

Abstract
Introduction
The mitogen-activated protein kinase (MAPK) pathway comprises several key signaling components and phosphorylation events that play important role in tumorigenesis. These activated kinases transmit extracellular signals that regulate cell growth, differentiation, proliferation, apoptosis and migration functions. Alteration of the RAS-RAF-MEK-ERK-MAPK (RAS-MAPK) pathway has frequently been reported in human cancer as a result of abnormal activation of receptor tyrosine kinases or gain-of-function mutations mainly in the RAS or RAF genes. Accordingly, these pathways are considered a potential therapeutic target for cancer treatment. Recently, several small-molecule inhibitors targeting this pathway have been developed and are currently being tested in clinical trials.
Areas covered
This paper focuses on the biological role of the RAS-MAPK pathway, the consequence of its disregulation, and the development of small-molecule inhibitors. The rationale for targeting the RAS-MAPK pathway will be reviewed here along with a discussion of the application and the results of various inhibitory molecules as anticancer agents in clinical trials.
Expert opinion
The RAS-MAPK pathway mediates cellular responses to growth signals and is often deregulated in human cancer. Activating mutations in the RAS and BRAF genes have been frequently identified in a wide range of cancers. Inhibitors of MEK and particularly of RAF kinases, have been effective in clinical trials with manageable side effects. RAS and BRAF genes need to be analyzed for mutations as markers of response to treatments and to avoid paradoxical effects. Further characterization of the RAS-MAPK molecular mechanisms regulation in malignant cells or underlying the acquired resistance to RAF inhibitors will facilitate development of novel combination therapies.
Keywords: mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK), MAP kinase kinase (MEK), RAS, RAF, inhibitors, targeted therapies
1.0 Introduction
The Mitogen-activated protein kinase (MAPK) pathway encompasses different signalling cascades of which the Ras-Raf-Mek-extracellular signal-regulated kinase 1 and 2 (ERK1/2) is one of the most dysregulated in human cancer. This pathway regulates multiple critical cellular functions including proliferation, growth and senescence (1). The Ras is an important component of the large family of GTPases. The ras genes are transforming oncogenes that have initially been recognized as murine sarcoma viruses by Jennifer Harvey (Harvey-Ras [H-RAS], and Werner Kirsten (Kirsten-Ras [K-Ras]) in 1960s (2, 3). The association of activated and transforming RAS genes in human cancer was concurrently reported by several authors in 1982 (4–6). Subsequent studies led to the identification of a third human RAS gene, designated NRAS in human neuroblastoma cells (Neuroblastoma-Ras [N-Ras]). The three human RAS genes encode four highly related 188 to 189 amino acid proteins, designated as H-RAS, N-RAS and K-RAS (K-RAS4A and K-RAS4B). Ras proteins function as binary molecular switches that control intracellular signaling pathways involved in fundamental cellular processes such as cell polarity, proliferation, differentiation, adhesion, migration, and apoptosis. Ras and ras-related proteins are often dysregulated in cancers by activating mutations of Ras isoforms or its effectors in nearly one-third of all human cancers (7). Ras activates several pathways, including the RAF-MEK-ERK/MAPK cascade, which transmits signals downstream and results in the transcription of genes involved in controlling several cellular mechanisms. The Ras family members are anchored to the cytoplasmic side of the plasma membrane by carboxyl-terminal farnesylation. This localization places the Ras in close proximity to adaptors, the growth factor receptor bound protein 2 (Grb2) and the nucleotide exchange factor son of sevenless (SOS), to mitigate the exchange of nucleotide guanosine diphosphate (GDP) bound to Ras with guanosine triphosphate (GTP) in the cytosol (8). This exchange activates Ras conformationally, allowing its interaction with a number of downstream effectors. Accordingly, Ras communicates external cellular signals to the nucleus, and its altered activation leads to inappropriate cellular activities including enhanced cell growth, differentiation and survival and ultimately to cancer (1,8,9). The RAS-RAF-MEK-ERK pathway is activated by several known growth factors and cytokines that act through receptor tyrosine kinase signals and by activating mutations mainly in the RAS and RAF genes (1–3).
Aberrations in the RAS-MAPK complex are implicated in several human cancers, render them an attractive therapeutic targets (10–13). Several drugs including humanized monoclonal antibodies (e.g. EGFR inhibitors), and small molecule inhibitors (i.e. RAS, RAF and MEK inhibitors) are currently being tested in clinical trials.
2.0 RAS-MAPK Pathway Functions
Ras (H-, K-, N-isotypes) (guanine nucleotide-binding protein), is a single GTPase molecule related in structure to the Gα subunit of heterotrimeric G proteins. G proteins act as molecular switches and timers that cycle from inactive GDP-bound to active GTP-bound states (14). In normal quiescent cells, Ras is bound to GDP and is inactive (“off” state), while upon extracellular stimuli, Ras bind to GTP (“on” state), which has an extra phosphate group than GDP. This extra phosphate holds the two switch regions in a “loaded-spring” configuration (switch I includes Threonine-35, switch II Glycine-60). Upon the release of this phosphate, the switch regions relax leading to conformational modifications and return to the inactivate state. Therefore, the activation and the inactivation of Ras and several other small G proteins are controlled by a cycling switching between the active GTP-bound and inactive GDP-bound forms (15).
The cyclic process of GDP/GTP is facilitated by guanine nucleotide exchange factors (GEFs) and the GTPase activating proteins (GAPs). The Ras intrinsic GTPase activity, hydrolyze the GTP into GDP. However, this process is inefficient and requires additional GAPs for binding, stabilizing, and accelerating the Ras catalytic activity. This is achieved by additional catalytic residues, “arginine fingers”, where a H2O molecule is positioned for nucleophilic attack on the gamma-phosphate of GTP, leading to the release of the inorganic phosphate molecule with a subsequent binding of Ras to GDP (15).
GEFs catalyze a “push and pull” process that unhinges the GDP from Ras by positioning close to the P-loop and the magnesium cation binding site to block the interaction with the gamma phosphate anion. Acidic (negative) residues in switch II “pull” a lysine in the P-loop away from the GDP which “pushes” switch I away from the guanine. The contacts holding GDP in place break, leading to its release in cytoplasm. Because intracellular GTP is abundant relative to GDP, it predominantly re-enters the nucleotide binding pocket of Ras and reloads the spring. Thus, the GEFs and GAPs balance underlie and facilitate Ras activation and inactivation, respectively (16).
The Ras binding domain is found in many effectors and invariably binds to one of the switch regions. Activated Ras-GTP has a high affinity for numerous downstream effectors and other small GTPases such as arfaptin or second messenger systems such as adenylyl cyclase as well.
Typically, ligand binding to receptor tyrosine kinases induces dimerization of the receptor and autophosphorylation of specific tyrosine residues in the C-terminal region. This generates binding sites for adaptor proteins e.g. growth factor receptor-bound protein 2 (GRB2), that recruit the GEF Sos at the plasma membrane, and in turn activates the membrane-bound Ras by catalyzing the GDP to GTP. In its GTP-bound conformation, Ras combines with Raf and mobilizes the inactive protein from the cytoplasm recruiting the Raf kinases (ARAF, BRAF and CRAF) to the plasma membrane (17, 18). Once the Ras-Raf complex is translocated to the cell membrane, Ras activates the serine/threonine kinase function of Raf isoforms. Upon activation of Ras, Raf acts as a MAP kinase kinase kinase (MAPKKK) to activate MEK1 and MEK2, which, in turn, catalyze the activation of the effector ERK1 and ERK2 kinases, and their translocation into the nucleus. Once activated, ERK1/ERK2 broadly phosphorylate several nuclear and cytoplasmic effector genes involved in diverse cellular responses such as cell proliferation, survival, differentiation, motility, and angiogenesis (19–21). Although RAF can also be activated by RAS-independent activators, (22) considerable experimental evidence indicates that the RAF-MEK-ERK cascade is a major mediator of Ras-induced oncogenesis. Recent data have clearly shown that Ras can activate other downstream signaling pathways including phosphatidylinositol 3-kinase (PI3K) and Rac and Rho proteins, associated with the regulation of the cytoskeleton and invasiveness of tumor cells. Through RAS, other signals may be activated such as p38 MAPK, and the stress-activated protein kinase pathway, c-Jun N-terminal [JNK] pathway (Figure 1) (23–25).
Figure 1.
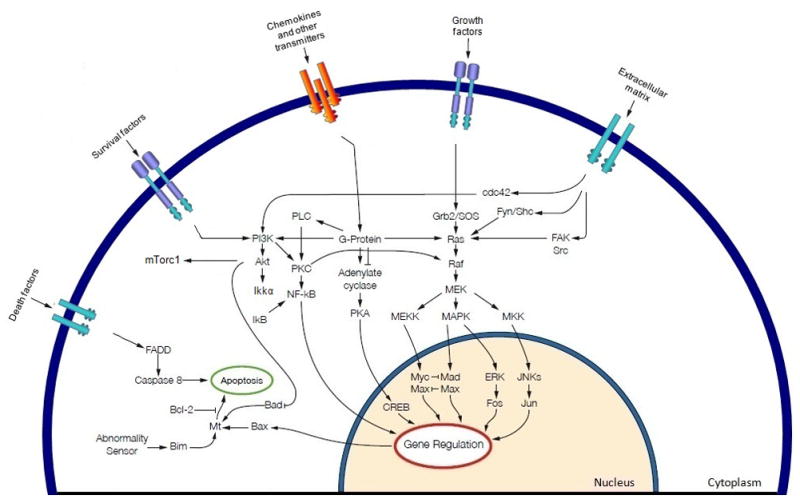
Schematic representation of the MAPK cascade activation and potential cross talk signals. Growth factors binding, and causing activation of, tyrosine kinase receptors, activating mutations of BRAF and RAS, and additional external cell membrane stimuli may cause persistent activation of the MAPK cascade in human cancers. Numerous effectors signals converge on RAF, activated RAF phosphorylates MEK in the cytoplasm, which in turn phosphorylates ERKs that translocates to the nucleus where they phosphorylate and regulate various nuclear and cytoplasmic substrates involved in diverse cellular responses, such as cell proliferation, survival, differentiation, motility, and angiogenesis. RAS may cross-talk with different signalling pathways, e.g. PI3K, to enhance tumorigenesis in cancer cells.
3.0 Deregulation of RAS-MAPK Pathway in Human Cancer
3.1) Ras activation
Activating point mutations of the RAS family genes (H-RAS, K-RAS, and N-RAS) are not uncommon and comprise up to 30% of all human cancers (7). A summary of the frequencies of RAS mutations in tumours of various sites is shown in Table 1. It has been reported that 90% of pancreatic adenocarcinomas harbour a RAS mutation and the majority of these mutations involve the codon 12 of the K-RAS gene in the majority of cases. These findings link mutation in the K-RAS proto-oncogene to the development of pancreatic cancer. K-RAS mutations are also a frequent event in colorectal, lung, and biliary tract carcinogenesis, while mutations at the H-RAS and N-RAS gene have rarely been detected. Conversely, melanomas present a quite high percentage of N-RAS mutations while salivary gland tumours present a majority of H-RAS mutations (Table 1). Of note, thyroid tumours based on their histotype origins may present different types of RAS alterations with K-RAS mutations mainly associated with papillary thyroid carcinoma (PTC) and N-RAS with follicular thyroid carcinomas, follicular variant PTCs, and poorly differentiated thyroid carcinomas (26–28); whereas medullary thyroid carcinomas present rare mutations of RAS genes (29). Ras has also been shown to be a key downstream effector in a number of activated receptor tyrosine kinases, including the epidermal growth factor receptor (EGFR)(30). Moreover, ERK activation can induce over-expression of EGFR ligands, promoting an autocrine growth loop critical for tumor growth (1). It is, therefore, conceivable to target these genes for new therapeutic strategies in cancer bearing these alterations.
Table 1.
Frequency of RAS (K-RAS, N-RAS, H-RAS) mutations in different types of tumors
| Tumors | Frequency of K-RAS mutations | Total samples analyzed |
|---|---|---|
| pancreas | 57% | 5169 |
| large intestine | 33% | 30793 |
| biliary tract | 31% | 1555 |
| small intestine | 20% | 316 |
| lung | 17% | 15080 |
| endometrium | 14% | 2143 |
| ovary | 14% | 2956 |
| gastrointestinal tract (site indeterminate) | 8% | 49 |
| cervix | 7% | 637 |
| prostate | 7% | 1126 |
| soft tissue | 7% | 1070 |
| peritoneum | 6% | 86 |
| stomach | 6% | 2571 |
| haematopoietic and lymphoid tissue | 5% | 5762 |
| liver | 5% | 450 |
| urinary tract | 5% | 868 |
| breast | 4% | 544 |
| eye | 4% | 90 |
| genital tract | 4% | 25 |
| oesophagus | 4% | 359 |
| penis | 4% | 28 |
| testis | 4% | 432 |
| autonomic ganglia | 3% | 63 |
| salivary gland | 3% | 170 |
| skin | 3% | 1422 |
| upper aerodigestive tract | 3% | 1554 |
| thymus | 2% | 186 |
| thyroid | 2% | 4710 |
| bone | 1% | 231 |
| central nervous system | 1% | 1032 |
| kidney | 1% | 617 |
| Tumors | Frequency of N-RAS mutations | Total samples analyzed |
|---|---|---|
| skin | 18% | 4759 |
| NS | 17% | 282 |
| haematopoietic and lymphoid tissue | 10% | 8548 |
| thyroid | 7% | 4206 |
| autonomic ganglia | 6% | 102 |
| adrenal gland | 5% | 170 |
| ovary | 4% | 133 |
| soft tissue | 4% | 481 |
| liver | 3% | 310 |
| testis | 3% | 283 |
| upper aerodigestive tract | 3% | 807 |
| breast | 2% | 330 |
| cervix | 2% | 132 |
| large intestine | 2% | 1056 |
| pancreas | 2% | 248 |
| prostate | 2% | 530 |
| stomach | 2% | 215 |
| urinary tract | 2% | 655 |
| biliary tract | 1% | 213 |
| central nervous system | 1% | 995 |
| endometrium | 1% | 314 |
| eye | 1% | 103 |
| lung | 1% | 2851 |
| Tumors | Frequency of H-RAS mutations | Total samples analyzed |
|---|---|---|
| salivary gland | 15% | 161 |
| urinary tract | 10% | 1499 |
| cervix | 9% | 264 |
| upper aerodigestive tract | 9% | 1083 |
| penis | 7% | 28 |
| prostate | 6% | 500 |
| skin | 6% | 2100 |
| soft tissue | 5% | 712 |
| stomach | 4% | 384 |
| testis | 4% | 130 |
| thyroid | 4% | 3681 |
| pituitary | 3% | 300 |
| bone | 2% | 199 |
| thymus | 2% | 46 |
| adrenal gland | 1% | 135 |
| breast | 1% | 542 |
| endometrium | 1% | 291 |
| oesophagus | 1% | 161 |
In bold all somatic mutations that are higher than 10% of frequency. (Sanger mutation database: http://www.sanger.ac.uk/genetics/CGP/cosmic/).
The most prevalent mutations in Ras genes occur at residue G12 in the P-loop and the catalytic residue Q61, which are commonly in tumors associated with Ras mutations (Table 1). This mutation leads to clycin to valine substitution at residue 12 that renders the GTPase domain of Ras insensitive to inactivation by GAP and autonomous activation (“on state”). However, residue 61 is responsible for stabilizing the transition state for GTP hydrolysis and because enzyme catalysis in general is achieved by lowering the energy barrier between substrate and product, this mutation reduces the rate of intrinsic Ras GTP hydrolysis to physiologically low levels (31, 32).
3.2) Raf activation
The RAF family members, (A-Raf, B-Raf, and C-Raf or Raf-1), are highly conserved serine/threonine kinases of the MAPK [RAS-RAF-MEK-ERK] pathway (Figure 2). The RAF proteins activate the MAPK pathway (33) where inappropriate and/or persistent activation leads to abnormal differentiation, proliferation and apoptosis, and cancer development. BRAF is frequently mutated in a variety of human tumors, especially in malignant melanoma, thyroid and colon carcinomas (34) (Table 2). The incidence of activating RAF gene mutations in cancer is presented in Table 2. Among different BRAF mutations, a single-base missense substitution (T to A at nucleotide 1,799) that substitute valine for glutamic acid at codon 600 (V600E) of the kinase domain (exon 15) is the most prevalent in melanomas and papillary thyroid carcinomas (35, 36). The mutant V600E BRAF protein results in impaired kinase activity, which induces hyperactivity of the MAPK pathway in a RAF1(CRAF)-dependent manner promoting the development of these tumors (37). Activation results from conformational change in the protein structure caused by glutamic acid acting as a phosphomimetic between the Thr598 and Ser601 phosphorylation sites that disrupts the interaction between the P-loop and the activation segment of the BRAF protein (38). In vitro and/or in vivo studies have demonstrated the oncogenic potential for BRAF V600E mutation in different settings.
Figure 2.

Schema of the domain structure of human A-Raf, B-Raf, and C-Raf. The three mammalian RAF serine/threonine protein kinases, ranging from 70 to 100 kDa in size, mediate the transduction of proliferative and differentiative signals from cell surface receptors to the nucleus catalyzing the phosphorylation of hydroxyl groups on specific Ser and Thr residues. A-Raf gene (Location: Xp11.4-p11.2). This isoform is the weakest activator of MEK, and can only activate MEK1 but not MEK2. B-Raf gene (Location: 7q34); BRAF point mutations within the kinase domain of the protein occur in several tumor types including, melanomas, papillary thyroid and colorectal carcinomas. C-Raf gene (Location: 3p25); CRAF is the cellular homolog of viral raf gene (v-raf). The encoded protein is a MAP kinase kinase kinase (MAP3K), which functions downstream of the Ras family of membrane associated GTPases to which it binds directly. Among 3 RAF isoforms, BRAF displays the highest basal kinase activity.
S, serine; T, threonine; Y, tyrosine: amino acid phosphorylation sites regulating the Raf kinases. Abbreviations: C, carboxyl terminus; N, amino terminus; RBD, Ras-binding domain; CRD, cysteine-rich domain; KSR, kinase suppressor of Ras; MEK, mitogen-activated protein kinase kinase; PAK, p21-activated kinase; PKA, protein kinase A; PKCα, protein kinase C alpha; SGK, steroid glucocorticoid kinase; Src, soluble nonreceptor tyrosine kinase.
Table 2.
Frequency of RAF (ARAF, BRAF, CRAF) mutations in different types of tumors
| Tumors | Frequency of ARAF mutations | Total samples analyzed |
|---|---|---|
| ovary | 4% | 27 |
| large intestine | 2% | 157 |
| Tumors | Frequency of BRAF mutations | Total samples analyzed |
|---|---|---|
| NS | 57% | 547 |
| skin^ | 39% | 7731 |
| thyroid | 38% | 19675 |
| genital tract | 12% | 33 |
| large intestine | 12% | 36546 |
| biliary tract | 11% | 242 |
| eye | 11% | 493 |
| ovary | 11% | 1898 |
| gastrointestinal tract | 6% | 31 |
| meninges | 5% | 19 |
| small intestine | 5% | 101 |
| central nervous system | 4% | 1312 |
| endometrium | 4% | 825 |
| prostate | 4% | 1130 |
| bone | 3% | 147 |
| breast | 3% | 424 |
| testis | 3% | 235 |
| adrenal gland | 2% | 154 |
| haematopoietic and lymphoid tissue | 2% | 1235 |
| lung | 2% | 3539 |
| oesophagus | 2% | 170 |
| pancreas | 2% | 503 |
| soft tissue | 2% | 1172 |
| upper aerodigestive tract | 2% | 723 |
| autonomic ganglia | 1% | 149 |
| cervix | 1% | 451 |
| liver | 1% | 145 |
| pituitary | 1% | 115 |
| salivary gland | 1% | 90 |
| stomach | 1% | 955 |
| urinary tract | 1% | 345 |
NS, nervous system;
BRAF V600E mutation has been demonstrated in a higher percentage of melanomas.
This percentage of mutations deserves further investigations; only 1 case of this type of cancer has been analyzed and demonstrated to be positive for CRAF mutations. In bold all somatic mutations that are superior to 10% of frequency. A high percentage of BRAF V600E mutations has recently been reported at American Society of Clinical Oncology 2011 in 47 consecutive patients with hairy cell leukemia (also published by Tiacci E. et al. on NEJM 2011 “BRAF Mutations in Hairy-Cell Leukemia”).(Sanger mutation database: http://www.sanger.ac.uk/genetics/CGP/cosmic/).
Marked activation of the MAPK pathway can also suppress cellular growth in a wide variety of normal and cancer cells by inducing cellular senescence (39, 40). This mechanism of senescence is usually modulated by cyclin dependent kinase inhibitors (e.g. p27Kip1), and it is adopted by normal cells to overcome oncogene activation (40, 41). In contrast, moderate levels of MAPK pathway activation could induce abnormal cellular functions (42). These different effects were found to cause transformation and immortalization of mouse melanocytes, increased in vitro colony formation, and elevated Erk1/2 activities (43). It has also been shown that BRAF V600E activating mutation initially promotes nevi development but further tumor progression is inhibited by marked activation of the MAPK. In this setting, subsequent genetic abnormalities such as loss of p16INK4a, or elevation in Akt3 activity, are required for the senescent melanocytic cells to reenter the cell cycle. Recent studies show that Akt3 and mutant V600E BRAF cooperate to promote early melanoma development; specifically Akt3 is shown to phosphorylate V600E B-Raf to lower its activity as well as that of the downstream MAP kinase pathway promoting cell proliferation (44).
BRAF codon 600 (V600E) mutations also play important roles in thyroid tumorigenesis although additional signals may be needed for thyroid follicular cells to acquire full metastatic capabilities (36, 45). This mutation has been reported in ~45% of papillary thyroid carcinomas (PTC) and in ~25% of undifferentiated thyroid cancers (35, 36, 46). Of interest, diverse types of BRAF mutations have been reported in the follicular variant of PTC (28). Recently, high BRAF mutations were reported in pleomorphic xanthoastrocytomas and gangliogliomas (47), colon carcinomas and, in other tumor types (Table 2). The frequency of mutational changes of ARAF and CRAF in human cancers is low (Table 2) except for a truncated form of CRAF (11).
4.0 Anticancer agents targeting the RAS-MAPK pathway
Strategic focus on the development of novel biologically-based treatment has gained remarkable momentum. Target-based therapies are widely considered to be the future of cancer treatment and much attention has been focused on developing inhibitors of the RAS-RAF-MEK-ERK/MAPK signaling pathway and its upstream activators. In this context, several MEK1/2 and RAF inhibitors have been tested clinically or are currently undergoing clinical trial evaluation (Table 3 and 4). RAS inhibition did not achieve the expected results in clinical trials probably due to the fact that these inhibitors are not able to hit specific target proteins. Inhibition of RAS remains an interesting target although challenging.
Table 3.
MEK inhibitors
| Compound | Tumor | Primary Target(s) | Potential side effects |
|---|---|---|---|
| CI-1040 (Pfizer Inc.) | Breast cancer, Lung cancer, Pancreatic cancer, Colorectal cancer, Breast Neoplasm, Non small cell lung cancer, Pancreatic Cancers | MEK | Diarrhea, Asthenia, Rush, Nausea, Vomiting |
| PD-0325901 (Pfizer Inc.) | Carcinoma, Non small cell lung cancer, Melanoma, Breast cancer | MEK | Diarrhea, Nausea, Fatigue, Rash, Reversible visual disturbances, Vomiting |
| ARRY-438162 (Array BioPharma) | Metastatic Biliary Cancer, and Metastatic Colorectal Cancer | MEK | Recruiting Phase |
| AZD6244 (AstraZeneca) | Breast cancer, Colon cancer, Lung cancer, Kidney cancer, Metastatic colorectal cancer, Advanced/metastatic Melanoma, Pancreatic cancer, Non small cell lung cancer, Hepatocellular carcinoma | MEK | Papulopustular rash, Xerosis, Fissures, Pruritus, Telangiectasias, Hyperpigmentation, Dermatitis, Alopecia, Angular chelitis, Paronychea, Fatigue |
| RDEA119/BAY 86-9766 (Bayer/Ardea Biosciences) | Advanced cancer, cancer, Thyroid cancer Pancreatic | MEK | Low toxicity, further studies are needed |
| GSK1120212 (GlaxoSmithKline) | Solid tumors, Melanoma, Non small cell lung cancer, Pancreatic cancer | MEK, C-Raf, B-Raf, V600E, BRAF wt | Fatigue, Diarrhea |
| TAK-733 (Millennium Pharmaceuticals) | Advanced Non-hematologic malignancies, Metastatic melanoma | MEK | Recruiting |
| GDC-0973/XL581 (Genentech/Exelixis) | Metastatic Melanoma, Solid cancers | MEK | High toxicity, Diarrhea, Fatigue, Rash, Nausea, Vomiting |
| AZD8330/ARRY-424704 (AstraZeneca) | Advanced malignancies | MEK | Ongoing trial |
| RO5126766 (Hoffmann-La Roche) | Metastatic or advanced solid tumors | MEK | Recruiting |
| RO4987655 (Hoffmann-La Roche) | Advanced and/or metastatic solid tumors | MEK | Recruiting |
| RO5068760 (Hoffmann-La Roche) | Melanoma, Colorectal cancer | MEK | Preclinical Phase Studies |
| AS703026 (EMD Serono) | Acute Myeloid Leukemia, and Hematological Malignancies | MEK | Recruiting |
Table 4.
RAF inhibitors
| Compound (Pharmaceutical Company) | Tumors | Primary Target(s) | Potential side effects |
|---|---|---|---|
| Bay 43-9006* Sorafenib (Bayer) | Squamous cell carcinoma, Non small cell lung, Leukemia, Hepatocellular carcinoma, Kidney cancer, Pancreatic cancer, Bladder cancer, Lung cancer, Urothelial cancer, Neuroendocrine tumors, Thyroid cancer, Renal cell carcinoma | C-Raf B-Raf B-Raf V600E |
Fatigue, Anorexia, Diarrhoea, Nausea, Skin reaction, Alopecia, Rash, Stomatitis, Elevated bilirubin, Pancreatitis |
| LErafAON (NeoPharm, Inc) | Different advanced malignancies | C-Raf | Back Pain, Chills Fatigue, Hypertension, Fever, Flushing, Dyspnea |
| ISIS 5132 (Isis Pharmaceuticals) | Ovarian cancer, Breast cancer | C-Raf | Minimal side effects |
| PLX4720 (Plexxikon/Roche) | Melanoma | B-Raf V600E | Minimal side effects, further studies are needed |
| PLX4032 (Plexxikon/Roche) | Malignant melanoma, Colorectal carcinoma | B-Raf B-Raf V600E |
Cutaneous side effects, Arthralgia, Fatigue |
| Raf-265 (Novartis Oncology) | Metastatic Melanoma | Raf | Recruiting phase |
| XL281 (Exelixis) | Non small cell lung cancer, Colorectal cancer, Melanoma, Papillary thyroid cancer | Raf | Recruiting phase |
| GDC-0879 (Genentech) | Melanoma | Raf | Preclinical Phase Studies |
| GSK2118436 (GlaxoSmithKline) | Melanoma | B-Raf V600E | Nausea, Fatigue, Fever, Rash, SCC, Headache |
WT, wild type,
sorafenib is a multityrosine kinase inhibitors, which inhibits additional targets such as, VEGFR2, drugs target additional markers. SCC, squamous cell carcinoma. Another novel multikinase inhibitor is BAY 73-4506 (Regorafenib), which potently inhibits endothelial cell kinases in biochemical and cellular kinase phosphorylation assays. Furthermore, regorafenib inhibits additional angiogenic kinases such as VEGFR1/3, platelet-derived growth factor receptor-β and FGFR 1 and the mutant oncogenic kinases KIT, RET and BRAF. This compound exhibited potent dose-dependent tumor growth inhibition in various preclinical human xenograft models in mice, with tumor shrinkages observed in breast MDA-MB-231 and renal carcinoma models.
4.1) MEK inhibitors
Blocking MAPK via small-molecule inhibitors has become a biologically viable model for targeted cancer therapeutics. PD 098059, the first MEK inhibitor described, was identified through screening a compound library of inhibitors by assessing the phosphorylation of an ERK target protein in the presence of both MEK1 and ERK. The compound inhibited MEK with an IC50 ~10 nmol/L but had no inhibitory effects when tested against a panel of other serine/threonine kinases (48). Another second MEK inhibitor, U0126, was also identified by screening a library of molecules using an assay designed to identify antagonist for activator protein-1–driven (c-Fos and c-Jun) transcription without blocking the glucocorticoid response elements (49). U0126 inhibited MEK1 and MEK2 with IC50 ~5 to 7 nmol/L but had little effect on other kinases and the inhibition was noncompetitive with respect to the MEK substrates ATP and ERK (49). Both PD 09859 and U0126 have shown in vitro antiproliferative effects on transformed cell lines. Although U0126 and PD 098059 have been extremely useful in animal studies of MAPK signaling, they have not been targeted for clinical development because of poor pharmacologic properties.
CI-1040 (PD 184352), a potent MEK inhibitor (IC50 of 17 nM on purified MEK1) that progressed to clinical testing, was identified during the screening of the PD 098059 (50). CI -1040 selectively inhibited MEK1 in a noncompetitive manner with respect to ATP. The drug also inhibited MEK in cell-based assays and human colon cancer xenograft cell growth in a noncompetitive mechanism, suggesting an allosteric inhibitory effect (51). CI-1040 binds to specific hydrophobic pocket of the MEK1 and MEK2 that has low homology for other kinases, supporting the high degree of specificity. A phase I study of orally administered CI-1040 in 77 patients with advanced cancers showed that this drug was well tolerated at doses resulting in a median 73% inhibition of phospho-ERK1/2 expression in tumor biopsies. Approximately 60% of patients experienced adverse effects, mostly grade 1 or 2, with no patient having drug-related grade 4 events. The most common toxicities included diarrhea, asthenia, rash, nausea, and vomiting. Interestingly, a single patient with pancreatic cancer achieved a partial response with significant symptom improvement that lasted 12 months. Moreover, 19 patients with different cancers had stabilization of the disease lasting from 4 to 17 months (50). A phase II study was initiated in patients with advanced pancreatic, breast, colon and non-small cell lung cancers (52). Unfortunately, none of the patients achieved a complete, partial response, and only 8 patients achieved stabilization of disease (median of 4.4 months). The poor antitumor activity, low bioavailability and solubility of this drug precluded further clinical studies.
Two additional MEK inhibitors have been developed and have advanced to clinical testing. PD 0325901 is a second generation analogue of CI-1040 with an IC50 of 1 nmol/L that is more potent and soluble than CI-1040 in vivo, with a single oral dose providing >50% inhibition at 24 h. The anticancer activity of this drug has also been demonstrated in a variety of human tumor xenografts bearing BRAF mutations (53). The clinical activity of PD 0325901 was first evaluated in a phase I–II dose-escalating study of 35 patients with advanced solid tumors with doses ≥ 2 mg BID that efficiently suppressed ERK1/2 phosphorylation (average of 84%) and Ki67 expression in tumor biopsies and resulted in two partial responses and eight stable disease that lasted 3–7 months of 27 patients. An extension of a phase I study documented, 3 partial responses and 24 stable diseases (22 melanoma and 2 non-small cell lung cancer) in 66 patients (54). However, PD 0325901 was associated with more toxicity than CI-1040, including acute neurotoxicity and blurred vision and retinal vein occlusion in patients receiving more than 15 mg BID leading to the termination of the clinical trial.
AZD6244 (ARRY-142886, Selumetinib) is an oral potent second-generation MEK1/2 inhibitor with similar structure to the CI-1040, but with improved pharmacologic properties (55). Similar to other MEK inhibitors, AZD6244 is ATP non-competitive with no significant inhibitory effects when tested against a broad range of serine/threonine kinases. Preclinical evaluation has shown anticancer activity in a variety of human tumor xenograft models with remarkable target inhibition (55–58). AZD6244 antitumor activity was found to correlate with suppression of ERK activation, in a phase I clinical trial, the pharmacokinetics and pharmacodynamics of AZD6244 in 57 patients with advanced cancer showed that the 50% maximal effective dose (100 mg BID) was well tolerated with cutaneous rash being the most frequent and dose-limiting toxicity. Most other adverse events were of grade 1 or 2. In this trial seven patients developed transient and reversible blurred vision, nine patients showed disease stabilization lasting for at least 5 months and a strong reduction in ERK1/2 phosphorylation (mean inhibition of 79%) in tumor biopsies (59). Preliminary results from four randomized phase II clinical trials of AZD6244 have recently been reported. A first trial compared AZD6244 to the alkylating agent temozolomide in advanced melanoma patients. AZD6244 showed a potent antitumor activity, but there was no significant difference in progression-free survival between the two treatment arms (60). In a second study, the efficacy of AZD6244 was compared with the antimetabolite pemetrexed as second- or third-line treatment of patients with non-small cell lung cancer. The study showed evidence for single agent antitumor activity, but failed to demonstrate a difference in primary disease progression endpoint (61). In a third study, AZD6244 versus capecitabine in patients with metastatic colorectal cancer, who had failed prior irinotecan and/or oxaliplatin treatments, showed no difference between the two treatments in the number of patients with disease progression (62). Another Phase II study of Selumetinib in 28 patients (thirty-nine percent of whom had received one prior systemic therapy) with metastatic biliary cancers, had a confirmed objective response in three patients (12%). Another 17 patients (68%), experienced stable disease (SD), 14 of whom (56%) experienced prolonged SD (> 16 weeks) (63). Finally, the results of a phase II study of AZD6244 in patients with advanced or metastatic hepatocellular carcinoma, showed lack of radiographic improvement that led to the termination of the trial (64).
Based on their high potency, oral bioavailability, and preclinical evidence of anticancer activity, PD 0325901 and AZD6244 were the first candidates MEK inhibitors for treatment of human cancers.
GDC-0973/XL518 (Exelixis/Genentech) is an oral active inhibitor of MEK1/2 (IC50 of <1 nM). Cell-based studies using this compound demonstrated an inhibition of ERK1/2 phosphorylation at subnanomolar concentrations, and antiproliferative effects in multiple tumor cell lines harboring K-RAS or BRAF mutations. Animal model studies have shown that a single dose of GDC-0973 inhibits phosphorylation of ERK1/2 in tumors for up to 48 hours strongly inhibiting tumor growth in human xenograft models (65). Remarkably, GDC-0973 has minimal side effects on the central nervous system. A phase I dose-escalating study of GDC-0973 was initiated in patients with solid tumors. Preliminary results of 13 patients’ trial indicate that GDC-0973 is well tolerated with no drug-related severe adverse events being reported (66). A patient with non-small cell lung cancer had stabilization of disease for 7 months. Another phase I trial of GDC-0973 in combination with the PI3K inhibitor GDC-0941 of 30 patients showed decreases in RECIST measurable target lesions in 5 patients, 2 melanoma, 1 prostate cancer, 2 NSCLC and three patients had prolonged stable disease (> 6 months) (67). Another phase Ib, dose-escalation study of GDC-0973 in combination with PLX4032/RO5185426 in patients with BRAFV600E-positive metastatic melanoma, is ongoing.
GSK1120212 (GlaxoSmithKline) is an orally bioavailable, allosteric and selective inhibitor of MEK1/2 enzymes. Cell-based assays and xenografts mouse models showed an important antitumor activity of this drug (68, 69). A phase I study of GSK1120212 patients with solid tumors and lymphoma and the preliminary evaluation of 6 patients treated at four dose levels, was well tolerated with no dose-limiting toxicity reported (70). Two other phase I/II studies of GSK1120212 have started to enroll patients with relapsed or refractory leukemias, and in combination with everolimus in patients with solid tumors. Randomized Phase III study comparing single agent GSK1120212 to chemotherapy (either dacarbazine or paclitaxel) in subjects with stage IIIc or stage IV cutaneous melanoma, is being conducted on patients with BRAF mutation-positive tumor.
RDEA119 (BAY 869766, Ardea Biosciences/Bayer) is an oral bioavailable, allosteric in vitro inhibitor of MEK1 (IC50 of 19 nM) and MEK2 (IC50 of 47 nM) in a non-ATP competitive manner. In vitro assays showed that RDEA119 inhibits ERK1/2 phosphorylation and cell proliferation (IC50 from 2.5 to 16 nM) in human cancer cell lines (71). In vivo, RDEA119 exhibits potent antitumor activity in xenograft models of melanoma and colon carcinoma. Interestingly, this compound has low central nervous system penetration and it is currently being evaluated as single agent in a phase I study in advanced cancer patients and in a phase I/II study in combination with the multikinase Raf inhibitor sorafenib.
4.1.1 Other MEK1/2 Inhibitors
Other MEK1/2 inhibitors are currently being evaluated in phase I clinical trials of advanced cancer patients. These are AZD8330 (Array BioPharma/AstraZeneca), RO5126766 and RO4987655 (Hoffmann La Roche), TAK-733 (Millenium Pharmaceuticals) and AS703026 (EMD Serono), XL518 (Genentech/Exelixis). Other novel MEK1/2 inhibitors such as RO4927350, RO5068760 and PD318088 have recently been reported on preclinical models (72).
4.2) RAF inhibitors
Efforts to develop antisense inhibitors of Raf expression and activity are being made (Table 4). ISIS-5132, a 20base phosphorothioate DNA oligonucleotide, blocks mainly the c-Raf-1 protein expression and showed antitumor activity in preclinical xenograph models and early clinical trials. Further trials, however, have been halted due to the lack of clinical responses (73). A similar approach, using a liposome encapsulated antisense c-raf-1 oligonucleotide LErafAON, showed antitumor activity in xenograft models (74) but in phase I clinical trials as a single agent and in combination with radiation or chemotherapy resulted in modest results (75, 76).
To date, the most successful anti-Raf inhibitor has been Sorafenib (tosylate salt of BAY 43-9006; Nexavar), an orally administered compound used for treatment of advanced renal cell carcinoma (RCC). Sorafenib is a bi-aryl urea compound that was originally developed as an inhibitor of Raf-1 (77). Subsequent studies revealed that sorafenib is also a potent inhibitor of both wild-type and mutant B-Raf V600E kinases in vitro, and has a potent activity for other protein kinases, such as, the proangiogenic tyrosine kinase receptors and VEGFR-2, VEGFR-3, FGFR-1, PDGFR-b, Flt-3, and c-Kit (77). Sorafenib may also inhibit angiogenesis-related kinases as well as other non-Raf kinases as evidenced through antitumor activity independent of RAS or BRAF mutation status in different studies. However, trials of sorafenib have not provided evidence to conclude that inhibition of Raf has clinical value. Furthermore, although preclinical data showed that continued expression of mutant Braf is critical for melanoma growth and tumorigenicity, phase II clinical trials with sorafenib (400 mg BID) showed only modest or no antitumor activity (78). Phase II trial of sorafenib and chemotherapy showed early response in melanoma irrespective of the BRAF mutational status. Moreover, a phase III trial of sorafenib in combination with carboplatin and paclitaxel failed to achieve improvement in overall survival (79). These results may lend credence to the concept that the primary mechanism of activity of sorafenib in solid cancer is likely anti-angiogenic and not RAF related. It’s worth noting, however, that phase II/III clinical trials of sorafenib in patients with advanced hepatocellular carcinoma have shown promising results (80). Clinical trials using sorafenib in combination with other agents for the treatments of advanced or metastatic breast and thyroid cancers are ongoing (81, 82).
The limited activity of sorafenib in tumors with BRAF mutation prompted the development of second-generation RAF inhibitors with greater selectivity for mutant BRAF. PLX4032/R7204 (Vemurafenib) and its analog PLX4720 have shown potent antiproliferative effects in several preclinical models specifically in cell lines harboring BRAF V600E mutations (83). In a recent phase 1 clinical trial of PLX4032, high (30 to 50 μM) steady state serum levels of the drug were tolerated with modest toxicity and intense inhibition of ERK signaling in tumors. The antitumor activity observed was 78% response rate by RECIST and tumor shrinkage in almost all patients (84). Notable toxicities included the development of squamous cell carcinomas and skin rash in approximately one third of patients. The average duration of response in phase I trial was approximately 9 months. A recent randomized phase III clinical trial comparing PLX4032 to dacarbazine as first-line therapy in patients with previously untreated BRAF V600E mutation positive melanoma patients showed that PLX4032 improved overall and progression-free survival in comparison to dacarbazine group. Common adverse events associated with PLX4032 were arthralgia, rash, fatigue, alopecia, keratoacanthoma or squamous-cell carcinoma, photosensitivity, nausea, and diarrhea (85). Despite the important impact made by these inhibitors for the treatment of melanoma patients bearing BRAF V600E mutations, early studies of these agents in patients with BRAF V600E colorectal carcinoma have thus far been disappointing (86). This data could be explained by the activation of different and/or redundant activating cellular mechanisms of the MAPK pathway in colorectal cancers.
It has been reported that PLX4032 inhibited ERK activation only in cancer cells with BRAF mutations, while in cells with wild-type BRAF, it induced ERK phosphorylation and activation (87). The underlying mechanism for this paradoxical effect is likely related to the allosteric effect of the drug, which enforces the dimerization of endogenous BRAF with CRAF or ARAF (88–90). Within these dimers, only one active component is required for activation of MEK–ERK pathway. At non-saturating concentrations, the inhibitors activate rather than inhibit the pathway, particularly in the presence of RAS mutations or activated upstream receptor kinases (by Ras activation). It is also possible that the rapid development of skin tumors in patients treated with RAF inhibitors might be enhanced by such mechanism (91, 92). The paradoxical activation of ERK by PLX4032 in wild type BRAF can also be explained by the low RAS activity, and that dimer formation is not required for the activation. It’s worth noting that such paradoxical activation of ERK signaling is not unique to PLX4032 since it is also observed with other ATP-competitive inhibitors of RAF and sorafenib. These results suggest that the clinical activity of PLX4032 will be restricted to tumors harboring activating mutations of BRAF.
GSK2118436 (another ATP competitive BRAF inhibitor), has recently shown a dramatic effect, with response rates of 70–80% as single agent in metastatic melanoma patients (84, 93, 94). A phase I/II clinical trial of patients with metastatic brain melanoma showed that ninety percent of the patients had reductions in size of metastases. The overall reductions ranged from 20 to 100% of brain metastases that were 3mm or larger in diameter before treatment. These drugs are currently being tested in other clinical trials in patients with other solid tumors with BRAF V600E mutations, such as thyroid carcinomas or colon cancers (95, 96). Potential risk during treatment of thyroid cancer patients with inhibitors that target mutants BRAF is the multifoci nature of PTCs and the heterogeneity of the BRAF V600E mutation in these lesions. Additional small molecule inhibitors of RAF are also in their early clinical testing. XL281 (BMS-908662) has shown modest biological activity and modulation of MAPK pathway activity in tumor tissue: clinical benefit (partial response or stable disease) was observed in 43% (13 of 30) of patients in the dose-escalation phase of a recent phase 1 study (97).
4.2.1 Other RAF Inhibitors
RAF265 is an oral highly selective inhibitor of RAF, including B-RAF and C-RAF and mutant B-RAFs. RAF265 has also an important anti-angiogenic activity through inhibition of vascular endothelial growth factor receptor 2 (VEGFR-2). Interestingly, growth inhibitory effects have been demonstrated in combination of RAF265 and mammalian target of rapamycin inhibitor RAD001 (everolimus) (98). In a BRAF mutant xenograft mouse model, RAF-265 induced dose dependent tumor regression and is currently being tested in Phase I clinical trials in patients with advanced malignant melanoma.
AZ628 is a selective RAF inhibitor of CRAF (Raf-1) as well as BRAF wild type and the V600E BRAF. Cytotoxic activity in many tumor cell lines, especially those with V600E BRAF mutations have been achieved using this agent. It has been demonstrated that CRAF overexpression is associated with both primary and acquired resistance to BRAF inhibition by AZ628 in in vitro melanoma model. Interestingly, AZ628 resistant melanoma cells are sensitive to HSP90 inhibitors, which promote CRAF degradation (99), suggesting that both drugs may overcome resistance in a subset of patients with CRAF overexpression.
SB-590885 is another selective smallmolecule inhibitor of the B-Raf kinase. It has been usedto overcome the dependence of MAPK signaling and tumor cellgrowth on B-Raf kinase activity. In melanoma and colorectal cancer cell lines with B-Raf V600E mutation, an effective inhibition of cell proliferation was achieved at concentrations that inhibited ERK phosphorylation. Both cytostatic and cytotoxicmechanisms of growth inhibition were observed based upon compound concentration and exposure time (100). The clinical safety and toxicity of this compound, however, requires further investigations.
GDC-0879 is a highly selective and orally available BRAF inhibitor that is effective in BRAF V600E containing cells in various in vitro and cell-based assays. For this compound, subnanomolar enzyme potency translated into very effective reduction of cellular viability of BRAF-mutant Malme3M cell lines (EC50 values were 0.75 μmol/L for GDC-0879) (101).
5.0 Expert Opinion & Discussion
Extensive preclinical data support an important role for the RAS-MAPK signaling pathway in cancer biology and its potential as a therapeutic target in human cancers. Molecular and pharmacologic studies have shown that MEK and ERK are required for the transforming activities of Ras and different oncogenes. More recently, mutationally activated Braf and Ras have been identified in several tumors suggesting a tight link between Braf activation and Ras function. The RAF–MEK–ERK cascade, therefore, constitutes a key node in the complex signalling network, with multiple converging signals affecting Raf activation.
In general, MEK inhibitors appear to be well tolerated with only rash, edema, and transient blurred vision as common side effects. Importantly, suppression for each compound in tumor tissue with plasma concentrations corresponding to those sufficient to inhibit MEK in vitro has been demonstrated. Despite the preclinical data, the objective response rate in these studies has been modest and could be attributed to several factors among which are: i) unknown concentration for tumor cytotoxicity, ii) the existence of alternative pathways that compensate for the effects of the MEK inhibitors iii) alterations of multiple signaling pathways in cancer, and iv) inhibition of only a single pathway may not be sufficient to promote apoptosis or cell growth arrest. Given the excellent safety records of these drugs, it is likely that combinations of cytotoxic agents and/or other targeted therapies will be tested in the future as evidence by preclinical data support the potential development of MEK inhibitor combinations. Targeted therapies are likely to benefit only a subset of patients as single agents. The inhibition of a single signal (e.g. RAS) is not able to block pathways downstream of RAS-RAF, which can be regulated by other signals. Some cellular pathways can be activated by both RAS-dependent and RAS-independent mechanisms. Preclinical studies, increased knowledge of the complexity of the Ras-MAPK pathways, and their intersection with other oncogenic pathways, provide the opportunity to design better therapeutic strategies. Combining different RAS-MAPK pathway inhibitors (e.g. RAS-, RAF-, MEK-, PI3K inhibitors), the use of different RAS-signaling inhibitors applied sequentially, or interfering with its upstream supportive signals (e.g. RTKs) might tailor drug combinations, which could have a significantly higher potential for killing tumor cells reducing the side effects on normal cells.
Raf-kinase inhibitors also remain an attractive therapeutic target, and additional preclinical and clinical studies are warranted in order to evaluate the clinical activity and benefit of these types of compounds. Further studies are also needed to better understand the specific biological mechanism of these inhibitors. In particular the identification of the “subpopulation” of patients that may likely benefit.
Efforts to identify predictive molecular markers or gene signatures of response are needed to advance tailored therapy. The identification of EGFR mutations that predict for response to EGFR inhibitors is a good example. BRAF V600E and RAS mutations may also represent viable biomarkers to predict clinical responsiveness to RAF and MEK inhibitors. Therefore, analysis of BRAF and RAS mutations should always be required in future therapeutic trials using these new classes of inhibitors. Particularly using BRAF inhibitors, which have been shown to enhance ERK phosphorylation and tumor growth in BRAF wild type cancer cells. Second line continue treatments with BRAF inhibitors may lead to resistance of cancer cells therefore proper combination therapies should be developed to overcome acquired and adaptive resistance to these agents. A model of resistance to BRAF inhibitors developed by chronic treatment of BRAF V600E melanoma cells with the BRAF inhibitor SB-590885 has recently been described where resistance was attributed to the flexible switching among the three RAF isoforms, underscoring the ability of melanoma cells to adapt to pharmacological challenges. It has been suggested that the activation of other pathways in particular IGF-1R/PI3K signaling, underlie this resistance. Only combined treatment with IGF-1R/PI3K and MEK inhibitors induced death of BRAF inhibitor-resistant cells. An increased IGF-1R and pAKT level in a post-relapse human tumor sample was consistent with a role for IGF-1R/PI3K-dependent survival in the development of resistance to BRAF inhibitors (99). The future design of potential drug combination therapies and the follow-up of their outcome will undoubtedly be facilitated by gene profilings. As the clinical trials of these inhibitors progress, more efforts should be directed to further unravel the complex biology and genetics, and the crosstalk signals of the cancer cells. We contend that combinational therapeutic approaches would be the best approach to address cancer cells escaping drug inhibition and developing resistance.
Acknowledgments
Dr. El-Naggar is supported in part by the NIH National Institute of Dental and Craniofacial Research (NIDCR) and Rare Disease Research (ORDR) Grant Number U01DE019756, the Head and Neck SPORE program Grant Number P50 CA097007 and The Kenneth D. Muller professorship and the NCI-CA-16672 grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institute of Health. Dr. Lippman is supported by the Head and Neck SPORE program Grant Number P50 CA097007. Dr. Santarpia is supported by Associazione Italiana per la Ricerca sul Cancro (AIRC - Grant Number 6251) and American Thyroid Association (ThyCa-Grant).
References
- 1.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey JJ. An unidentified virus which causes the rapid production of tumours in mice. Nature. 1964;204:1104–5. doi: 10.1038/2041104b0. [DOI] [PubMed] [Google Scholar]
- 3.Kirsten WH, Schauf V, McCoy J. Properties of a murine sarcoma virus. Bibl Haematol. 1970;(36):246–9. doi: 10.1159/000391714. [DOI] [PubMed] [Google Scholar]
- 4.Cooper GM. Cellular transforming genes. Science (journal) 1982;217:801–6. doi: 10.1126/science.6285471. [DOI] [PubMed] [Google Scholar]
- 5.Santos E, Tronick SR, Aaronson SA, et al. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. 1982;298:343–7. doi: 10.1038/298343a0. [DOI] [PubMed] [Google Scholar]
- 6.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982 Jun;297:474–8. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 7.http://www.sanger.ac.uk/genetics/CGP/cosmic
- 8.Shields JM, Pruitt K, McFall A, et al. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–54. doi: 10.1016/s0962-8924(00)01740-2. Review. [DOI] [PubMed] [Google Scholar]
- 9.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 10.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. Review. [DOI] [PubMed] [Google Scholar]
- 11.Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–90. doi: 10.1200/JCO.2005.08.036. Review. [DOI] [PubMed] [Google Scholar]
- 12.Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. Review. [DOI] [PubMed] [Google Scholar]
- 13.Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010;16:3329–34. doi: 10.1158/1078-0432.CCR-09-3064. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science (journal) 2001;294:1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 15.Díez D, Sánchez-Jiménez F, Ranea JA. Evolutionary expansion of the Ras switch regulatory module in eukaryotes. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr154. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–57. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–9. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 18.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 19.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–80. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 20.Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–39. doi: 10.1038/sj.onc.1210414. Review. [DOI] [PubMed] [Google Scholar]
- 21.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. 2009;8:1168–75. doi: 10.4161/cc.8.8.8147. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roskoski R., Jr RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399:313–7. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 23.Smeal T, Binetruy B, Mercola DA, et al. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–6. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 24.Binetruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351:122–7. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 25.Pulverer BJ, Kyriakis JM, Avruch J, et al. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–4. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 26.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasko V, Ferrand M, Di Cristofaro J, et al. Specific pattern of RAS oncogene mutations in follicular thyroid tumors. J Clin Endocrinol Metab. 2003;88:2745–52. doi: 10.1210/jc.2002-021186. [DOI] [PubMed] [Google Scholar]
- 28.Santarpia L, Myers JN, Sherman SI, et al. Genetic alterations in the RAS/RAF/mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways in the follicular variant of papillary thyroid carcinoma. Cancer. 2010;116:2974–83. doi: 10.1002/cncr.25061. [DOI] [PubMed] [Google Scholar]
- 29.Volante M, Rapa I, Gandhi M, et al. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J Clin Endocrinol Metab. 2009;94:4735–41. doi: 10.1210/jc.2009-1233. [DOI] [PubMed] [Google Scholar]
- 30.Rapa I, Saggiorato E, Giachino D, et al. Mammalian Target of Rapamycin Pathway Activation Is Associated to RET Mutation Status in Medullary Thyroid Carcinoma. J Clin Endocrinol Metab. 2011 doi: 10.1210/jc.2010-2655. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. Review. [DOI] [PubMed] [Google Scholar]
- 32.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim Biophys Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. Review. [DOI] [PubMed] [Google Scholar]
- 33.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011 doi: 10.1038/onc.2011.160. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 35.Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19:11–5. doi: 10.1016/j.ccr.2011.01.008. Review. [DOI] [PubMed] [Google Scholar]
- 36.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742–62. doi: 10.1210/er.2007-0007. Review. [DOI] [PubMed] [Google Scholar]
- 37.Garnett MJ, Rana S, Paterson H, et al. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20:963–9. doi: 10.1016/j.molcel.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Wan PT, Garnett MJ, Roe SM, et al. Cancer Genome Project. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 39.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 40.Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–7. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- 41.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–76. doi: 10.1016/j.biocel.2004.10.013. Review. [DOI] [PubMed] [Google Scholar]
- 42.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. Review. [DOI] [PubMed] [Google Scholar]
- 43.Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol. 2010;80:624–37. doi: 10.1016/j.bcp.2010.04.029. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–39. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santarpia L, El-Naggar AK, Cote GJ, et al. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278–84. doi: 10.1210/jc.2007-1076. [DOI] [PubMed] [Google Scholar]
- 46.Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer. 2009;16:17–44. doi: 10.1677/ERC-08-0154. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 48.Alessi DR, Cuenda A, Cohen P, et al. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 49.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 50.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–93. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 51.Sebolt-Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 52.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 53.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 55.Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 56.Davies BR, Logie A, McKay JS, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 57.Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6:138–46. doi: 10.1158/1535-7163.MCT-06-0436. [DOI] [PubMed] [Google Scholar]
- 58.Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 59.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drummer R, Robert C, Chapman P, et al. AZD6244 (ARRY-142886) vs Temozolomide in Patients With Advanced Melanoma: an Open-Label, Randomized, Multicenter, Phase II Study. J Clin Oncol (abstract) 2008;26:9033. [Google Scholar]
- 61.Tzekova V, Cebotaru C, Ciuleanu TE, et al. Efficacy and safety of AZD6244 (ARRY-142886) as second/third-line treatment of patients (pts) with advanced non-small cell lung cancer (NSCLC) J Clin Oncol (abstract) 2008;26:8029. [Google Scholar]
- 62.Lang I, Adenis A, Boer K, et al. AZD6244 (ARRY-142886) Versus Capecitabine in Patients With Metastatic Colorectal Cancer Who Have Failed Prior Chemotherapy. J Clin Oncol (abstract) 2008;26:4114. [Google Scholar]
- 63.Bekaii-Saab T, Phelps MA, Li X, et al. Multi-Institutional Phase II Study of Selumetinib in Patients With Metastatic Biliary Cancers. J Clin Oncol. 2011;29:2357–63. doi: 10.1200/JCO.2010.33.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Neil BH, Goff LW, Kauh JS, et al. Phase II Study of the Mitogen-Activated Protein Kinase 1/2 Inhibitor Selumetinib in Patients With Advanced Hepatocellular Carcinoma. J Clin Oncol. 2011;29:2350–6. doi: 10.1200/JCO.2010.33.9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnston S. XL518, a potent, selective orally bioavailable MEK1 inhibitor, down-regulates the Ras/Raf/MEK/ERK pathway in vivo, resulting in tumor growth inhibition and regression in preclinical models. 19th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2007. p. Abstract C209. [Google Scholar]
- 66.Rosen LS, Galatin P, Fehling JM, et al. A phase 1 dose-escalation study of XL518, a potent MEK inhibitor administered orally daily to subjects with solid tumors. J Clin Oncol (abstract) 2008;26:14585. [Google Scholar]
- 67.Shapiro G, LoRusso P, Kwak EL, et al. ASCO J Clin Oncol. 2011;29 (suppl; abstr 3005^) [Google Scholar]
- 68.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 69.Yamaguchi T, Kakefuda R, Tajima N, et al. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39:23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 70.Thompson D, Flaherty K, Messersmith W, et al. A three-part, phase I, dose-escalation study of GSK1120212, a potent MEK inhibitor, administred orally with solid tumors or lymphoma. J Clin Oncol (abstract) 2009;27:e14584. [Google Scholar]
- 71.Iverson C, Larson G, Lai C, et al. RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009;69:6839–47. doi: 10.1158/0008-5472.CAN-09-0679. [DOI] [PubMed] [Google Scholar]
- 72.Daouti S, Higgins B, Kolinsky K, et al. Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Molecular cancer therapeutics. 2010;9:134–44. doi: 10.1158/1535-7163.MCT-09-0601. [DOI] [PubMed] [Google Scholar]
- 73.Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther. 2005;4:677–85. doi: 10.1158/1535-7163.MCT-04-0297. Review. [DOI] [PubMed] [Google Scholar]
- 74.Gokhale PC, Zhang C, Newsome JT, et al. Pharmacokinetics, toxicity, and efficacy of ends-modified raf antisense oligodeoxyribonucleotide encapsulated in a novel cationic liposome. Clin Cancer Res. 2002;8:3611–21. [PubMed] [Google Scholar]
- 75.Dritschilo A, Huang CH, Rudin CM, et al. Phase I study of liposome-encapsulated c-raf antisense oligodeoxyribonucleotide infusion in combination with radiation therapy in patients with advanced malignancies. Clinical Cancer Research. 2006;12:1251–9. doi: 10.1158/1078-0432.CCR-05-1260. [DOI] [PubMed] [Google Scholar]
- 76.Rudin CM, Marshall JL, Huang CH, et al. Delivery of a liposomal c-raf-1 antisense oligonucleotide by weekly bolus dosing in patients with advanced solid tumors: a phase I study. Clin Cancer Res. 2004;10:7244–51. doi: 10.1158/1078-0432.CCR-04-0642. [DOI] [PubMed] [Google Scholar]
- 77.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 78.Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–6. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–30. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 80.Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69:223–40. doi: 10.2165/00003495-200969020-00006. Review. [DOI] [PubMed] [Google Scholar]
- 81.Sherman SI. Targeted therapies for thyroid tumors. Mod Pathol. 2011;24(Suppl 2):S44–52. doi: 10.1038/modpathol.2010.165. Review. [DOI] [PubMed] [Google Scholar]
- 82.Moreno-Aspitia A. Clinical overview of sorafenib in breast cancer. Future Oncol. 2010;6:655–63. doi: 10.2217/fon.10.41. Review. [DOI] [PubMed] [Google Scholar]
- 83.Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107:14903–8. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chapman PB, Hauschild A, Robert C, et al. The BRIM-3 Study Group. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011 doi: 10.1056/NEJMoa1103782. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 88.Kopetz S, Desai J, Chan E, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(suppl):15s, abstract 3534. [Google Scholar]
- 89.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010 Mar 18;464(7287):427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robert C, Soria JC, Spatz A, et al. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol. 2005;6:491–500. doi: 10.1016/S1470-2045(05)70243-6. Review. [DOI] [PubMed] [Google Scholar]
- 91.Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011;23:177–82. doi: 10.1097/CCO.0b013e3283436e8c. Review. [DOI] [PubMed] [Google Scholar]
- 92.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kefford R, Arkenau H, Brown MO, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol. 2010;28:15s. (suppl; abstr 8503) [Google Scholar]
- 94.Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer. 2011;104:392–8. doi: 10.1038/sj.bjc.6606030. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Puzanov I, Burnett P, Flaherty KT. Biological challenges of BRAF inhibitor therapy. Mol Oncol. 2011;5:116–23. doi: 10.1016/j.molonc.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schwartz GK, Robertson S, Shen A, et al. A phase I study of XL281, a selective oral RAF kinase inhibitor, in patients (Pts) with advanced solid tumors. J Clin Oncol. 2009;27:3513. [Google Scholar]
- 97.Mordant P, Loriot Y, Leteur C, et al. Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive the activity of RAF265, a novel RAF/VEGFR2 inhibitor, and RAD001 (Everolimus) in combination. Mol Cancer Ther. 2010;9:358–68. doi: 10.1158/1535-7163.MCT-09-1014. [DOI] [PubMed] [Google Scholar]
- 98.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.King AJ, Patrick DR, Batorsky RS, et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–5. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 100.Hoeflich KP, Herter S, Tien J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–51. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 101.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]