a, Reid et al., 2006).
The following sections present recent examples of the application of functional genomics approaches to study highly pathogenic influenza and Ebola virus infection that showed the global relationships between regulation of type I IFN responses, viral replication, pathogenesis and viral disease outcome. How expression functional genomics studies can be further developed to provide more detailed knowledge of the contribution of aberrant immune response activation to development of severe pathology will also be discussed. By elucidating underlying molecular mechanisms of pathogenesis and identifying common host pathways for different viral infections, the goal of these experimental approaches is to identify novel host targets for broad spectrum antiviral therapies to reduce disease severity and improve human health.
3. Expression microarray studies of severe viral infections
3.1. 1918 H1N1 influenza
The influenza viruses are members of the Orthomyxoviridae family of negative sense, segmented RNA viruses. They replicate primarily in the epithelial cells of the upper and lower respiratory tract, but have also been shown to replicate in alveolar macrophages in humans (Suarez et al., 1998, Van Campen et al., 1989). Influenza A virus is responsible for approximately 36,000 deaths in the United States annually, with periodic epidemics and pandemics causing significantly higher mortalities (Thompson et al., 2003). The influenza pandemic of 1918–19, known as the Spanish influenza, was responsible for an estimated 40–60 million deaths worldwide and approximately 675,000 deaths in the United States (Morens et al., 2008, Taubenberger, 2006, Taubenberger et al., 2007). Histologic analysis of autopsy samples from human influenza cases from 1918 has shown that deaths were associated with significant damage to the lungs with acute, focal bronchitis and alveolitis (Taubenberger and Morens, 2008). Human cases also showed that 1918 influenza viral pneumonia was further associated with massive pulmonary edema, hemorrhage and destruction of the respiratory epithelium that in a majority of cases examined was also associated with severe secondary bacterial pneumonia (Morens et al., 2008).
Recently, highly pathogenic H5N1 viruses have emerged as human pathogens with very high case fatality rates. Based on evidence that a “cytokine storm” may contribute to the lethality of these infections, it has been hypothesized that the severity of 1918 influenza viral pneumonia could have resulted from similarly exaggerated inflammatory responses. With the renewed threat of a modern pandemic with catastrophic health and economic consequences, understanding the contribution of host immune responses to the severity of influenza virus infection is essential for developing new prognostic indicators and treatment modalities. However, the contributions of viral and host inflammatory responses in the development of severe lung pathology and disease caused by the 1918 influenza virus was unknown until the virus could be reconstructed and studied.
The reconstruction of the 1918 influenza virus was accomplished after the genes of the 1918 (H1N1) influenza virus were sequenced from lung tissue collected from human fatalities during 1918 pandemic (Basler et al., 2001, Reid et al., 1999, Reid et al., 2002, Reid et al., 2003, Reid et al., 2004, Taubenberger et al., 1997, Taubenberger et al., 2005, Tumpey et al., 2005). Sequence analysis suggested that the 1918 pandemic virus was likely a direct introduction of an avian virus into humans. The 1918 influenza virus was constructed using reverse genetics and initial experiments showed that the reconstructed 1918 virus (r1918) replicated to high titer in cell culture and was lethal in mice and embryonated chicken eggs (Tumpey et al., 2005).
3.1.1. Host responses and immunopathology during 1918 virus infection in mice
To study the activation of the host response to r1918 virus infection, infection of Balb/c mice with a seasonal human H1N1 virus A/Texas/36/91 (Tx91) was compared to infection with the reconstructed 1918 influenza virus (Kash et al., 2006c). Lungs from infected mice were analyzed for viral replication by plaque assay and for pathology by hematoxylin and eosin (H&E) staining. The host response was determined by expression microarrays, quantitative real-time PCR and ELISA of key immune mediators. The goal of these experiments was to determine the relationship between the severe pathology induced by 1918 infection with the activation of host immune responses to help understand why the influenza pandemic of 1918–19 was so severe.
Mice infected with Tx91 virus had minimal to mild diffuse histiocytic alveolitis without neutrophils or lesions in the bronchi or bronchioles. In contrast, mice infected with r1918 showed rapid and severe weight loss with very high levels of viral replication in the lungs with death by day 4–5 post-infection (p.i.). Lungs from these animals displayed severe pulmonary lesions and severe necrotizing bronchitis with accompanying severe alveolitis and edema. To study activation of host responses during infection, expression microarray analysis was performed by comparing equal masses of total RNA isolated from lungs of mice infected with Tx91 or r1918 influenza viruses to a common reference sample prepared from lungs of mock-infected mice (Kash et al., 2006c).
Bioinformatic analysis of the microarray data showed that Tx91 infection resulted in a host gene expression response that was limited in intensity and duration and mirrored the kinetics of viral replication and pathology. Interestingly, lungs from Tx91-infected mice also showed activation of cell cycle, glutathione metabolism and ATP generation related genes. This observation suggested that activation of these metabolic pathways could be important in preventing damage from reactive oxygen species and in stimulating tissue remodeling. In contrast, r1918 virus infection resulted in significant increases in expression of inflammatory response genes, such as IL6, IL12, IFN-gamma, TNF-alpha, CXCL1 and CXCl10, and additional genes associated with activation of immune cell populations during r1918 virus infection, including TH1 cells, NK cells, macrophages and neutrophils. These gene expression changes persisted in intensity from 24 h until death at day 4–5 p.i. and mirrored the severity of lung pathology. Additionally, r1918 virus infection resulted in the marked activation of many cell death-related genes, particularly those related to death receptor responses, including TRAIL, Fas, caspase-8 and caspase-9. These results suggested that inflammatory responses, reactive oxygen species generation and activation of cell death pathways might be important contributors to severe immunopathology associated with r1918 influenza virus infections.
This experimental approach has also used to study how different combinations of 1918 genes contribute to virulence (Kash et al., 2004, Kash et al., 2006c). For example, mice were additionally infected a Tx91 chimeric virus containing the 1918 HA and NA genes (2:6 1918), or the 1918 HA, NA, M, NP and NS genes (5:3 1918). Although all three 1918-related viruses caused lethal infections, mice infected with r1918 showed the most severe weight loss and lung pathology with the earliest mean time of death with the most rapid peak in viral replication. As shown in Fig. 1 , mice infected with 5:3 1918, 2:6 1918 or r1918 viruses displayed a continuum of mild to severe necrotizing bronchitis with accompanying moderate to severe alveolitis and alveolar edema. The character of the alveolitis also varied between the chimeric and fully reconstructed 1918 viruses with day 3 r1918-infected mice displaying acute moderate to severe diffuse alveolitis with a prominent neutrophilic component and severe alveolar edema that was not observed with the other viruses.
Fig. 1.
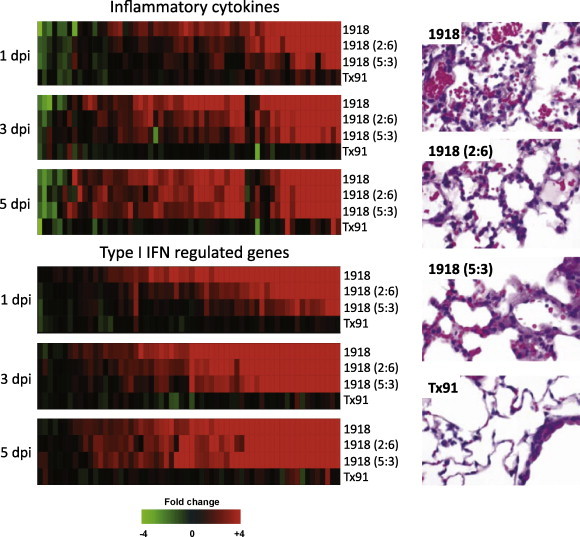
Activation of inflammatory and type I IFN-regulated genes in 1918 influenza virus-infected mouse lung tissue. Gene expression profiles in the lungs of mice infected with equivalent doses the reconstructed 1918 virus (r1918), a chimeric virus expressing either the 1918 HA and NA genes (2:6) or the 1918 HA, NA, M, NP and NS genes (5:3) compared to a contemporary H1N1 human-adapted influenza virus A/Texas/36/91 (Tx91). (Top panel) expression of inflammatory cytokine genes, (bottom panel) expression of type I IFN-regulated genes. For each infection point, the data presented are the error-weighted average expression changes calculated from four technical replicate arrays performed on three individual mice (n = 12 total). Genes shown in red were up-regulated and genes shown in green were down-regulated in infected relative to mock-infected mouse lung. At right is shown the lung pathology at 3 days post-infection. Modified from Kash et al. (2006c) (data available at http://viromics.washington.edu/publications.html).
As also shown in Fig. 1, the expression patterns of cytokine, chemokine and cell death-related genes paralleled the pathology findings and showed the most significant and earliest activation occurred during r1918 infection. While infection with r1918 induced significant expression of immune response genes that occurred early (by 24 h) and persisted until death (day 5), Tx91 infection resulted in very limited expression of these genes in both intensity and duration. Moreover, the appearance and magnitude of up-regulated immune-related mRNAs could be correlated with virulence because the r1918 virus induced the highest and earliest expression of these genes, followed in magnitude and occurrence by 2:6 1918 then 5:3 1918. Moreover, pathway analysis of the expression array data revealed that infection with r1918 virus resulted in the most significant activation of death receptor, interleukin-6, Toll-like receptor and type I interferon responses. Previous studies had shown that the 1918 NS1 protein was a very potent antagonist of the type I IFN response (Geiss et al., 2002), and thus, it was surprising that significant induction of the type I IFN response, was observed as shown in Fig. 1. This robust expression of IFN-regulated genes in the lungs of infected mice, however, failed to inhibit viral replication and was unable to protect the host from the effects of 1918 influenza virus infection. It was hypothesized that this increased antiviral response gene expression associated with increased viral replication might be explained by (i) the heterogeneity of cell type and infection status of cells present in the lung, (ii) that only a small percentage of the cells are susceptible for sustained influenza virus infection and (iii) that a significant part of the host response gene expression would derive from the marked infiltration of activated immune cells. Taken together, these results suggested that over-activation of inflammatory responses may be contributing to the severity of highly pathogenic influenza virus infections and also that protecting cells from damage by immune cells, hypercytokinemia and oxygen radicals may limit tissue destruction and facilitate repair.
However, these experiments were performed from intact lung tissue and the gene expression patterns were representative of “average” gene expression of all cells in the lung tissue, including large numbers of infiltrating immune cells. Thus, to more clearly understand the relationship between viral regulation of antiviral genes and 1918 influenza virus virulence, a study of infected tissue isolated from infected animals where tissue dissection could be used to deconvolute the gene expression pattern by limiting cell heterogeneity would be needed.
3.1.2. Aberrant activation of host responses to 1918 virus infection in a nonhuman primate model
In a complementary study, the response of cynomolgus macaques (Macaca fascicularis) infected with r1918 influenza virus was compared to infection with a seasonal human-adapted H1N1 influenza virus A/Kawasaki/1/73 (K173) (Kobasa et al., 2007). Infection of macaques with r1918 influenza resulted in a uniformly lethal and severe viral pneumonia; while K173-infected animals developed greatly reduced symptoms and pathology and survived. To study the activation of antiviral responses during 1918 virus infection, bronchus tissue was chosen for expression microarray analysis because it allowed for a comparison of the host response of bronchial epithelial cells, which are a primary target cell for influenza virus replication, and reduced the confounding effects of immune cell derived gene expression compared to studies on intact lung. Microarray analysis performed on bronchus tissue isolated from r1918 and K173-infected animals showed significant differences in the intensity and duration of antiviral and other host response gene expression patterns.
As shown in Fig. 2 , it was observed that infection with the K173 virus induced a significant antiviral response, particularly the expression of type I IFN related genes, including many type I IFNs, that correlated with peak viral load (102–103 PFU/g tissue on day 3) that was subsequently repressed when K173 virus was no longer detected (days 6–8) (Kobasa et al., 2007). In contrast, microarray analysis of bronchus tissue isolated from r1918 virus infected macaques showed little expression of the IFNα genes observed during K173 infection. These tissues also possessed a sustained and mildly reduced activation of type I IFN related gene expression compared to K173 even though r1918 viral loads were approximately 1000–10,000 times higher than K173. Interestingly, r9181-infected samples showed significantly elevated expression of pro-inflammatory cytokines and chemokines, including IL6 and IL8 and neutrophil chemoattractants CXCL1, CXCL2, and CXCL6. These results indicated that the 1918 influenza virus was able to significantly suppress expression of at least some type I IFN genes and impair the protective host response to infection observed in the K173-infected animals; while inducing expression of neutrophilic chemokines.
Fig. 2.
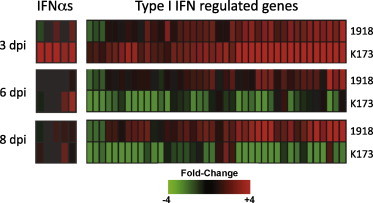
Expression of IFN-alpha and type I IFN-regulated genes in 1918 influenza virus-infected macaque bronchus. Gene expression profiles in bronchus tissue isolated from macaques infected with equivalent doses of the reconstructed 1918 virus (r1918) compared to a H1N1 human-adapted influenza virus A/Kawasaki/1/73 (K173). For each infection point, the data presented are the error-weighted average expression changes calculated from four technical replicate arrays performed on one to three individual macaques (n = 4–12 total). Genes shown in red were up-regulated and genes shown in green were down-regulated in infected relative to mock-infected macaque lung. Modified from Kobasa et al. (2007) (data available at http://viromics.washington.edu/publications.html).
Thus, significant viral antagonism of the type I IFN response and activation of chemokine expression appear to be a critical component of 1918 influenza virus virulence that leads to high replication rates, significant recruitment of inflammatory cells and severe lung pathology. Thus by focusing on upper respiratory parenchymal cells studied by expression microarray, an important insight into the viral control of antiviral and IFN-related responses by the 1918 influenza virus in vivo could be made. The advantages of utilizing approaches that minimize cell heterogeneity while preserving the benefits of studying infection in animals will be discussed in detail later.
3.2. H5N1 avian influenza
Highly pathogenic H5N1 viruses from Southeast Asia, have recently emerged as viruses that can cause rare, but rapidly fatal disease in humans with mortality rates exceeding 50% (Anon, 1997a, Anon, 1997b, Abdel-Ghafar et al., 2008, Claas et al., 1998, de Wit and Fouchier, 2008). Further studies have similarly demonstrated that these H5N1 viruses also cause rapid and lethal disease in many animal models, including mice and ferrets (Gao et al., 1999, Gubareva et al., 1998, Lu et al., 1999, Suarez et al., 1998, Subbarao et al., 1998). Similar to the experiments with 1918 influenza virus in mouse lung, David Kelvin's group has recently used functional genomics to show marked activation of type I IFN responses was associated with highly virulent H5N1 influenza virus infection in ferret lung (Cameron et al., 2008). In particular, these studies showed robust lung expression of the IFN-regulated gene CXCL10 and its cognate receptor CXCR3. Kelvin's group then used this observation from their genomics data to perform an additional study where they treated infected ferrets with a CXCR3 antagonist (Amgen; AMG487) to quantify the role of this type I IFN-regulated pathway. Treatment of H5N1-infected ferrets with AMG487 reduced disease severity and delayed mortality of H5N1-infected ferrets showing that unregulated IFN responses were at least partially response for H5N1 disease severity in this model. This study illustrates the potential of functional genomics to identify host pathways that may be over-activated during highly virulent infections for which current inhibitory drugs have already been developed.
3.3. The need to reduce the cellular complexity of tissue models
While providing the opportunity to characterize the relationship between activation of inflammatory responses in the lungs of infected animals, genomics studies that rely on whole lung tissues are heavily influenced by cell type heterogeneity, immune cell infiltration and infection status. The temptation is to try to interpret these microarray data from the perspective of the infected cell; however this is fundamentally incorrect. Because these microarray experiments were performed on RNA isolated from infected lung tissues, the results give the average gene expression of cells in the entire tissue. Thus, in the end we are left with a lack of detailed knowledge as to the contributions that individual cell types, both infected and uninfected, make to the aggregate gene expression changes identified in the intact lung. However, these experiments raise exciting and fundamental new questions. Which populations of genes are expressed in which populations of cells? What is the contribution of infected and uninfected individual immune cell types to pathogenesis? How are infected cells that surround infected cells (i.e. “bystander” cells) affected by high cytokine levels in the tissue? Are the observed necrotic and cell death phenotypes triggered by the viral cytopathic effect (CPE) and limited to end-stage infected cells? Or does excess killing of bystander cells by immune cells add to the effects of the viral CPE?
Studies on normal human lung have shown that at least five major cell types are present in the parenchyma. These cells include type I and II alveolar epithelial (15%), capillary epithelial (30%) and interstitial cells (37%) and a range of alveolar macrophages (5–20%) (Crapo et al., 1982). This cell type heterogeneity is further complicated during influenza virus infection, which results in the significant infiltration of numerous activated immune cells, including macrophages, neutrophils, NK cells and T and B lymphocytes (Perrone et al., 2008). Adding to this complexity is that approximately 15–20% of lung cells (i.e. the respiratory epithelial cells and alveolar macrophages) are susceptible to primary influenza virus infection. Therefore, the contribution of influenza viral replication in the respiratory epithelium to the overall transcriptional profile of whole infected lung is impossible to determine using an intact tissue model. Thus, reducing the cellular heterogeneity of experimental animal (or human) derived samples should be an important goal in understanding viral disease in a biologically relevant model system.
One simple approach to focus more closely on infected cell types or uninfected bystander cells is to use traditional surgical dissection to isolate distinct regions of the infected tissue or organ, as was performed in the 1918 virus macaque study. However, this can be difficult for small animal models such as mice and cannot allow for the discrimination of responses between infected and bystander cells. Another approach to reducing the confounding effects of cell heterogeneity is to isolate and study individual live cell populations using flow cytometry and fluorescent activated cell sorting (FACS). Similarly, laser capture microdissection (LCM) of fixed samples could be used to isolate specific cell populations to determine the gene expression profiles of infected cells and cells adjacent to infected and/or inflammatory cells. Such isolation approaches would be required to study effects of infection and robust inflammatory responses on bystander cells.
Why would the study of such bystander cells be critical to our understanding of how hypercytokinemia influences immunopathology and disease outcome? During infection, these cells would be exposed to the same microenvironment of antiviral, inflammatory and cell death soluble mediators produced to inhibit and kill nearby infected cells. However, in contrast to the infected cell expressing viral proteins that inhibit host responses, uninfected bystander cells do not contain such antagonists and are yet exposed to similarly high concentrations of cytokines, chemokines and activators of cells death. Thus, it is reasonable to hypothesize that a portion of the respiratory epithelial cell necrosis associated with influenza virus infection may result from immune response-mediated bystander cell death. Studies designed to address such issues should provide important insights into pathogenesis and how cytokine responses may contribute to disease severity. Moreover, by characterizing the host response pathways responsible for loss of bystander cells, new treatment modalities, such as reducing damage by reactive oxygen species produced by neutrophils and macrophages, could be developed.
In the following section, the application of microarray analysis to study the regulation of the type I IFN and inflammatory responses during primary Ebola and Marburg virus infection in vitro in cultured human liver cells and ex vivo in peripheral blood mononuclear cells (PBMCs) isolated from infected macaques will be discussed. These studies revealed inhibition of type I IFN stimulated gene expression in primary infected cells and the significant activation of inflammatory and IFN responses in the peripheral blood of infected hosts are interrelated and likely important components of the extreme human virulence of these viruses.
3.4. Filoviral hemorrhagic fever
Ebolavirus (EBOV) and Marburgvirus (MARV) are members of the Filoviridae family of non-segmented negative-strand RNA viruses, which are among the most deadly of human pathogens (Gonzalez et al., 2007, Hoenen et al., 2006, Mohamadzadeh et al., 2006). In humans, filovirus infections are associated with high viremia, increased endothelial cell permeability and severe tissue destruction, lymphopenia, and coagulopathy. Both EBOV and MARV can cause severe disease and viral hemorrhagic fever in humans and nonhuman primates. Disease outbreaks caused by the Zaire EBOV (ZEBOV) subtype and MARV have had case fatality rates of as high as 80–90%. In contrast, the Reston EBOV (REBOV) has not been associated with disease in humans, but is lethal for nonhuman primates (Groseth et al., 2002, Hutchinson et al., 2001, Jahrling et al., 1996, Morikawa et al., 2007, Sanchez et al., 1999). These observations raise an important question regarding virulence: do all filoviruses evoke similar responses in human cells? Because the ability of EBOV and MARV to cause severe disease appears to be associated with their capacity to replicate to high titers, is evasion of the type I IFN response a common feature? Does the host response to REBOV differ in significant ways that might explain its attenuation in humans? Given their ability to characterize the global host response to infection, expression microarrays and functional genomics have been used to address these questions.
3.4.1. IFN response antagonism and virulence
One hypothesis for the extreme virulence of EBOV and MARV virus for humans is that very high systemic replication rates are achieved through potent antagonism of global antiviral responses, particularly the type I IFN response. To test this hypothesis, the gene expression response of cultured human liver (Huh7) cells infected with ZEBOV or MARV were compared to mock-infected cells in the presence and absence of exogenous type I IFN (IFNα-2b) (Kash et al., 2006b). In the absence of exogenous IFN, bioinformatic analysis showed many similarities in the expression of antiviral, and pro-inflammatory, coagulation and acute phase-related responses during ZEBOV and MARV infection. Most significantly, examination of the expression of antiviral response genes during infection in the presence of exogenous IFNα, revealed that ZEBOV and MARV strongly antagonized IFN responses. Of these two viruses, ZEBOV was the most potent inhibitor of type I IFN receptor responses and blocked activation of approximately 90% of the nearly 200 genes induced by IFNα treatment of mock-infected cells; while MARV was able to inhibit approximately 75% of these IFNα-induced genes. These results showed that regulation of host responses and in particular antagonism of type I IFN responses were fundamental properties of these viruses and likely contribute to their ability to cause severe, acute disease in humans.
In contrast, identical studies using REBOV uncovered significant differences in the expression of antiviral and cellular defense response-related genes, compared to ZEBOV, and revealed that REBOV was significantly less efficient at controlling host responses in human cells (Kash et al., 2006b). Most dramatically, REBOV-infected cells treated with IFNα still expressed a majority of IFNα genes observed in treated mock-infected cells, including the increased expression of many MHC class I mRNAs, including PSME1, B2M, HLA-Cw2, HLA-C, HLA-A, and HLA-B. These experiments showed that REBOV has a greatly reduced ability to inhibit activation of antiviral responses in human cells, most notably the type I IFN response. These results put forth a molecular basis for the hypothesis that REBOV might be significantly attenuated in humans due to reduced IFN response antagonism leading to slower replication rates, increased expression of antigen presentation genes and immune cell clearance.
The VP35 protein of ZEBOV has been shown to be an important antagonist of IFN-related responses (Basler et al., 2000). In addition to functioning as a cofactor for the viral polymerase, VP35 inhibits activation of several key early antiviral response proteins, including IRF3 and PKR (Basler et al., 2003, Hartman et al., 2006, Hartman et al., 2006). Mutation of a key residue in the carboxy-terminus of VP35 (R312) has subsequently been found to abolish the IRF3 inhibitory activity of VP35 (Hartman et al., 2008a). The extent of the VP35-mediated antagonism of type I IFN responses has recently been explored using expression microarrays (Hartman et al., 2008b). In this study, Hartman et al, showed that infection of HepG2 cell cultures with a recombinant ZEBOV expressing the R312A mutant VP35 protein resulted in robust expression of many ISGs, including RIG-I, ISG15, IFIT1 and 2, MX1 and OAS that were not observed with wild-type ZEBOV. Thus, a single mutation in ZEBOV VP35 could abolish the global suppression of antiviral responses observed during wild-type Ebola virus infection in cell culture. However, an important limitation of these studies is that they were both performed in cell culture and could only address questions about the control and evasion of host and IFN responses during primary infection in an artificial environment and could not describe the true host environment of infection in the context of an intact immune response. Such experiments would require study of an infected animal model.
3.4.2. Profiling responses of peripheral blood mononuclear cells
In an attempt to understand how ZEBOV infection affects the global host response in infected animals, Rubins et al. examined the gene expression profiles of peripheral blood mononuclear cells (PBMC) serially collected from infected macaques (Rubins et al., 2007). These experiments showed marked expression of many inflammatory genes, including IL1α, IL1R and IL6; apoptosis related genes, including TNF and TRAIL, and innate immune response genes, including TLR1 and TLR4, in the PBMCs from infected cynomolgus macaques. There was also early and robust expression of many type I IFN-regulated genes, including INFAR1, ISG15, IP10, MX1, OAS1/2 and STAT1. Interestingly, expression of IFN-regulated genes persisted throughout the course of infection and up until death. The authors concluded that the ZEBOV IFN antagonists VP24 and VP35 acted to influence the antiviral state of the infected cell rather than to inhibit the systemic IFN response. Thus, the antiviral activities of EBOV IFN antagonists are limited to the infected cell and therefore uninfected bystander cells would still be capable of responding to the high circulating concentrations of IFN, inflammatory and cell death mediators.
These functional genomics studies have shown that robust activation of type I IFN and other antiviral and inflammatory responses did not appear to affect viral replication or in the case of the macaque experiments protect the host from the lethal effects of EBOV infection. Because of the localized activity of the VP24 and VP35 IFN antagonists to the infected cell, these studies further raise the question of whether the significant activation of immune responses might contribute to cell death and severe pathology associated with EBOV and MARB infections. However, as with the studies on influenza virus in intact animal tissue, the direct relationship of gene expression of infected cells in a mixture cell types and infection status cannot be discriminated. Analogous to the discussion of reducing cellular complexity during influenza virus infection, understanding how EBOV infection modulates gene expression programs of distinct cells in peripheral blood will require separation and analysis of individual cell types.
The studies described in this review highlight how the use of genomics technologies has added to our understanding of the extent of altered antiviral, type I IFN and inflammatory responses during highly virulent RNA virus infections. However, these studies have left many persistent questions about the role aberrant activation of host responses play in virulence and pathogenesis unanswered. Why are some exaggerated host immune responses not protective to the host? What are the consequences of high concentrations of immune mediators on cells neighboring infected cells and what is the contribution of by-stander cell responses to this stressful cytokine microenvironment? Are there common inflammatory pathways responsible for immunopathology? What is the relationship between inhibition of antiviral and IFN responses in infected cells and systemic activation of IFN responses? What is the extent of type I interferon and immune responses?
3.5. Identifying new drug targets
3.5.1. Novel genes, pathways and targets
Microarray profiling of virus-infected cells and tissues has been instrumental in revealing the complexity and character of IFN responses (de Veer et al., 2001). Such studies have shown that type I IFN signaling regulates the expression of hundreds of genes that collectively establish an intracellular antiviral state, but the functions of the vast majority of these genes remain unknown (Sen and Sarkar, 2007). It is also likely that individual ISGs differ in their antiviral activity against different families of viruses. Thus, the characterization of type I IFN responsive genes and pathways is critical to further our understanding of broad-ranging effects of this key family of antiviral cytokines.
Because of their high-throughput and discovery-based nature, functional genomics experiments can be very effective at elucidating the full repertoire of antiviral and infection response genes and identifying new host responses. Unfortunately, with the rapid advances in pathway-directed analysis tools, investigation of genomics data from infection models is more and more limited to genes with known identities and/or functions. In many published studies, genes of unknown function or limited annotation are not described or discussed. Thus, one underutilized asset of microarray studies is their use as an avenue for increasing our understanding of the host response during acute infections and discovering new genes and pathways.
For example, as shown in Fig. 3 , published studies on 1918 influenza virus infection in mice revealed approximately 600 gene sequences of unknown or unannotated function that were up- or down-regulated (Kash et al., 2006c). Moreover, many of these genes were only regulated during 1918 virus infection and not during A/Tx/36/91 (Tx91) infection, suggesting they are associated with aberrant activation of inflammatory responses and immunopathology. Similarly, functional genomics can identify novel members of know antiviral pathways, including the type I IFN response, as shown in Fig. 4 . This example, taken from published studies of type I IFN treatment in the absence or presence of Ebola or Marburg virus infection, reveals many mRNAs with unannotated or unknown identity/function whose abundance is increased upon type I IFN stimulation (Kash et al., 2006b). Moreover the IFN-induced expression of the many of these genes is inhibited during ZEBOV, but not REBOV infection in human cells suggesting these genes could be associated with human virulence of these viruses. These examples demonstrate that many immune-regulated genes and pathways remain to be identified and characterized and that microarrays are a useful method for uncovering new features of fulminant RNA viral infections.
Fig. 3.
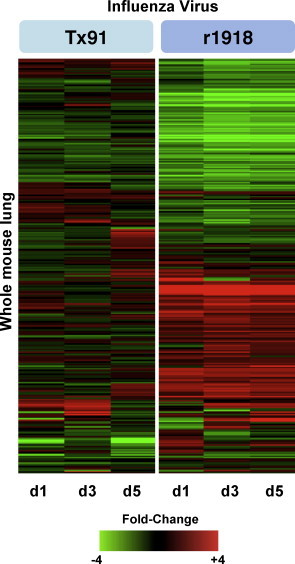
Expression of unannotated genes in lung tissue isolated from mice infected with equivalent doses the reconstructed 1918 virus (r1918) compared to a H1N1 human-adapted influenza virus A/Texas/36/91 (Tx91). For each infection point, the data presented are the error-weighted average expression changes calculated from four technical replicate arrays performed on three individual mice (n = 12 total). Adapted from Kash et al. (2006c) (data available at http://viromics.washington.edu/publications.html).
Fig. 4.
Expression of genes with unknown or unannotated function during Ebola and Marburg virus infection and type I IFN treatment. Expression of unannotated genes in mock, Zaire ebolavirus (ZEBOV), Marburvirus (MARV) or Reston ebolavirus (REBOV)-infected Huh7 cells treated with IFNα-2b for 24 h. For each infection time point, the data presented are the average expression from four technical replicate arrays performed on a pool of RNA from two independent experiments (n = 4 total). Adapted from Kash et al. (2006b) (data available at http://viromics.washington.edu/publications.html).
3.5.2. Using genomics for therapeutic target selection and validation
Analysis of microarray data in the presence and absence of a drug can indicate the range of gene expression programs affected by treatment and help to determine its efficacy. For example, numerous microarray studies have been performed to examine the responsiveness of patients with hepatitis C virus to therapy with interferon and ribavirin (reviewed in Walters and Katze, 2009). However, one major rate-limiting step in developing new host immune response targets for drug design is identification of new molecules/pathways whose activity reduces disease when modulated by therapeutic intervention, but does not have deleterious side effects. The high-throughput nature of expression microarrays allow for the identification of hundreds, if not thousands of infection responsive genes. The number of changes can be daunting, especially when viewed as potential drug targets. However, microarray studies can be used to refine selection of targets for further study and screen for primary and secondary gene expression changes affected by treatment.
A very simple experimental design is to monitor for changes in gene expression during drug treatment, so as to help determine how the drug exerts its down-stream effects. For example, Cao et al. used microarray analysis to show that inhibition of the mammalian target of rapamycin (mTOR) pathway by the immunosuppressive drug rapamycin suppressed expression of IFN and other immune response genes by plasmacytoid dendritic cells (Cao et al., 2008). Microarray analysis can also be used to examine the molecular effects of known antiretroviral drug therapies (Pacenti et al., 2006, Wu et al., 2008) and tradition medicines (Pan-Hammarstrom et al., 2006). The flexibility of global gene expression analysis has been applied to monitor for potential toxicity of drugs (Mendrick, 2008, Nuwaysir et al., 1999), vaccine candidates (Mizukami et al., 2008) and adjuvants (Mosca et al., 2008).
The discovery driven approach of functional genomics can unravel the complexity and dynamic range of responses associated with specific host response genes/pathways by delineating the gene expression response to infection of genetically modified gene knock-out (KO) animals. How does loss of a single cytokine or chemokine receptor affect host responses? For example, what is the effect on pathogenesis and host gene expression response to 1918 or H5N1 influenza virus in mice lacking expression of key antiviral response pathways, such as type I IFN, or inflammatory response receptors thought to be associated with development of immunopathology, such as Tnfr1? Are there redundant or alternate activation pathways, such as type III IFN stimulation of ISGs? Can microarray analysis of knock-out models be employed to identify any differences in the molecular mechanisms of host response antagonism and evasion of related pathogens?
A complementary genome-wide approach for identifying host pathways important for viral infection and new potential targets for antiviral therapy is using RNAi libraries to screen for host proteins that aid in viral replication. This approach has recently been used by Kawaoka and colleagues to identify cellular components necessary for the replication of influenza virus (Hao et al., 2008). Such studies, especially when combined with the high-throughput discovery driven nature of functional genomics studies will provide important new insights and open up new areas of research in the characterization of the host-pathogen dynamic and the identification of new host antiviral targets and development of novel therapies.
3.6. Conclusion
Understanding the molecular mechanisms of virulence and pathogenesis is critical to developing new antiviral therapies. This article has discussed how global gene expression studies have be used to detail the global relationship between antagonism of host antiviral responses during severe fulminant RNA viral infection and how selective tissue sampling considerations can be developed to better characterize the activation of inflammatory cascades and the development of severe pathology. As the importance of immune cell activation, killing and immunopathology become more prominent, isolation of infected and/or immune cells from infected tissue or blood samples using FACS or LCM as a method to isolate specific cell populations for genomics analysis should become more widely used. Discovery-based genomics approaches can aid in identifying the host response pathways associated with immunopathology and contribute to a more detailed understanding of how global host responses are regulated during viral infection that will be critical for the development of novel antiviral treatments. Thus, an important goal for developing new antiviral therapies is the development of multi-target approaches that both inhibit viral enzymatic activities and limit the activation of deleterious immune responses associated with the development of immunopathology. While it may seem counter-intuitive to block host responses to infection, a limited and temporary inhibition of some host inflammatory response could provide additional protection against the development of severe disease pathology and improve clinical outcome. The strength of genomics-based approaches lies in their ability to characterize the global effects of infection and to serve as a discovery-based tool to identify new host response components and pathways for further study, which may ultimately lead to novel targets of therapeutic intervention.
Acknowledgments
I would like to thank Drs. Mike Bray and Jeffery Taubenberger at NIH/NIAID for helpful discussions and suggestions. This work was supported by the Intramural Research Program of the NIH and the NIAID.
References
- Anon Influenza A virus subtype H5N1 infection in humans. Commun. Dis. Rep. Wkly. 1997;7:441. [PubMed] [Google Scholar]
- Anon Isolation of avian influenza A(H5N1) viruses from humans—Hong Kong, May–December 1997. Morb. Mortal. Wkly. Rep. 1997;46:1204–1207. [PubMed] [Google Scholar]
- Abdel-Ghafar A.N., Chotpitayasunondh T., Gao Z., Hayden F.G., Nguyen D.H., de Jong M.D., Naghdaliyev A., Peiris J.S., Shindo N., Soeroso S., Uyeki T.M. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 2008;358:261–273. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- Ank N., Iversen M.B., Bartholdy C., Staeheli P., Hartmann R., Jensen U.B., Dagnaes-Hansen F., Thomsen A.R., Chen Z., Haugen H., Klucher K., Paludan S.R. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Ank N., West H., Paludan S.R. IFN-lambda: novel antiviral cytokines. J. Interferon Cytokine Res. 2006;26:373–379. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- Baas T., Roberts A., Teal T.H., Vogel L., Chen J., Tumpey T.M., Katze M.G., Subbarao K. Genomic analysis reveals age-dependent innate immune responses to severe acute respiratory syndrome coronavirus. J. Virol. 2008;82:9465–9476. doi: 10.1128/JVI.00489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S., Barber G.N. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol. Biol. 2007;383:277–301. doi: 10.1007/978-1-59745-335-6_18. [DOI] [PubMed] [Google Scholar]
- Basler C.F., Mikulasova A., Martinez-Sobrido L., Paragas J., Muhlberger E., Bray M., Klenk H.D., Palese P., Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler C.F., Reid A.H., Dybing J.K., Janczewski T.A., Fanning T.G., Zheng H., Salvatore M., Perdue M.L., Swayne D.E., Garcia-Sastre A., Palese P., Taubenberger J.K. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2746–2751. doi: 10.1073/pnas.031575198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler C.F., Wang X., Muhlberger E., Volchkov V., Paragas J., Klenk H.D., Garcia-Sastre A., Palese P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden E.C., Sen G.C., Uze G., Silverman R.H., Ransohoff R.M., Foster G.R., Stark G.R. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M. The role of the type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001;82:1365–1373. doi: 10.1099/0022-1317-82-6-1365. [DOI] [PubMed] [Google Scholar]
- Cameron C.M., Cameron M.J., Bermejo-Martin J.F., Ran L., Xu L., Turner P.V., Ran R., Danesh A., Fang Y., Chan P.K., Mytle N., Sullivan T.J., Collins T.L., Johnson M.G., Medina J.C., Rowe T., Kelvin D.J. Gene expression analysis of host innate immune responses during Lethal H5N1 infection in ferrets. J. Virol. 2008;82:11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M., Muller M.P., Gold W.L., Richardson S.E., Poutanen S.M., Willey B.M., DeVries M.E., Fang Y., Seneviratne C., Bosinger S.E., Persad D., Wilkinson P., Greller L.D., Somogyi R., Humar A., Keshavjee S., Louie M., Loeb M.B., Brunton J., McGeer A.J., Kelvin D.J. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W., Manicassamy S., Tang H., Kasturi S.P., Pirani A., Murthy N., Pulendran B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claas E.C., Osterhaus A.D., van Beek R., De Jong J.C., Rimmelzwaan G.F., Senne D.A., Krauss S., Shortridge K.F., Webster R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- Crapo J.D., Barry B.E., Gehr P., Bachofen M., Weibel E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982;126:332–337. doi: 10.1164/arrd.1982.126.2.332. [DOI] [PubMed] [Google Scholar]
- de Lang A., Baas T., Teal T., Leijten L.M., Rain B., Osterhaus A.D., Haagmans B.L., Katze M.G. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV-infected macaques. PLoS Pathog. 2007;3:e112. doi: 10.1371/journal.ppat.0030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Veer M.J., Holko M., Frevel M., Walker E., Der S., Paranjape J.M., Silverman R.H., Williams B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- de Wit E., Fouchier R.A. Emerging influenza. J. Clin. Virol. 2008;41:1–6. doi: 10.1016/j.jcv.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enterlein S., Warfield K.L., Swenson D.L., Stein D.A., Smith J.L., Gamble C.S., Kroeker A.D., Iversen P.L., Bavari S., Muhlberger E. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob. Agents Chemother. 2006;50:984–993. doi: 10.1128/AAC.50.3.984-993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink J., Gu F., Ling L., Tolfvenstam T., Olfat F., Chin K.C., Aw P., George J., Kuznetsov V.A., Schreiber M., Vasudevan S.G., Hibberd M.L. Host gene expression profiling of dengue virus infection in cell lines and patients. PLoS Negl. Trop. Dis. 2007;1:e86. doi: 10.1371/journal.pntd.0000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornek J.L., Korth M.J., Katze M.G. Use of functional genomics to understand influenza–host interactions. Adv. Virus Res. 2007;70:81–100. doi: 10.1016/S0065-3527(07)70003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen B.L., Smith M., Katze M.G., Shi P.Y., Gale M., Jr. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P., Watanabe S., Ito T., Goto H., Wells K., McGregor M., Cooley A.J., Kawaoka Y. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 1999;73:3184–3189. doi: 10.1128/jvi.73.4.3184-3189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology. 2001;279:375–384. doi: 10.1006/viro.2000.0756. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A. Identification and characterization of viral antagonists of type I interferon in negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004;283:249–280. doi: 10.1007/978-3-662-06099-5_7. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A. Antiviral response in pandemic influenza viruses. Emerg. Infect. Dis. 2006;12:44–47. doi: 10.3201/eid1201.051186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus–host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Geiss G.K., Bumgarner R.E., An M.C., Agy M.B., van’t Wout A.B., Hammersmark E., Carter V.S., Upchurch D., Mullins J.I., Katze M.G. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266:8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- Geiss G.K., Salvatore M., Tumpey T.M., Carter V.S., Wang X., Basler C.F., Taubenberger J.K., Bumgarner R.E., Palese P., Katze M.G., Garcia-Sastre A. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl. Acad. Sci. U.S.A. 2002;99:10736–10741. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J.P., Pourrut X., Leroy E. Ebolavirus and other filoviruses. Curr. Top. Microbiol. Immunol. 2007;315:363–387. doi: 10.1007/978-3-540-70962-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groseth A., Stroher U., Theriault S., Feldmann H. Molecular characterization of an isolate from the 1989/90 epizootic of Ebola virus Reston among macaques imported into the United States. Virus Res. 2002;87:155–163. doi: 10.1016/s0168-1702(02)00087-4. [DOI] [PubMed] [Google Scholar]
- Gubareva L.V., McCullers J.A., Bethell R.C., Webster R.G. Characterization of influenza A/HongKong/156/97 (H5N1) virus in a mouse model and protective effect of zanamivir on H5N1 infection in mice. J. Infect. Dis. 1998;178:1592–1596. doi: 10.1086/314515. [DOI] [PubMed] [Google Scholar]
- Hale B.G., Randall R.E., Ortin J., Jackson D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Hao L., Sakurai A., Watanabe T., Sorensen E., Nidom C.A., Newton M.A., Ahlquist P., Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt B.H., Sanchez A., Offermann M.K. Ebola virus selectively inhibits responses to interferons, but not to interleukin-1beta, in endothelial cells. J. Virol. 1999;73:3491–3496. doi: 10.1128/jvi.73.4.3491-3496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Bird B.H., Towner J.S., Antoniadou Z.A., Zaki S.R., Nichol S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 2008;82:2699–2704. doi: 10.1128/JVI.02344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Dover J.E., Towner J.S., Nichol S.T. Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J. Virol. 2006;80:6430–6440. doi: 10.1128/JVI.00044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Ling L., Nichol S.T., Hibberd M.L. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008;82:5348–5358. doi: 10.1128/JVI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen T., Groseth A., Falzarano D., Feldmann H. Ebola virus: unravelling pathogenesis to combat a deadly disease. Trends Mol. Med. 2006;12:206–215. doi: 10.1016/j.molmed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Hutchinson K.L., Villinger F., Miranda M.E., Ksiazek T.G., Peters C.J., Rollin P.E. Multiplex analysis of cytokines in the blood of cynomolgus macaques naturally infected with Ebola virus (Reston serotype) J. Med. Virol. 2001;65:561–566. [PubMed] [Google Scholar]
- Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling P.B., Geisbert T.W., Jaax N.K., Hanes M.A., Ksiazek T.G., Peters C.J. Experimental infection of cynomolgus macaques with Ebola-Reston filoviruses from the 1989–1990 U.S. epizootic. Arch. Virol. Suppl. 1996;11:115–134. doi: 10.1007/978-3-7091-7482-1_11. [DOI] [PubMed] [Google Scholar]
- Jenner R.G., Young R.A. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Basler C.F., Garcia-Sastre A., Carter V., Billharz R., Swayne D.E., Przygodzki R.M., Taubenberger J.K., Katze M.G., Tumpey T.M. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J. Virol. 2004;78:9499–9511. doi: 10.1128/JVI.78.17.9499-9511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash J.C., Goodman A.G., Korth M.J., Katze M.G. Hijacking of the host-cell response and translational control during influenza virus infection. Virus Res. 2006;119:111–120. doi: 10.1016/j.virusres.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Muhlberger E., Carter V., Grosch M., Perwitasari O., Proll S.C., Thomas M.J., Weber F., Klenk H.D., Katze M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash J.C., Tumpey T.M., Proll S.C., Carter V., Perwitasari O., Thomas M.J., Basler C.F., Palese P., Taubenberger J.K., Garcia-Sastre A., Swayne D.E., Katze M.G. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobasa D., Jones S.M., Shinya K., Kash J.C., Copps J., Ebihara H., Hatta Y., Kim J.H., Halfmann P., Hatta M., Feldmann F., Alimonti J.B., Fernando L., Li Y., Katze M.G., Feldmann H., Kawaoka Y. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- Korth M.J., Kash J.C., Furlong J.C., Katze M.G. Virus infection and the interferon response: a global view through functional genomics. Methods Mol. Med. 2005;116:37–55. doi: 10.1385/1-59259-939-7:037. [DOI] [PubMed] [Google Scholar]
- Kotenko S.V., Gallagher G., Baurin V.V., Lewis-Antes A., Shen M., Shah N.K., Langer J.A., Sheikh F., Dickensheets H., Donnelly R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Liu M., Popper S.J., Rubins K.H., Relman D.A. Early days: genomics and human responses to infection. Curr. Opin. Microbiol. 2006;9:312–319. doi: 10.1016/j.mib.2006.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo Y.M., Gale M., Jr. Viral regulation and evasion of the host response. Curr. Top. Microbiol. Immunol. 2007;316:295–313. doi: 10.1007/978-3-540-71329-6_14. [DOI] [PubMed] [Google Scholar]
- Lu X., Tumpey T.M., Morken T., Zaki S.R., Cox N.J., Katz J.M. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J. Virol. 1999;73:5903–5911. doi: 10.1128/jvi.73.7.5903-5911.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrick D.L. Genomic and genetic biomarkers of toxicity. Toxicology. 2008;245:175–181. doi: 10.1016/j.tox.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Mizukami T., Imai J., Hamaguchi I., Kawamura M., Momose H., Naito S., Maeyama J., Masumi A., Kuramitsu M., Takizawa K., Nomura N., Watanabe S., Yamaguchi K. Application of DNA microarray technology to influenza A/Vietnam/1194/2004 (H5N1) vaccine safety evaluation. Vaccine. 2008;26:2270–2283. doi: 10.1016/j.vaccine.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Mohamadzadeh M., Chen L., Olinger G.G., Pratt W.D., Schmaljohn A.L. Filoviruses and the balance of innate, adaptive, and inflammatory responses. Viral Immunol. 2006;19:602–612. doi: 10.1089/vim.2006.19.602. [DOI] [PubMed] [Google Scholar]
- Mordstein M., Kochs G., Dumoutier L., Renauld J.C., Paludan S.R., Klucher K., Staeheli P. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens D.M., Taubenberger J.K., Fauci A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa S., Saijo M., Kurane I. Current knowledge on lower virulence of Reston Ebola virus (in French: Connaissances actuelles sur la moindre virulence du virus Ebola Reston) Comp. Immunol. Microbiol. Infect. Dis. 2007;30:391–398. doi: 10.1016/j.cimid.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Mosca F., Tritto E., Muzzi A., Monaci E., Bagnoli F., Iavarone C., O’Hagan D., Rappuoli R., De Gregorio E. Molecular and cellular signatures of human vaccine adjuvants. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10501–10506. doi: 10.1073/pnas.0804699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuwaysir E.F., Bittner M., Trent J., Barrett J.C., Afshari C.A. Microarrays and toxicology: the advent of toxicogenomics. Mol. Carcinog. 1999;24:153–159. doi: 10.1002/(sici)1098-2744(199903)24:3<153::aid-mc1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Onoguchi K., Yoneyama M., Takemura A., Akira S., Taniguchi T., Namiki H., Fujita T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- Osterlund P.I., Pietila T.E., Veckman V., Kotenko S.V., Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J. Immunol. 2007;179:3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- Pacenti M., Barzon L., Favaretto F., Fincati K., Romano S., Milan G., Vettor R., Palu G. Microarray analysis during adipogenesis identifies new genes altered by antiretroviral drugs. Aids. 2006;20:1691–1705. doi: 10.1097/01.aids.0000242815.80462.5a. [DOI] [PubMed] [Google Scholar]
- Pan-Hammarstrom Q., Wen S., Hammarstrom L. Cytokine gene expression profiles in human lymphocytes induced by a formula of traditional Chinese medicine, vigconic VI-28. J. Interferon Cytokine Res. 2006;26:628–636. doi: 10.1089/jir.2006.26.628. [DOI] [PubMed] [Google Scholar]
- Perrone L.A., Plowden J.K., Garcia-Sastre A., Katz J.M., Tumpey T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S., Krause C.D., Walter M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Randall R.E., Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Reghunathan R., Jayapal M., Hsu L.Y., Chng H.H., Tai D., Leung B.P., Melendez A.J. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A.H., Fanning T.G., Hultin J.V., Taubenberger J.K. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc. Natl. Acad. Sci. U.S.A. 1999;96:1651–1656. doi: 10.1073/pnas.96.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A.H., Fanning T.G., Janczewski T.A., Lourens R.M., Taubenberger J.K. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J. Virol. 2004;78:12462–12470. doi: 10.1128/JVI.78.22.12462-12470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A.H., Fanning T.G., Janczewski T.A., McCall S., Taubenberger J.K. Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J. Virol. 2002;76:10717–10723. doi: 10.1128/JVI.76.21.10717-10723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A.H., Janczewski T.A., Lourens R.M., Elliot A.J., Daniels R.S., Berry C.L., Oxford J.S., Taubenberger J.K. 1918 influenza pandemic caused by highly conserved viruses with two receptor-binding variants. Emerg. Infect. Dis. 2003;9:1249–1253. doi: 10.3201/eid0910.020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S.P., Leung L.W., Hartman A.L., Martinez O., Shaw M.L., Carbonnelle C., Volchkov V.E., Nichol S.T., Basler C.F. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins K.H., Hensley L.E., Wahl-Jensen V., Daddario DiCaprio K.M., Young H.A., Reed D.S., Jahrling P.B., Brown P.O., Relman D.A., Geisbert T.W. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 2007;8:R174. doi: 10.1186/gb-2007-8-8-r174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler A.J., Williams B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Gale M., Jr. Principles of intracellular viral recognition. Curr. Opin. Immunol. 2007;19:17–23. doi: 10.1016/j.coi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Sanchez A., Ksiazek T.G., Rollin P.E., Miranda M.E., Trappier S.G., Khan A.S., Peters C.J., Nichol S.T. Detection and molecular characterization of Ebola viruses causing disease in human and nonhuman primates. J. Infect. Dis. 1999;179(Suppl. 1):S164–S169. doi: 10.1086/514282. [DOI] [PubMed] [Google Scholar]
- Sen G.C., Sarkar S.N. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr. Top. Microbiol. Immunol. 2007;316:233–250. doi: 10.1007/978-3-540-71329-6_12. [DOI] [PubMed] [Google Scholar]
- Sommereyns C., Paul S., Staeheli P., Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez D.L., Perdue M.L., Cox N., Rowe T., Bender C., Huang J., Swayne D.E. Comparisons of highly virulent H5N1 influenza A viruses isolated from humans and chickens from Hong Kong. J. Virol. 1998;72:6678–6688. doi: 10.1128/jvi.72.8.6678-6688.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K., Klimov A., Katz J., Regnery H., Lim W., Hall H., Perdue M., Swayne D., Bender C., Huang J., Hemphill M., Rowe T., Shaw M., Xu X., Fukuda K., Cox N. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Taubenberger J.K. The origin and virulence of the 1918 “Spanish” influenza virus. Proc. Am. Philos. Soc. 2006;150:86–112. [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J.K., Hultin J.V., Morens D.M. Discovery and characterization of the 1918 pandemic influenza virus in historical context. Antivir. Ther. 2007;12:581–591. [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J.K., Morens D.M. The pathology of influenza virus infections. Annu. Rev. Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J.K., Reid A.H., Krafft A.E., Bijwaard K.E., Fanning T.G. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science. 1997;275:1793–1796. doi: 10.1126/science.275.5307.1793. [DOI] [PubMed] [Google Scholar]
- Taubenberger J.K., Reid A.H., Lourens R.M., Wang R., Jin G., Fanning T.G. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- Thompson W.W., Shay D.K., Weintraub E., Brammer L., Cox N., Anderson L.J., Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- Tumpey T.M., Basler C.F., Aguilar P.V., Zeng H., Solorzano A., Swayne D.E., Cox N.J., Katz J.M., Taubenberger J.K., Palese P., Garcia-Sastre A. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- Ubol S., Masrinoul P., Chaijaruwanich J., Kalayanarooj S., Charoensirisuthikul T., Kasisith J. Differences in global gene expression in peripheral blood mononuclear cells indicate a significant role of the innate responses in progression of dengue fever but not dengue hemorrhagic fever. J. Infect. Dis. 2008;197:1459–1467. doi: 10.1086/587699. [DOI] [PubMed] [Google Scholar]
- Van Campen H., Easterday B.C., Hinshaw V.S. Virulent avian influenza A viruses: their effect on avian lymphocytes and macrophages in vivo and in vitro. J. Gen. Virol. 1989;70(Pt 11):2887–2895. doi: 10.1099/0022-1317-70-11-2887. [DOI] [PubMed] [Google Scholar]
- Venter M., Myers T.G., Wilson M.A., Kindt T.J., Paweska J.T., Burt F.J., Leman P.A., Swanepoel R. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology. 2005;342:119–140. doi: 10.1016/j.virol.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Villinger F., Rollin P.E., Brar S.S., Chikkala N.F., Winter J., Sundstrom J.B., Zaki S.R., Swanepoel R., Ansari A.A., Peters C.J. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 1999;179(Suppl. 1):S188–S191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- Walters K.A., Katze M.G. Using high-throughput genomics to study hepatitis C: what determines the outcome of infection? Antiviral Res. 2009;81:198–208. doi: 10.1016/j.antiviral.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warke R.V., Xhaja K., Martin K.J., Fournier M.F., Shaw S.K., Brizuela N., de Bosch N., Lapointe D., Ennis F.A., Rothman A.L., Bosch I. Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J. Virol. 2003;77:11822–11832. doi: 10.1128/JVI.77.21.11822-11832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Q., Dwyer D.E., Dyer W.B., Yang Y.H., Wang B., Saksena N.K. Transcriptional profiles in CD8+ T cells from HIV+ progressors on HAART are characterized by coordinated up-regulation of oxidative phosphorylation enzymes and interferon responses. Virology. 2008;380:124–135. doi: 10.1016/j.virol.2008.06.039. [DOI] [PubMed] [Google Scholar]