

Abstract
Ribosomal RNA (rRNA) genes, essential to all forms of life, have been viewed as highly conserved and evolutionarily stable, partly because very little is known about their natural variations. Here, we explored large-scale variations of rRNA genes through bioinformatic analyses of available complete bacterial genomic sequences with an emphasis on formation mechanisms and biological significance. Interestingly, we found bacterial genomes in which no 16S rRNA genes harbor the conserved core of the anti–Shine-Dalgarno sequence (5′-CCTCC-3′). This loss was accompanied by elimination of Shine-Dalgarno–like sequences upstream of their protein-coding genes. Those genomes belong to 1 or 2 of the following categories: primary symbionts, hemotropic Mycoplasma, and Flavobacteria. We also found many rearranged rRNA genes and reconstructed their history. Conjecturing the underlying mechanisms, such as inversion, partial duplication, transposon insertion, deletion, and substitution, we were able to infer their biological significance, such as co-orientation of rRNA transcription and chromosomal replication, lateral transfer of rRNA gene segments, and spread of rRNA genes with an apparent structural defect through gene conversion. These results open the way to understanding dynamic evolutionary changes of rRNA genes and the translational machinery.
Keywords: rRNA, Shine–Dalgarno sequence, symbiosis, Mycoplasma, Flavobacteria, genomic rearrangement
Introduction
Ribosomal RNA (rRNA) genes have been widely used for estimating phylogeny because they are present in all cells, share a high similarity in their conserved regions (used for gene detection), but vary among different lineages in their less conserved regions (used for phylogeny classification) (Woese 1987; Doolittle 1999). There are often multiple rRNA operons within a genome (Lee et al. 2009). Intragenomic heterogeneity of rRNA genes has been observed (Acinas et al. 2004; Pei et al. 2010) in spite of homogenization processes of paralogous rRNA genes through gene conversion between rRNA operons on different loci (Hashimoto et al. 2003). Bacillus subtilis mutants that had only one rRNA operon showed varied sporulation abilities depending on which remained among the 11 rRNA operons (Nanamiya et al. 2010). Ribosomes with structural variations arising from the heterogeneous rRNA genes might play distinct roles, and diversity in the ribosomal functions may be advantageous to cells.
There is a functional region on 16S rRNAs that is highly conserved but unique to prokaryotes: the anti–Shine-Dalgarno (anti-SD) sequence on their 3′ tails (Shine and Dalgarno 1975). Direct binding of the SD sequence on the messenger RNA (mRNA) 5′-untranslated region (5′-UTR) with an anti-SD sequence initiates translation (fig. 1), which was first described in Escherichia coli as 5′-ACCUCCU-3′ (Shine and Dalgarno 1974). Its core sequence, 5′-CCUCC-3′, has been known to be universal among all the surveyed prokaryotes (Ma et al. 2002; Nakagawa et al. 2010) except for Candidatus Carsonella ruddii, which is a primary symbiont of insects (Thao et al. 2000). Degeneration of the anti-SD sequence suggests that the SD/anti-SD interaction is not the only mechanism for translation initiation. A well-known alternative mechanism is direct translation of leaderless mRNAs, which have no or an extremely short 5′-UTR. Such leaderless mRNAs account for 2.2% of the Helicobacter pylori transcriptome (Sharma et al. 2010). In Halobacterium salinarum, leaderless mRNAs were much more efficiently expressed than mRNAs containing the SD sequence (Sartorius-Neef and Pfeifer 2004). Escherichia coli MazF (an ACA-specific endoribonuclease) likely induces SD-independent translation by producing leaderless mRNAs and 16S rRNAs without the anti-SD sequence (Vesper et al. 2011).
Fig. 1.
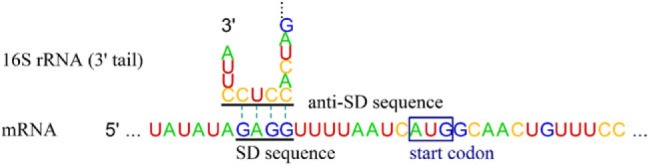
Interaction between SD and anti-SD sequences. Interaction is between the mRNA 5′-UTR (for rpsB gene) and the 16S rRNA 3′ tail in Escherichia coli.
Little effort has been made to understand variations and evolutionary changes of rRNA genes, which are possibly crucial proxies for cellular translation. In this study, variations that span a region larger than a single base pair on 16S rRNA genes in eubacteria were systematically surveyed. Some bacterial genomes do not possess the conserved core of the anti-SD sequences on any of their 16S rRNA genes and conceivable forces for the loss are inferred and discussed. We also describe other structural variations of rRNA genes, propose plausible underlying mechanisms through genomic comparison, and discuss their biological significance.
Materials and Methods
Genomic Sequences
Reference sequences of complete eubacterial genomes (supplementary table S1, Supplementary Material online) and their annotation information were downloaded from the National Center for Biotechnology Information website (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/) on 26 April 2011.
Search for 16S rRNA Genes without the Anti-SD Sequence
16S rRNA gene sequences were retrieved based on their registered annotations. We located corresponding regions of those sequences to the 3′ tail of the reference 16S rRNA gene (a sequence of the 16S rRNA gene of E. coli str. K-12 substr. DH10B) and assumed the located regions as the 3′ tail. The ClustalW algorithm (Larkin et al. 2007) was used for aligning the sequences. When the registered annotation did not include the 3′ tail sequence, we searched for it downstream. We surveyed the 3′ tail for the core anti-SD sequence, 5′-CCTCC-3′.
Phylogenetic Tree
Sequences were aligned by ClustalW with default parameters, and trees were constructed using the Molecular Evolutionary Genetics Analysis (MEGA5) alignment tool (Tamura et al. 2011) with the following implementations: maximum likelihood method, Tamura–Nei model (Tamura and Nei 1993), and bootstrap replication of 1,000 times.
Analysis of SD/Anti-SD Interaction
For every protein-coding gene, we retrieved the region −20 to −5 nt from the start codon (based on the registered annotations), which was designated as the SD region. Schurr et al. (1993) quantified the change in free energy (ΔG) in duplex formation between the SD region and the 3′ tail of 16S rRNA to select favorable SD sequences. Following this approach, other previous studies set a cutoff ΔG value for determining SD-like sequences, which was −3.4535 kcal/mol using the calculation by Starmer et al. (2006), but was −4.4 kcal/mol using the calculation by Ma et al. (2002). In this study, both parameters were considered for the prediction of SD-like sequences. ΔG was measured by the FREE_SCAN algorithm (Starmer et al. 2006).
Using this strategy, we estimated the gene fraction carrying the SD-like sequences among all protein-coding genes for a given genome by the equation: FSD = (Number of protein-coding genes with the SD-like sequences)/(Number of total protein-coding genes). FSD of each genome was adjusted to the SD index (dFSD) as Nakagawa et al. (2010) described by the equation: dFSD = FSD − rFSD, where rFSD was the fraction of the SD-like sequences among 20,000 artificial sequences (16 nt) constructed using a background fraction of each nucleotide as the probability for generating a particular nucleotide. The background nucleotide fraction was defined as the overall fraction of each nucleotide in sequences collected from −21 to −100 nt of all protein-coding genes in the given genome.
Analysis of Nucleotide Bias in the SD Region
We retrieved the upstream sequences (from −50 to −1) of every protein-coding gene in a given genome based on the registered annotations. Nucleotide fractions at specific positions of the retrieved sequences were compared with the background fractions (described above), and dfN (N = A, C, G, or T) denotes its change by the equation: dfN = (fraction of nucleotide N at the specific position) − (the background fraction of nucleotide N).
Search for Imperfect rRNA Genes and Reconstruction of Rearrangement History
We searched for sequences with partial homology to the E. coli 16S rRNA gene using the Blastn algorithm (Altschul et al. 1990) against the complete genomic sequences of Eubacteria. The detected sequence and its flanking 2-kb regions were compared with other homologous counterparts in the same genome or closely related genomes using the MEGA5 with the ClustalW algorithm implemented within. Although our primary targets were 16S rRNA genes, we expanded our analysis when a rearrangement event involved other adjacent genes. A dotplot was drawn to visualize genomic rearrangements using Gepard (Krumsiek et al. 2007) with a word length of 10 nt.
Results and Discussion
Degeneration of the Anti-SD Sequence
Unusual Sequences on 3′ Tails of 16S rRNA Genes
As asserted in many publications (Ma et al. 2002; Chang et al. 2006; Nakagawa et al. 2010), SD-like sequences are rarely seen upstream of protein-coding genes in some prokaryotic genomes despite the conserved anti-SD sequence on their 16S rRNA genes. This may imply that the SD/anti-SD interaction functions in an extremely minor portion of genes in those genomes. Evidence for loss of such interaction was reported previously in Candidatus Carsonella ruddii, as polymerase chain reaction sequencing of 16S–23S spacer regions revealed the loss of the core anti-SD sequence, 5′-CCTCC-3′ (referred to as the anti-SD motif) (Thao et al. 2000). Accelerated accumulation of complete genomic sequences allowed us to confirm the loss of the anti-SD motif not only in Candidatus Carsonella ruddii but also in many other bacterial genomes (table 1). Fifteen among 1,182 complete genomes of Eubacteria do not have the anti-SD motif in any of their 16S rRNA genes (herein, we refer to the 15 genomes as non–anti-SD genomes for simplicity).
Table 1.
Bacterial Strains with Unusual 16S rRNA 3′ Tail Sequences.
| Group | Class | Strain | Number of 16S rRNA Genes |
Genomic Size (nt) | Note | |
|---|---|---|---|---|---|---|
| Total | w/o Anti-SD Motif | |||||
| 1 | Flavobacteria | Flavobacteriaceae bacterium 3519-10 | 2 | 2 | 2,768,102 | Psychrophile |
| Flavobacteria | Riemerella anatipestifer DSM 15868 | 3 | 3 | 2,155,121 | Pathogen of poultry | |
| Flavobacteria | Weeksella virosa DSM 16922 | 5 | 5 | 2,272,954 | Isolated from human urine | |
| 2 | Flavobacteria | Blattabacterium sp. str. Bge | 1 | 1 | 636,850 | Primary symbionts of insects |
| Flavobacteria | Blattabacterium sp. str. BPLAN | 1 | 1 | 636,994 | ||
| Flavobacteria | Candidatus Sulcia muelleri CARI | 1 | 1 | 276,511 | ||
| Flavobacteria | Candidatus Sulcia muelleri DMIN | 1 | 1 | 243,933 | ||
| Flavobacteria | Candidatus Sulcia muelleri GWSS | 1 | 1 | 245,530 | ||
| Flavobacteria | Candidatus Sulcia muelleri SMDSEM | 1 | 1 | 276,984 | ||
| 3 | Alphaproteobacteria | Candidatus Hodgkinia cicadicola Dsem | 1 | 1 | 143,795 | Primary symbionts |
| Betaproteobacteria | Candidatus Zinderia insecticola CARI | 1 | 1 | 208,564 | of insects | |
| Gammaproteobacteria | Candidatus Carsonella ruddii PV | 1 | 1 | 159,662 | ||
| 4 | Mollicutes | Mycoplasma haemofelis str. Langford 1 | 1 | 1 | 1,147,259 | Hemotropic mycoplasmas |
| Mollicutes | Mycoplasma suis str. Illinois | 1 | 1 | 742,431 | ||
| Mollicutes | Mycoplasma suis KI3806 | 1 | 1 | 709,270 | ||
We categorized the non–anti-SD genomes into four groups (table 1) in terms of phylogeny and life style: Group 1 consists of three strains that harbor multiple 16S rRNA genes and that belong to the class Flavobacteria (Raymond et al. 2008; Lang et al. 2011; Mavromatis et al. 2011); Group 2 consists of six Flavobacteria strains which are primary symbionts (obligate and mutualistic bacteria with an ancient history of host association) of insects (McCutcheon et al. 2009a; Sabree et al. 2009); Group 3 members are also primary symbionts of insects, but belong to the phylum Proteobacteria (Nakabachi et al. 2006; McCutcheon et al. 2009b; McCutcheon and Moran 2010); Group 4 consists of three Mycoplasma strains living in erythrocytes (Barker et al. 2011; Guimaraes et al. 2011).
In figures 2–4, we present phylogenetic trees based on full-length 16S rRNA genes of these non–anti-SD genomes and other references to show phylogenetic contexts of the loss. Groups 1 (fig. 2A), 2 (fig. 2A), and 4 (fig. 4A) clustered into distinctive clades, individually, implying that the first mutation within the anti-SD motif possibly took place in an ancestor of each group. Members of Group 1 share a variation: the anti-SD motif, 5′-CCTCC-3′, was changed to 5′-TCTCA-3′ (fig. 2B). In the other groups, the variation is diverse within a group. In Group 2, the anti-SD motif, 5′-CCTCC-3′, was changed to 5′-TCTCT-3′ or 5′-TTTCT-3′ (fig. 2B). There is divergence even within the strains in Candidatus Sulcia muelleri: 5′-TCTCT-3′ from CARI and SMDSEM and 5′-TTTCT-3′ from DMIN and GWSS. Extensively degenerated 3′ tail sequences were found in Group 3; 5′-CCTCC-3′ was changed to 5′-TTTGA-3′, 5′-CATTT-3′, or 5′-TTTTT-3′ (fig. 3B). In Group 4, Mycoplasma haemofelis has a degenerated sequence, 5′-TCTTC-3′, and the two M. suis strains have 5′-CTTTT-3′, instead of the anti-SD motif (fig. 4B).
Fig. 2.

Comparative analysis of the anti-SD genomes and the non–anti-SD genomes in the class Flavobacteria. (A) Maximum likelihood phylogenetic tree. Groups 1 and 2 non-anti-SD genomes are shown in gray. (B) Predicted 16S rRNA 3′ tail sequences. The regions corresponding to the Escherichia coli anti-SD motif are shaded in gray. (C) SD indexes (dFSD). Triangle: cutoff value < −3.4535 kcal/mol; Dot: cutoff value < −4.4 kcal/mol. (D) Fractions of the four nucleotides (dfA, dfC, dfG, and dfT) at specific positions between −50 and −1 nt from all protein-coding genes in each genome. The background fraction was subtracted. For details, see Materials and Methods.
Fig. 3.

Comparative analysis of the anti-SD genomes and the non-anti-SD genomes in the phylum Proteobacteria. (A) Maximum likelihood phylogenetic tree. Group 3 non–anti-SD genomes are shown in gray. (B) Predicted 16S rRNA 3′ tail sequences. The regions corresponding to the Escherichia coli anti-SD motif are shaded in gray. (C) SD indexes (dFSD). Triangle: cutoff value < −3.4535 kcal/mol; Dot: cutoff value < −4.4 kcal/mol. (D) Fractions of the four nucleotides (dfA, dfC, dfG, and dfT) at specific positions between −50 and −1 nt from all protein-coding genes in each genome. The background fraction was subtracted. For details, see Materials and Methods. (i) The class Gammaproteobacteria. (ii) The class Betaproteobacteria. (iii) The class Alphaproteobacteria.
Fig. 4.

Comparative analysis of the anti-SD genomes and the non–anti-SD genomes in the genus Mycoplasma. (A) Maximum likelihood phylogenetic tree. Group 4 non–anti-SD genomes are shown in gray. (B) Predicted 16S rRNA 3′ tail sequences. The regions corresponding to the Escherichia coli anti-SD motif are shaded in gray. (C) SD indexes (dFSD). Triangle: cutoff value < −3.4535 kcal/mol; Dot: cutoff value < −4.4 kcal/mol. (D) Fractions of the four nucleotides (dfA, dfC, dfG, and dfT) at specific positions between −50 and −1 nt from all protein-coding genes in each genome. The background fraction was subtracted. For details, see Materials and Methods.
Multiple Cs in the anti-SD sequence possibly are pivotal elements for firm RNA–RNA binding by forming C-G hydrogen bonds, which are stronger than the A-T bonds (Freier et al. 1986). The degeneration of the anti-SD motif, mentioned above, predominantly resulted in substitutions from C to T or A, thereby likely decreasing the SD/anti–SD-binding capacity.
Lack of SD-Like Sequences in Genomes with a Defect in the Anti-SD Motif
Next, we looked for SD-like sequences in these non–anti-SD genomes. The widely accepted strategy to determine SD-like sequences is to test whether the SD region (defined as the region −20 to −5 nt from the start codon) is able to form a duplex with the host's 16S rRNA 3′ tail. There will be many combinations of duplexes. Starmer et al. (2006) assumed that a given SD region had an SD-like sequence when the lowest ΔG value among the combinations was lower than −3.4535 kcal/mol, whereas Ma et al. (2002) applied a more stringent parameter, −4.4 kcal/mol. Using both parameters, we obtained fraction of protein-coding genes with SD-like sequences (FSD). Based on the work of Nakagawa et al. (2010), we used FSD after substituting the SD fraction in artificial sequences generated based on its background nucleotide fraction (for details, see Materials and Methods). Thus, the adjusted value (dFSD) indicates a gene fraction with the SD/anti-SD interaction relative to a fraction with random intergenic region/anti-SD interaction. A dfSD value < 0 indicates that fewer SD regions than random intergenic regions have capacities for binding to 16S rRNA 3′ tails.
Another method we applied directly showed nucleotide bias in the SD region. We calculated changes in the nucleotide fraction (dFN; N = A, C, G, or T) at specific positions before the start codon. Because the conserved anti-SD motif is C-rich, the functional SD region should be G-rich, which is clearly seen in the E. coli SD regions (fig. 3D(i)).
Signal intensities of the SD-like sequences have not been studied in Groups 1 and 2 non–anti-SD genomes belonging to the class Flavobacteria (table 1). To our surprise, we found that members of this class showed dFSD values < 0 and mean ΔG values > −1 kcal/mol with the standard deviations ranging from 1.4 to 2.12, regardless of the presence of an anti-SD motif (fig. 2C and supplementary table S2, Supplementary Material online). A possible explanation may be due to A-rich signals at the corresponding areas of the SD region in all surveyed Flavobacteria (fig. 2D), as opposed to the G-rich signal in E. coli, which may aid ribosomal protein S1 to bind and assist in translation initiation as an alternative to the anti-SD sequence. In E. coli, ribosomal protein S1 co-contributes to translation initiation complex formation through its high affinity to AU-rich regions often observed on 5′-UTRs (Draper et al. 1977; Boni et al. 1991; Sengupta et al. 2001; Salah et al. 2009). The protein seems to assist firm binding between mRNA and the ribosome because it was dispensable when the SD/anti-SD interaction was strong (Farwell et al. 1992). The ability of ribosomal protein S1 to initiate translation by itself without the SD/anti-SD interaction has not been demonstrated. Another conceivable possibility is that a stretch of T-rich sequences right after the anti-SD sequence (fig. 2B) interacts with the mRNA A-rich region for translation initiation. Our 16S rRNA 3′ tail prediction was based on sequence comparison with the reference E. coli 16S rRNA. Thus, Flavobacteria 16S rRNA 3′ tails could be longer (which may support the A-T interaction hypothesis) or shorter (which may dim the hypothesis) than the predicted length. Accurate annotation of 16S rRNA ends in the non–anti-SD strains, which include Flavobacteria and other taxonomic groups, remains to be achieved, which would give insight to processes underlying anti-SD motif degeneration.
Figure 3 shows the non–anti-SD genomes (Group 3) and the reference genomes in the phylum Proteobacteria. Various dFSD values (range: 0.09–0.41 with a cutoff value of −4.4 kcal/mol) are seen in the reference genomes, whereas those ≤0 were observed in the non–anti-SD genomes (fig. 3C, supplementary table S2, Supplementary Material online). This result is consistent with the nucleotide bias analysis (fig. 3D): G peaks were observed in the references but not in Group 3 non–anti-SD genomes. Mean ΔG values of Group 3 non–anti-SD genomes (range: −1.43 to −0.73) were higher than those of the references (range: −5.81 to −1.78) (supplementary table S2, Supplementary Material online). The ΔG standard deviation values more clearly distinguish Group 3 (range: 1.07–1.60) from the references (range: 2.64–3.18). It is noteworthy that many reference genomes we chose were host-obligate bacteria with similar features, in terms of host association, to Group 3 strains. For example, Buchnera aphidicola str. 5A, Baumannia cicadellinicola str. Hc, and Candidatus Blochmannia vafer str. BVAF are primary symbionts of insects, as are all Group 3 strains. Our result supports the loss of the SD/anti-SD interaction among Group 3 members and also indicates that being a primary symbiont of insects is not necessarily indicative of anti-SD motif loss.
The phylum Mollicutes, which includes the Mycoplasma genus, is diverse in SD-like signals (Nakagawa et al. 2010). Three recently sequenced hemotropic mycoplasmas (Group 4) that lost anti-SD motifs (fig. 4B) did not indicate SD/anti-SD interactions; there were no G-rich signals or any indicators within the SD regions (fig. 4C and D). Among Mycoplasma strains with the anti-SD motif, Mycoplasma genitalium and Mycoplasma pneumonia, which are phylogenetically closely related (fig. 4A), showed no G-rich signals within the SD regions and had very low dFSD and high ΔG values (fig. 4C and D and supplementary table S2, Supplementary Material online). The ΔG standard deviation values (range: 2.60–3.47) of these two strains, however, were distinct from Group 4 (range: 1.69–1.73). In other Mycoplasma species, the SD/anti-SD interaction appears to largely involve translation initiation, as considerable SD-like signals were observed.
These results substantiate our assumption that the loss of the anti-SD motif equates to loss of SD/anti-SD interactions. Conservation of the anti-SD motif, however, does not always correspond to a high frequency in SD-like sequences, as dramatically seen in Flavobacteria (with the anti-SD motif) with an A-rich signal prior to the start codon as well as in M. genitalium and M. pneumonia. We assume that SD-led and non–SD-led mechanisms (direct translation of leaderless mRNA or other unknown mechanisms) for translation initiation coexist in most bacteria, and SD-led mechanisms appear in a very small number of Flavobacteria (with the anti-SD motif), M. genitalium, and M. pneumonia genes or not at all in the non–anti-SD strains.
Factors Responsible for the Anti-SD Motif Loss
The loss of the anti-SD motif in primary symbionts (Groups 2 and 3) and hemotropic mycoplasmas (Group 4) might be related to their intracellular characteristics. An intracellular lifestyle restricts the effective population size, causes frequent population bottlenecks at the time of transmission, and maintains copious metabolites, which promote deleterious mutations and massive genomic downsizing (Toft and Andersson 2010). Primary symbionts and Mycoplasma are thought to be at the extreme of this adaptation, as they lack genes most free-living bacteria have (including those for DNA repair, recombination, and transfer) and carry the smallest genomes among the sequenced to date as a consequence (Moran 2002; Dale and Moran 2006). The SD/anti-SD interaction, which is useful but not essential for life, might have degenerated in response to genomic minimization. If we assume that ancestors of obligate non–anti-SD strains (Groups 2, 3, and 4) in a free-living state had multiple mechanisms for translation initiation, the evolutionary forces toward large-scale gene/function loss during a period of host association may have forced the strains to sacrifice some genetic elements for the SD-led mechanism.
Loss of the SD/anti-SD interaction, however, is not strongly correlated with reduced genomic size. Although Groups 2 and 3 non–anti-SD genomes are members of the smallest genomes among those used in this study (table 1 and supplementary table S1, Supplementary Material online), SD-led translation initiation appears to work in B. aphidicola str. 5A, B. cicadellinicola str. Hc and Ca. Blochmannia vafer str. BVAF (fig. 3), which are also primary symbionts of insects and have genomic sizes as small as those in Groups 2 and 3 (supplementary table S1, Supplementary Material online). Moreover, the genomic size of M. haemofelis str. Langford 1 (Group 4) is the third largest among 26 Mycoplasma strains we analyzed (supplementary table S1, Supplementary Material online and fig. 4). Each bacterium is on its own evolutionary stage and path, which makes it difficult to simplify causes for anti-SD motif loss. Other evolutionary forces related with host interactions, such as immune evasion, unique intracellular environments (e.g., within an erythrocyte), and lineage-specific inherited features of translation processes, may have played a role in this loss.
Another feature relevant to anti-SD motif loss is being a member of the class Flavobacteria. No primary symbionts in this class that has been completely sequenced to date possess an anti-SD motif. Three non–anti-SD genomes that do not show extreme genomic reduction and have multiple rRNA operons (Group 1) also fall within this class (table 1). The distinctive A-rich pattern before a start codon (fig. 2D) represents an unknown alternative mechanism for translation initiation (which might be enabled by ribosomal protein S1 or a novel form of rRNA–mRNA interaction as described above) that is superior to the SD-led mechanism in Flavobacteria. Flavobacteria seem prone to the loss of the anti-SD motif due to the weak dependence on the SD/anti-SD interaction compared with other bacterial groups.
Inferred Mechanisms of Rearranged rRNA Genes
We also analyzed structural variants of rRNA genes with an emphasis on their underlying formation mechanisms and possible biological significance to elucidate rRNA gene evolution. Among 1,182 complete eubacterial genomes we surveyed, 15 (1.3%) carried rearranged 16S rRNA genes formed by various mechanisms (supplementary table S3, Supplementary Material online).
Inversion of the rRNA Operon
Splitting of 16S rRNA genes associated with inversion was found. Yersinia pestis biovar Microtus 91001 has undergone extensive genomic rearrangements and disruptions when compared with the genome of Y. pestis KIM 10 (fig. 5A) (Song et al. 2004).
Fig. 5.

Inversion of rRNA operons. (A) Dotplot between genomes. (B) Reconstruction. (C) Recombination sites in one operon. (D) Recombination sites in the other operon. Amino acid names indicate the corresponding tRNA genes.
The KIM 10 genome (fig. 5B(i)) harbored rRNA operons in the same direction as chromosomal DNA replication, as do many bacterial genomes (Rocha 2002; Price et al. 2005), which is probably because head-to-head encounters of replisomes and RNA polymerases disturb genomic replication (Liu and Alberts 1995; Wang et al. 2007; Srivatsan et al. 2010), while bacteria seem to have acquired mechanisms to cope with a codirectional collision (Pomerantz and O'Donnell 2008). A chromosomal region of approximately 220 kb carrying two rRNA operons was somehow translocated to the other “arm” of the chromosome across the replication origin in the same orientation (fig. 5B(ii)). (A possible mechanism for this translocation is two rounds of inversion.) Then, the two rRNA operons lay in opposite direction of DNA replication. Inversion of each operon within this region corrected their relative orientation and avoided the conflict in biovar Microtus 91001-type genomes (fig. 5B(iii)).
The above scenario is based on our dotplot analyses (fig. 5A). Further sequence comparison (fig. 5C and D) revealed that the inversion was likely mediated by recombination at short (3–5 nt) similar or identical DNA sequences. One recombination site is located at the 5′ end of the 16S rRNA gene, and the product has split of the 5′ end (fig. 5C and D). We assumed that the split resulted in a negative functional effect on these 16S rRNA genes because the short split region (about 20 nt from the 5′ end) plays an important role in connecting the 16S rRNA's 5′ domain to the 3′ major domain (Wimberly et al. 2000), hence the split region (the shorter one) is highly conserved and frequently used as a universal priming site (Turnbaugh et al. 2009; Unno et al. 2010).
Partial Duplication
We observed a novel rearrangement pattern that resembles internal duplications within an rRNA operon with an insertion of a short DNA between the duplicates (fig. 6).
Fig. 6.

Partial duplication of rRNA genes. (A) Gene rearrangement in Actinobacillus pleuropneumoniae L20. (i) A proposed mechanism for the rearrangement: insertion with long target duplication. (ii) The other mechanism: tandem duplication followed by homologous recombination. (iii) The rearrangement. (B) Gene rearrangement in Bacillus thuringiensis BMB171. (i) A proposed mechanism for the rearrangement. (ii) The rearrangement.
Figure 6A illustrates the rearrangements (fig. 6A(iii)) and two possible underlying mechanisms (fig. 6A(i) and (ii)). In the first pathway (fig. 6A(i)), a short (21 nt) DNA segment (or its larger ancestral form) (black box) was inserted into the genome with a duplication of a long sequence (white arrow) on the target. Such mechanisms of “insertion with long target duplication” have been recognized for restriction-modification systems and other DNAs (Nobusato et al. 2000; Furuta et al. 2010, 2011), but not yet for short DNAs. In the second possible pathway (fig. 6A(ii)), tandem duplication of a DNA segment (black box and white arrow) on a 16S rRNA gene occurs first and is followed by homologous recombination with the same locus on another genome (or with another 16S rRNA gene locus on the same genome or a different genome). If the recipient DNA in the homologous recombination is so diverged from the donor DNA that it lacks the sequence corresponding to the black box (fig. 6A(ii)), the final product will show the observed pattern: direct repeats (white arrows) interrupted with a short DNA (black box).
The likely end product was observed in Actinobacillus pleuropneumoniae L20, where two direct duplicates (270 nt) were detected with a DNA of unknown origin (21 nt) between them (fig. 6A(iii)). Because the origin of the 21-nt DNA was not identified through our homology search, we put more weight on the first hypothesis of insertion with long target duplication for this rearrangement.
Figure 6B(i) illustrates an extended version of the mechanism described in figure 6A(ii). Two events of tandem duplication nearby (A∼B to AA∼BB) are followed by a substitution in the inner duplicates (AA∼BB) with its corresponding region in another rRNA operon that has partial divergence at the regions homologous to the endpoints of the recipient's duplicated region. The reconstruction was inferred from a rearrangement pattern in the Bacillus thuringiensis BMB17 rRNA operons (fig. 6B(ii)). One duplication event occurred at the 16S rRNA gene 3′ end and the other at the 23S rRNA gene 5′ end. Short diverging DNAs (white and black box) did not show any significant similarity with any region in the B. thuringiensis Al Hakam genome, but the DNAs and their flanking regions exactly matched to rRNA operons of Bacillus cereus ATCC 14579 (fig. 6B(ii)). This suggests an interspecific lateral transfer of an rRNA operon fragment. Our hypothesis states that two tandem duplications occurred at an rRNA operon in the B. cereus ATCC 14579-type genome, and the duplicated region was substituted by an rRNA operon of the Al Hakam–type genome, as we described above, to produce the BMB171-type operon.
An Incomplete 16S rRNA Gene on a Plasmid
Figure 7A illustrates a plausible scenario of lateral transfer of a partial 16S rRNA gene via a plasmid, followed by its partial incorporation into a homologous region in the chromosome through homologous recombination. Such a scenario was proposed for Lactococcus lactis cremoris SK11, which carries a partial 16S rRNA gene in plasmid 3 (fig. 7B) (Siezen et al. 2005). The gene on the plasmid and those on the chromosome differ in two regions (fig. 7B and C). Figure 7C compared the diverged positions with their homologous sequences on another genome (L. lactis cremoris MG1363) in the same subspecies cremoris and on a genome (L. lactis lactis I11403) in a different subspecies lactis. On “region 1,” there were nine diverged positions among the genes, through which two classes can be distinguished (fig. 7C). All cremoris MG1363 16S rRNA loci and five of six loci on the cremoris SK11 chromosome comprise one class with complete sequence identity, whereas one locus on the chromosome and one on the cremoris SK11 plasmid comprise the other class with all lactis I11403 loci. This suggests that partial gene was transferred from the lactis I11403-type to the cremoris MG1363-type genome by the plasmid, and homologous recombination replaced the region 1 on a locus with that in the partial gene. “Region 2” in the partial gene is also identical to that in the lactis I11403-type but not to that of the other cremoris-type genes (fig. 7C), suggesting that homologous recombination took place outside of region 2. Significance of the variation in the translation process and in the biology of the plasmid has yet to be examined.
Fig. 7.

An incomplete 16S rRNA gene on a plasmid. (A) Reconstruction. (B) The rearrangement. Two diverged regions between the two genes are boxed. (C) Sequence comparison in the diverged regions.
Transposon Insertion
Recent transposon insertions into 16S rRNA genes retaining duplicated target sequences and imperfect inverted repeats at the transposon ends were found in two distantly related species (fig. 8). The insertion in Corynebacterium aurimucosum ATCC 700975 occurred near the boundary between the 5′ and central domains (fig. 8A), whereas that in Thermus scotoductus SA-01 (fig. 8B) occurred near the boundary between the central and 3′ major domains. We do not know whether these split genes are functional or expressed.
Fig. 8.
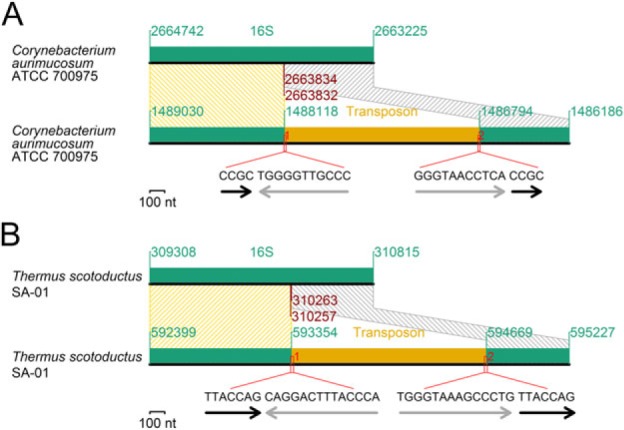
Transposon insertion. (A) An insertion into a 16S rRNA gene. (B) Another insertion into a 16S rRNA gene. Black arrows: target duplication. Gray arrows: incomplete inverted repeats at the transposon ends.
Deletion
One of ten 16S rRNA genes in Clostridium difficile CD196 has lost a segment of 101 nt near its 3′ end. Sequence similarity (9 nt) within the gene seems responsible for the recombination-mediated deletion (fig. 9A). The lost region comprises about a half of the major projection in the 16S rRNA 3′ minor domain, which dictates 30S and 50S subunit interactions. The region that includes 3/4 of a 23S rRNA gene and 1/2 of a 16S rRNA gene has been deleted together with the intergenic region in the Candidatus Protochlamydia amoebophila UWE25 rRNA operon (fig. 9B), as previously reported (Pei et al. 2010).
Fig. 9.

Other modes of rRNA gene rearrangements. (A) Internal deletion in a 16S rRNA gene. (B) Deletion in an rRNA operon. (C) Substitution. An amino acid name indicates the corresponding tRNA gene. (D) Inversion within an rRNA operon.
Substitution
One Bacillus amyloliquefaciens FZB42 16S rRNA gene is already known for being partial (Pei et al. 2010). By genomic comparison with B. amyloliquefaciens DSM7 and a homology search against its own genome, we revealed that this could be explained by DNA substitution (fig. 9C). Substitution through recombination requires sequence similarity at two locations, 5′ and 3′. In our scenario, similarity at the 5′ end is provided by transfer RNA (tRNA) genes (Arg-Pro tRNA) and that at the 3′ end by a short DNA sequence, 5′-GAG-3′. The region enclosed by the similar sequences was about more than a half of a 16S rRNA gene in one duplex and Ala-Met tRNA in the other duplex (fig. 9C). The substitution brought about addition of the Ala and Met tRNA genes and partial deletion of the 16S rRNA gene in FZB42.
Another Inversion Event
In Lactobacillus reuteri JCM 1112, inversion separated the majority of the 16S rRNA gene from its short part (about 30 nt) at the 3′ end (fig. 9D) and also separated the majority of the 23S rRNA gene from its short part (about 10 nt) at the 5′ end. The former short part constitutes the minor projection of 3′ minor domain. Sequence similarity at the break points is not clear. Biological significance of this event is also not clear.
Spread of rRNA Gene Variants through Gene Conversion
Structural variants with nearly identical sequences are found in multiple rRNA operons within a genome (table 2), suggesting that these rRNA gene variants have spread to other rRNA operon loci through gene conversion. The rearrangement in the B. thuringiensis BMB171 genome (fig. 6B) leaves intact RNA-coding regions, and all the rRNA operons in the genome share the same mutant form (table 2). The rearrangement in A. pleuropneumoniae L20 resulted in a large structural alteration of the gene (fig. 6A), and this mutant allele has spread to only two more loci out of five other rRNA operon loci (table 2). The mutant form in L. reuteri JCM 1112 (fig. 9D) was found in two of six rRNA operon loci (table 2).
Table 2.
Distribution of Structural Variants among Multiple rRNA Genes within a Genome.
Gene conversion between rRNA operons was proposed to be responsible for homogenizing the operons and retaining conserved rRNA structures (Hashimoto et al. 2003). Gene conversion may also help in incorporating foreign rRNA genes (Yap et al. 1999). Our report implies that gene of rRNA gene conversion can propagate mutants with a structural defect to other loci, which was up to a half of the loci within a genome in this study.
Inactivation of some rRNA genes within a genome may not be crucial for cell growth. In E. coli, only five among seven rRNA operons were sufficient to compete against a wild-type strain under stable growth conditions (Condon et al. 1995). The smaller number of available rRNA genes, however, could be disadvantageous to bacterial adaptation in fluctuating growth conditions (Klappenbach et al. 2000; Stevenson and Schmidt 2004). It would be a good subject for experimental studies to investigate the fate and phenotypic relevance of those rearranged rRNA genes.
Conclusion
In the present study, we revealed novel evolutionary histories and variations of bacterial rRNA genes. The loss of the anti-SD motif in all of 16S rRNA genes within some bacterial genomes (table 1) indicated that the SD/anti-SD interaction is not a universal translation initiation mechanism in prokaryotes. The loss is seen in many minute genomes that are obligate to eukaryotic hosts (hemotropic Mycoplasma and primary symbionts of insects), implying some relationship between the loss and extreme genomic reduction of the obligate bacteria. Flavobacteria rarely rely on the SD-led translation initiation (fig. 2), thus the anti-SD motif might start degenerating even in non–host-associated bacteria. We also described how the rRNA genes rearrange. One of the rearrangements (fig. 5) supports the earlier concept of necessity of co-orientation of rRNA operon with chromosomal replication. Other reconstructions (fig. 6) suggested partial rRNA gene duplication followed by gene conversion to other loci, some of which occurred across the special boundary. rRNA genes, likely laterally transferred via a plasmid, can play a role in intragenomic rRNA sequence heterogeneity (fig. 7). We showed that gene conversion sometimes spreads defective forms of rRNA genes to other loci (table 2), thereby reducing available numbers of rRNA operons. These structural variants could provide novel targets for evolutionary and functional studies of rRNAs and translation.
Supplementary Material
Supplementary tables S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Saaya Lim Tsutsué and Riu Yamashita for critical review and helpful suggestions. We thank Hor-Gil Hur and Tatsuya Unno for providing perspective on rRNA genes. This work was supported by grants to I.K. from the global Center of Excellence project of “Genome Information Big Bang” and “Grant-in-Aid for Scientific Research on Innovative Areas” (24113506) from the Ministry of Education, Culture, Sports, Science and Technology-Japan (MEXT) and the “Grant-in-Aid for Scientific Research” from the Japan Society for the Promotion of Science (JSPS) (21370001). This research was conducted at the Department of Medical Genome Sciences, Graduate School of Frontier Sciences, University of Tokyo, Tokyo, Japan.
References
- Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol. 2004;186:2629–2635. doi: 10.1128/JB.186.9.2629-2635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barker EN, Helps CR, Peters IR, Darby AC, Radford AD, Tasker S. Complete genome sequence of Mycoplasma haemofelis, a hemotropic mycoplasma. J Bacteriol. 2011;193:2060–2061. doi: 10.1128/JB.00076-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni IV, Isaeva DM, Musychenko ML, Tzareva NV. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 1991;19:155–162. doi: 10.1093/nar/19.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Halgamuge S, Tang SL. Analysis of SD sequences in completed microbial genomes: non-SD-led genes are as common as SD-led genes. Gene. 2006;373:90–99. doi: 10.1016/j.gene.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Condon C, Liveris D, Squires C, Schwartz I, Squires CL. rRNA operon multiplicity in Escherichia coli and the physiological implications of rrn inactivation. J Bacteriol. 1995;177:4152–4156. doi: 10.1128/jb.177.14.4152-4156.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale C, Moran NA. Molecular interactions between bacterial symbionts and their hosts. Cell. 2006;126:453–465. doi: 10.1016/j.cell.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Draper DE, Pratt CW, von Hippel PH. Escherichia coli ribosomal protein S1 has two polynucleotide binding sites. Proc Natl Acad Sci U S A. 1977;74:4786–4790. doi: 10.1073/pnas.74.11.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell MA, Roberts MW, Rabinowitz JC. The effect of ribosomal protein S1 from Escherichia coli and Micrococcus luteus on protein synthesis in vitro by E. coli and Bacillus subtilis. Mol Microbiol. 1992;6:3375–3383. doi: 10.1111/j.1365-2958.1992.tb02205.x. [DOI] [PubMed] [Google Scholar]
- Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH. Improved free-energy parameters for predictions of RNA duplex stability. Proc Natl Acad Sci U S A. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Abe K, Kobayashi I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010;38:2428–2443. doi: 10.1093/nar/gkp1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Kawai M, Yahara K, et al. (12 co-authors) Birth and death of genes linked to chromosomal inversion. Proc Natl Acad Sci U S A. 2011;108:1501–1506. doi: 10.1073/pnas.1012579108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes AM, Santos AP, SanMiguel P, Walter T, Timenetsky J, Messick JB. Complete genome sequence of Mycoplasma suis and insights into its biology and adaption to an erythrocyte niche. PLoS One. 2011;6:e19574. doi: 10.1371/journal.pone.0019574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto JG, Stevenson BS, Schmidt TM. Rates and consequences of recombination between rRNA operons. J Bacteriol. 2003;185:966–972. doi: 10.1128/JB.185.3.966-972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klappenbach JA, Dunbar JM, Schmidt TM. rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol. 2000;66:1328–1333. doi: 10.1128/aem.66.4.1328-1333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23:1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- Lang E, Teshima H, Lucas S, et al. (37 co-authors) Complete genome sequence of Weeksella virosa type strain (9751) Stand Genomic Sci. 2011;4:81–90. doi: 10.4056/sigs.1603927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. (13 co-authors) Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lee ZM, Bussema C, 3rd, Schmidt TM. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 2009;37:D489–D493. doi: 10.1093/nar/gkn689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Alberts BM. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995;267:1131–1137. doi: 10.1126/science.7855590. [DOI] [PubMed] [Google Scholar]
- Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromatis K, Lu M, Misra M, et al. (35 co-authors) Complete genome sequence of Riemerella anatipestifer type strain (ATCC 11845) Stand Genomic Sci. 2011;4:144–153. doi: 10.4056/sigs.1553862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci U S A. 2009a;106:15394–15399. doi: 10.1073/pnas.0906424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, McDonald BR, Moran NA. Origin of an alternative genetic code in the extremely small and GC-rich genome of a bacterial symbiont. PLoS Genet. 2009b;5:e1000565. doi: 10.1371/journal.pgen.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol. 2010;2:708–718. doi: 10.1093/gbe/evq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583–586. doi: 10.1016/s0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA, Hattori M. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Niimura Y, Miura K, Gojobori T. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc Natl Acad Sci U S A. 2010;107:6382–6387. doi: 10.1073/pnas.1002036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanamiya H, Sato M, Masuda K, Sato M, Wada T, Suzuki S, Natori Y, Katano M, Akanuma G, Kawamura F. Bacillus subtilis mutants harbouring a single copy of the rRNA operon exhibit severe defects in growth and sporulation. Microbiology. 2010;156:2944–2952. doi: 10.1099/mic.0.035295-0. [DOI] [PubMed] [Google Scholar]
- Nobusato A, Uchiyama I, Ohashi S, Kobayashi I. Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene. 2000;259:99–108. doi: 10.1016/s0378-1119(00)00456-x. [DOI] [PubMed] [Google Scholar]
- Pei AY, Oberdorf WE, Nossa CW, et al. (16 co-authors) Diversity of 16S rRNA genes within individual prokaryotic genomes. Appl Environ Microbiol. 2010;76:3886–3897. doi: 10.1128/AEM.02953-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O'Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762–766. doi: 10.1038/nature07527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Alm EJ, Arkin AP. Interruptions in gene expression drive highly expressed operons to the leading strand of DNA replication. Nucleic Acids Res. 2005;33:3224–3234. doi: 10.1093/nar/gki638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JA, Christner BC, Schuster SC. A bacterial ice-binding protein from the Vostok ice core. Extremophiles. 2008;12:713–717. doi: 10.1007/s00792-008-0178-2. [DOI] [PubMed] [Google Scholar]
- Rocha E. Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes? Trends Microbiol. 2002;10:393–395. doi: 10.1016/s0966-842x(02)02420-4. [DOI] [PubMed] [Google Scholar]
- Sabree ZL, Kambhampati S, Moran NA. Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc Natl Acad Sci U S A. 2009;106:19521–19526. doi: 10.1073/pnas.0907504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah P, Bisaglia M, Aliprandi P, Uzan M, Sizun C, Bontems F. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009;37:5578–5588. doi: 10.1093/nar/gkp547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorius-Neef S, Pfeifer F. In vivo studies on putative Shine-Dalgarno sequences of the halophilic archaeon Halobacterium salinarum. Mol Microbiol. 2004;51:579–588. doi: 10.1046/j.1365-2958.2003.03858.x. [DOI] [PubMed] [Google Scholar]
- Schurr T, Nadir E, Margalit H. Identification and characterization of E. coli ribosomal binding sites by free energy computation. Nucleic Acids Res. 1993;21:4019–4023. doi: 10.1093/nar/21.17.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta J, Agrawal RK, Frank J. Visualization of protein S1 within the 30S ribosomal subunit and its interaction with messenger RNA. Proc Natl Acad Sci U S A. 2001;98:11991–11996. doi: 10.1073/pnas.211266898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, et al. (12 co-authors) The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Shine J, Dalgarno L. The 3'-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A. 1974;71:1342–1346. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- Siezen RJ, Renckens B, van Swam I, Peters S, van Kranenburg R, Kleerebezem M, de Vos WM. Complete sequences of four plasmids of Lactococcus lactis subsp. cremoris SK11 reveal extensive adaptation to the dairy environment. Appl Environ Microbiol. 2005;71:8371–8382. doi: 10.1128/AEM.71.12.8371-8382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Tong Z, Wang J, et al. (29 co-authors) Complete genome sequence of Yersinia pestis strain 91001, an isolate avirulent to humans. DNA Res. 2004;11:179–197. doi: 10.1093/dnares/11.3.179. [DOI] [PubMed] [Google Scholar]
- Srivatsan A, Tehranchi A, MacAlpine DM, Wang JD. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010;6:e1000810. doi: 10.1371/journal.pgen.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer J, Stomp A, Vouk M, Bitzer D. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput Biol. 2006;2:e57. doi: 10.1371/journal.pcbi.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson BS, Schmidt TM. Life history implications of rRNA gene copy number in Escherichia coli. Appl Environ Microbiol. 2004;70:6670–6677. doi: 10.1128/AEM.70.11.6670-6677.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao ML, Moran NA, Abbot P, Brennan EB, Burckhardt DH, Baumann P. Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl Environ Microbiol. 2000;66:2898–2905. doi: 10.1128/aem.66.7.2898-2905.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft C, Andersson SG. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 2010;11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, et al. (15 co-authors) A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno T, Jang J, Han D, Kim JH, Sadowsky MJ, Kim OS, Chun J, Hur HG. Use of barcoded pyrosequencing and shared OTUs to determine sources of fecal bacteria in watersheds. Environ Sci Technol. 2010;44:7777–7782. doi: 10.1021/es101500z. [DOI] [PubMed] [Google Scholar]
- Vesper O, Amitai S, Belitsky M, Byrgazov K, Kaberdina AC, Engelberg-Kulka H, Moll I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell. 2011;147:147–157. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JD, Berkmen MB, Grossman AD. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:5608–5613. doi: 10.1073/pnas.0608999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap WH, Zhang Z, Wang Y. Distinct types of rRNA operons exist in the genome of the actinomycete Thermomonospora chromogena and evidence for horizontal transfer of an entire rRNA operon. J Bacteriol. 1999;181:5201–5209. doi: 10.1128/jb.181.17.5201-5209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.