

Abstract
We previously demonstrated the therapeutic effects of MHC class II derived recombinant T cell receptor ligands (RTL), single-chain two domain complexes of the α1 and β1 domains of MHC class II molecules genetically linked with an immunodominant peptide, in experimental autoimmune encephalomyelitis. In the current study, we produced a monomeric murine I-Aq-derived RTL construct covalently linked with bovine collagen type II peptide (bCII257–270) suitable for use in DBA/1LacJ mice that develop collagen-induced arthritis (CIA), an animal model of human rheumatoid arthritis, after immunization with bCII protein in CFA. In this study, we demonstrate that the I-Aq-derived RTLs reduced the incidence of the disease, suppressed the clinical and histological signs of CIA and induced long-term modulation of T cells specific for arthritogenic Ags. Our results showed that the I-Aq/bCII257–270 molecule could systemically reduce proinflammatory IL-17 and IFN-γ production and significantly increase anti-inflammatory IL-10, IL-13, and FoxP3 gene expression in splenocytes. Moreover, I-Aq/bCII257–270 molecule could also selectively inhibit IL-1β, IL-6, and IL-23 expression in local joint tissue. This is the first report demonstrating effective prevention of joint inflammation and clinical signs of CIA with an I-Aq-derived RTL, thus supporting the possible clinical use of this approach for treating rheumatoid arthritis in humans.
Rheumatoid arthritis (RA)3 is characterized by chronic synovitis with erosive pannus formation and destruction of articular cartilage and other periarticular structures (1). Although the etiology of the disease remains unknown, cumulative findings suggest an important role for the immune system in the inflammatory phase of the disease, and both genetic predisposition and environmental factors influence the risk of developing disease (2–4). The susceptibility of the disease appears to be associated with several polymorphic loci of HLA genes, including HLA-DR and -DQ loci (4). The HLA genes encode MHC class II molecules, which are heterodimeric proteins that selectively bind antigenic peptide for presentation to T lymphocytes and also have a major role in positive and negative selection in the thymus of the TCR repertoire released to the periphery (5, 6). The MHC class II linkage suggests a potential role for T cell-mediated immune response in RA. Early studies with SKG mice, a spontaneous mouse model with a T cell dependent inflammatory arthritis that has clinical and histological similarities with human RA, suggested a central role of T cells in the pathogenesis of RA (7, 8). Recent studies (9) of type II collagen (CII) autoimmunity in RA patients suggest that CII-reactive T cells were significantly present in synovial fluid, especially in the early stage of the disease. When CII-reactive T cells isolated from RA patients were cultured with fibroblast-like synoviocytes, there was a significant increase in the production of proinflammatory cytokines and inflammatory chemokines (9), which suggests that CII-reactive T cells could play a crucial role in RA development in a genetically predisposed host. However, it is unclear whether CD4+ T cells may directly mediate the pathogenesis of RA.
The studies of collagen-induced arthritis (CIA), an animal model of autoimmune disease that shares many characteristic features of RA, showed that passive transfer of CII-specific T cells to susceptible strains only induced minor pathological changes in the synovial joints of the recipients (10, 11), whereas transfer of immune sera from arthritic mice (12, 13) or from RA patients (14) to naive mice induced a moderate to severe arthritic inflammation regardless of the genetic background. These studies suggest that autoantibodies may also be a major pathogenic factor in this disease. Interestingly, this Ab-transferred disease is transient and mostly does not lead to cartilage destruction, which is a prominent clinical sign of RA. T cells, in contrast, can transfer arthritis that can become destructive, but clinical signs of disease, in this case, are not as severe as seen in CII-induced CIA (11). Thus, it seems likely that T cells not only induce inflammation in synovial tissues, but also serve another important role in RA, modulating B cell differentiation and autoantibody production (15, 16). Moreover, a recent study (17) showed that the activation of Th type 17 (Th17) cells, a highly pathogenic T cell subset that produces the proinflammatory IL-17 cytokine, could directly enhance osteoclastogenesis and result in bone destruction.
Autoimmunity of CII, a major component of human cartilage, has been suggested (9) to play an important role in the pathogenesis of RA. Immunization of susceptible mice strains (e.g., DBA1/J mice expressing the I-Aq class II gene) with human CII leads to the development of CIA (18–20). CIA is the most widely used model of RA, and has provided considerable insights into the pathogenesis of this human disease. Immunization with collagen proteins in CFA induces a TNF-α regulated production of IL-1β, IL-6, IL-8, and GM-CSF leading to inflammation of synovial tissue and destruction of cartilage and bone, resulting in loss of joint function (18, 19, 21). Analysis of inbred mouse strains demonstrates that specific MHC haplotypes (I-Aq and I-Ar) confer susceptibility to CIA (22, 23). Unlike the “shared epitope” expressed by human HLA-DR alleles (24, 25), there are no distinctive sequence similarities between the β-chain polymorphic regions of these two class II molecules (19). An immunodominant peptide derived from the CII molecule bound to the CIA susceptible I-Aq molecule has been described. This peptide was found to be located between positions 256 to 270 of CII (26, 27) with a determinant core-binding motif between positions 260 to 267. Interestingly, a very similar binding motif of the CII261–273 peptide has also been identified in DR1 and DR4 Transgenic (Tg) mice (28–30), in which the determinant core binds to DR1 or DR4 molecule with the same major MHC anchor residues at positions 263 and 264 as compared with the binding of the same peptide to the I-Aq molecule (30). Moreover, the CII molecule is posttranslationally modified, and the majority of CII256–270 specific T cells from mice bearing the I-Aq class II molecule seem to recognize the lysine (K) residue at position 264, whose side chain can be modified (e.g., hydroxylated, galactosylated, and glycosylated) (31, 32).
It has been demonstrated that partial activation of pathogenic T cells through the context in which the peptide/MHC complex interacts with the Ag-specific TCR without costimulators could induce anergy (33, 34) or apoptosis (35). A direct approach toward Ag-driven immunosuppression would be to present the minimal TCR ligand, Ag plus MHC, in the absence of costimulatory signals that are normally provided by specialized APCs. Thus, we designed MHC class II derived recombinant TCR ligands (RTLs), which consist of only the minimal Ag/TCR interface in which α1 and β1 domains of MHC class II molecules were genetically linked into a single polypeptide chain (36–39). Our previous studies showed that RTLs containing a single chain two-domain murine I-As or HLA-DR2 class II moiety covalently linked to the immunodominant peptides of proteolipid protein (PLP139–151) or myelin oligodendrocyte glycoprotein (MOG35–55), respectively, could reverse clinical and histological signs of experimental autoimmune encephalomyelitis (EAE), an animal model of human multiple sclerosis (MS), and induce long-term tolerance in SJL mice (40) and HLA-DR2 Tg mice (41). In the current study, we produced a monomeric murine I-Aq-derived RTL containing a single chain two-domain I-Aq MHC class II molecule covalently linked to the immunogenic peptides of bCII257–270. The results demonstrated that I-Aq-derived RTLs reduced the incidence and clinical and histological signs of CIA, and induced long-term T cell tolerance against arthritogenic Ags. This study may contribute to the current understanding of the role of pathogenic T cells in autoimmune diseases and potentially add a target-specific drug to the list of interventional therapies against RA.
Materials and Methods
Animals
DBA/1LacJ mice were obtained from The Jackson Laboratory were maintained in microisolators at the animal facility of the Portland Veterans Affairs Medical Center. All protocols were conducted in accordance with institutional guidelines.
Collagen
Bovine collagen was obtained from Chondrex. The collagen peptide 257– 274 was synthesized by Fmoc chemistry using an automated peptide synthesizer (PE Applied Biosystems).
RTL construction, modification, and production
General methods for the design, cloning and expression of RTLs have been described previously (37, 39, 40). In brief, mRNA was isolated from the splenocytes of DBA1/LacJ mice using an Oligotex Direct mRNA mini kit (Qiagen). cDNA of the Ag binding/TCR recognition domain of murine I-Aq MHC class II β1 and α1 chains was derived from mRNA using two pairs of PCR primers. The two chains were sequentially linked by a 5 amino acid linker (GGQDD) in a two-step PCR with NcoI and XhoI restriction sites being added to the amino terminus of the β1 chain and to the carboxyl terminus of the α1 chain respectively, to create RTL2000. A linker (GSGSGSGSGSGSGS) and bCII257–270 peptide with modification at E257A, G265A, G268A, and P269A were covalently linked to the 5′ end of the β1 domain of RTL2000 to form RTL2001MII. The murine I-Aq β1α1 insert was then ligated into pET21d+ vector and transformed into Nova blue E. coli host (Novagen) for positive colony selection and sequence verification. RTL2000 and RTL2001MII plasmid constructs were then transformed into E. coli strain BL21 (DE3) expression host (Novagen). The purification of proteins has been described previously (37, 40). The final yield of purified protein varied between 30 to 40 mg/L of bacterial culture.
I-Aq-derived RTL treatment and arthritis induction
Before arthritis induction, male DBA/1LacJ mice between 7 and 9 wk of age and body weight 20–25 g were randomly divided into four treatment groups. The animals were given a daily i.v. injection for 5 days of 100 μl containing 100 μg of RTL2000 protein, 100 μg of RTL2001MII protein, an equimolar dose of 10 μg of bCII257–274 peptide, or buffer. Then, the disease was induced by intradermal injection at the base of tail of with 100 μg of bCII protein in CFA containing 100 μg of Mycobacterium tuberculosis in a 100-μl injection volume. Animals were monitored for onset and progression of disease 3–10 wk postimmunization. The arthritis severity of mice was evaluated with a grading system for each paw according to the following scale: 0 = no redness or swelling; 1 = slight swelling in ankle or redness in foot; 2 = progressive swelling/inflammation and redness from ankle to mid foot; 3 = swelling/inflammation of entire foot; 4 = swelling and inflammation of entire foot including toes. The Arthritis Score for each mouse was determined by adding the severity scores for each of the four paws.
bCII-specific IgG1 and IgG2a detection
Sera were collected from each group of mice at day 36 and day 70 post immunization and bCII-specific IgG1 and IgG2a Ab levels were measured by ELISA. Ninety-six-well plates were coated with 5 μg/ml whole bCII in Jerry Gross buffer (127 mM K2HPO4, 3.4 mM KH2PO4, pH 7.6) and incubated overnight at 4°C. Plates were blocked with blocking buffer (1× PBS, 2% BSA, 0.05% Tween 20) for 2 h at 4°C. Plates were washed once and then 200 μl of diluted serum sample in ELISA buffer containing 2% normal goat serum (NGS)/well was added in triplicate wells. Samples were incubated at 4°C overnight. One hundred microliters of 1/7500 diluted anti-IgG1 or IgG2a-biotin was added to the samples and incubated at 4°C overnight. Plates were washed three times and then incubated with 100 μl of 1/2000 diluted goat anti-mouse streptavidin conjugate HRP (Sigma-Aldrich) for 30 min at 4°C. Samples were washed followed by addition of 100 μl of 3,3′,5,5′-tetramethylbenzidine/chromogen solution. The plates were allowed to develop for ~30 min, and then the reaction was stopped by adding 100 μl stop solution. The OD was measured at 490 nm (42).
Cytokine analysis by Bio-Plex cytokine assays
Spleen cells were cultured at 4 × 106 cells/well in a 24-well flat-bottom culture plate in stimulation medium with 25 μg/ml bCII257–274 peptide for 48 h. Supernatants were then harvested and stored at −80°C until testing for cytokines. A customized mouse Bio-Plex cytokine kit was used to detect IFN-γ, IL-1β, IL-6, IL-17, TNF-α, IL-10, and IL-13 simultaneously (Bio-Rad) as described by the manufacturer’ protocol. The signals were analyzed on the Luminex 200 (Bio-Rad), and the data were analyzed using the Bio-Plex Management software (Bio-Rad).
RNA isolation and RT-PCR
Total RNA was isolated from the joint tissues and spleen cells using the RNeasy mini kit protocol (Qiagen) and then converted to cDNA using oligo(dT), random hexamers and Superscript RT II enzyme (Invitrogen). Real-time PCR was performed using Quantitect SYBR Green PCR master mix (Qiagen) and primers (synthesized by ABI). Reactions were conducted on the ABI Prism 7000 Sequence Detection System (Applied Biosystems) to detect the following genes: L32 (forward, GGAAACCCAGAGGCAT TGAC; reverse, TCAGGATCTGGCCCTTGAAC); IFN-γ (forward, TG CTGATGGGAGGAGATGTCT; reverse, TGCTGTCTGGCCTGCTGT TA); TNF-α (forward, CAGCCGATGGGTTGTACCTT; reverse, GGCA GCCTTGTCCCTTGA); IL-1β (forward, TTGACGGACCCCAAAAGA; reverse, TGGACAGCCCAGGTCAAAGTG); IL-6 (forward, CCACGGCC TTCCCTACTTC; reverse, TGGGAGTGGTATCCTCTGTGAA); IL-17 (forward, CCCTTGGCGCAAAAGTGA; reverse, CGTGGAACGGTTGA GGTAGTC); IL-23 (forward, CCGTTCCAAGATCCTTCGAA; reverse, GACCCGGGCAGCTATGG); IL-10 (forward, GATGCCCCAGGCAGAG AA; reverse, CACCCAGGGAATTCAAATGC); IL-13 (forward, ACTG CTCAGCTACACAAAGCAACT; reverse, TGAGATGCCCAGGGATG GT); and FoxP3 (forward, GGCCCTTCTCCAGGACAGA; reverse, GC TGATCATGGCTGGGTTGT). Expression of each gene was calculated relative to the expression of the housekeeping gene, L32 (40).
Histology
Fixed tibiotarsal joints were decalcified in formic acid and processed for paraffin embedding. Tissue sections (5 γm) were stained with hematoxylin and eosin for histopathological scoring. Joints were scored for synovial proliferation and articular inflammation based upon the scoring methodology of Leung et al. (43). Synovial proliferation was scored as follows: Grade 0 proliferation was absent. Grade 1 proliferation was mild with two to four layers of reactive synoviocytes. Grade 2 proliferation was moderate with four plus layers of reactive synoviocytes, increased mitotic activity and mild or absent synovial invasion of adjacent bone and connective tissue. Grade 3 proliferation was severe and characterized by invasion and effacement of joint space and adjacent cartilage, bone and connective tissue. Articular inflammation was scored as follows. Grade 0 inflammation lacked significant leukocyte infiltrates or aggregates. Grade 1 inflammation was mild with one aggregate or minimal diffuse leukocyte infiltrates. Grade 2 inflammation was moderate with two or more aggregates of leukocytes. Grade 3 inflammation was severe with significant coalescing to diffuse infiltrates of leukocytes.
Statistical analyses
Significant differences in the incidence between control and treated mice were assessed by Fisher’s exact test. Differences in the day of disease onset, Ab and cytokine production, and cytokine mRNA expression were determined using Student’s t test. Differences in the peak score and cumulative disease index were assessed by Mann-Whitney U test and/or ANOVA. Values of p ≤ 0.05 were considered to be significant.
Results
RTL2001MII treatment reduced the incidence and clinical signs of CIA and induced a long term modulation of T cell and Ab responses against arthritogenic Ags in DBA/1LacJ mice
In the current study, we constructed and produced a set of monomeric murine I-Aq-derived RTLs containing a single chain two-domain I-Aq MHC class II molecule (RTL2000) covalently linked to the immunogenic peptide of bCII257–270 (RTL2001MII). These novel constructs were used to test our hypothesis that the specific targeting of Ag-specific TCR by the RTLs can regulate pathogenic T cell activation. As shown in Fig. 1, RTL2001MII pretreatment reduced the severity of clinical signs of CIA compared with the controls. The animals were pretreated i.v. with 100 μg RTL daily for 5 days, and then immunized with 100 μg bCII/ mouse with CFA. Our results showed significant protection starting as early as day 34 after immunization (Fig. 1) and lasting for 70 days after disease induction without further boosting, which suggests that the RTLs could induce a long term tolerance against the arthritogenic Ags. Similar results were reported in our previous studies with the EAE model. As shown in Fig. 1, the average arthritis scores (Fig. 1A) were significantly reduced in the RTL2001MII pretreated group in comparison to the control, the “empty” RTL2000 treated group, and the CII 257–270 peptide treated group ( p < 0.01). The disease incidence (Fig. 1B) was also significantly reduced in the RTL2001MII treated group (22.2%) vs the control (84.6%), “empty” RTL2000 (83.2%), and CII peptide 257–270 (66.7%). Moreover, the cumulative disease index that monitors the severity of the disease throughout the course of disease showed that there was a significant difference between RTL2001MII treated group vs the control group (data not shown). These results strongly suggest that only the cognate peptide/MHC complex but not the MHC moiety alone could modulate arthritogenic T cell responses, consistent with our previous studies (40, 41, 44).
FIGURE 1.

Intravenous administration of RTL2001MII ameliorates CIA in DBA/1LacJ mice. For each treatment group, DBA1/J mice between 7 and 9 wk of age were given a daily i.v. injection for 5 days of 100 μl containing 100 μg RTL2000 or RTL2001MII proteins, an equimolar dose of 10 μg of the bCII257–274 peptide, or buffer before induction of CIA. The disease was then induced by intradermal injection at the base of the animal’s tail with 100 μg of bCII protein in CFA containing 100 μg of Mycobacterium tuberculosis (MTB) in 100 μl injection volume. Animals were monitored for onset and progression of disease 3–10 wk post immunization. The disease severity was scored with a grading system for each paw as described previously (42) and presented as mean score of the disease for each treatment group (A). α, Significance between the control and treatment groups was determined using the Mann-Whitney U test (p < 0.05). The incidence of CIA was recorded daily for each treatment group (B). α, Significance between the control and treatment groups was determined using Fisher’s Exact test (p < 0.05). Data represent combined results from two complete experiments involving 10–20 mice per group.
RTL2001MII treatment led to an Ag-specific Ab profiling switch from proinflammatory to anti-inflammatory responses
Even though the RTLs were designed specifically to target pathogenic T cells, the effect of the RTLs on the B cell response was also evaluated. Sera were collected from the control, RTL2000 and RTL2001MII treated mice on days 36 and day 70 post immunization and assayed for IgG1 and IgG2a Ab levels that were specific for the bCII257–273 peptide. As shown in Fig. 2, there was a significant change in IgG1 and IgG2a production following the RTL treatment. Our data show that RTL treatment results in a significant increase in IgG1 isotype, which reflects an increase in anti-inflammatory responses. Reduction of IgG2a level may indicate a decrease in proinflammatory responses. A recent study reported that posttranslational modification of a lysine residue (K264) within bCII257–273 could reduce clinical signs of the disease. The treatment of the complex of I-Aq class II with glycosylated bCII259–273 peptide also significantly reduced the CII-specific IgG Abs (45), which is in agreement with our results.
FIGURE 2.

RTL2001MII treatment significantly increased the bCII-specific IgG1 response and reduced the bCII-specific IgG2a response. Sera were collected on day 36 and 70 after immunization and incubated in serial dilutions in bCII-coated wells. Levels of IgG1 and IgG2a anti-CII Abs were measured by ELISA. A and B, Represent the levels of IgG1 and IgG2a, respectively. α, Significance between the control and experimental groups was determined using Student’s t test (p < 0.05).
RTL2001MII treatment significantly reduced the proliferation and the histological signs of CIA in synovial joint tissue
The hallmark of RA is local joint inflammation and bone destruction. The histopathological examination of local joint tissue could provide important information for us to understand the mechanism of the RTL treatment. We performed histopathological analyses of joint tissues obtained from the control, RTL2000 and RTL2001MII treated mice at the end of the experiment. As shown in Fig. 3, there was a significant decrease in joint inflammation following treatment with RTL2001MII. There were significantly fewer infiltrating cells in RTL2001MII-treated joints compared with the control or “empty” RTL2000 that showed severe inflammation and synovial hyperplasia with several layers of reactive synovial tissue. The histological arthritis score was also determined in a blinded fashion for proliferative and inflammatory changes and graded from 0 to 3 for each limb as described previously (42). There were significant proliferative and inflammatory changes throughout the stifle and tibio-tarsal joints in the control and “empty” RTL2000 treated groups. In comparison, there were only mild proliferative and inflammatory changes in the joint tissues of RTL2001MII treated animals, which may be due to the CFA adjuvant during the induction (Table I). It appears that the RTL treatment could prevent the infiltration of the peripheral pathogenic T cells into the local joint tissue and block synovial T cell activation.
FIGURE 3.
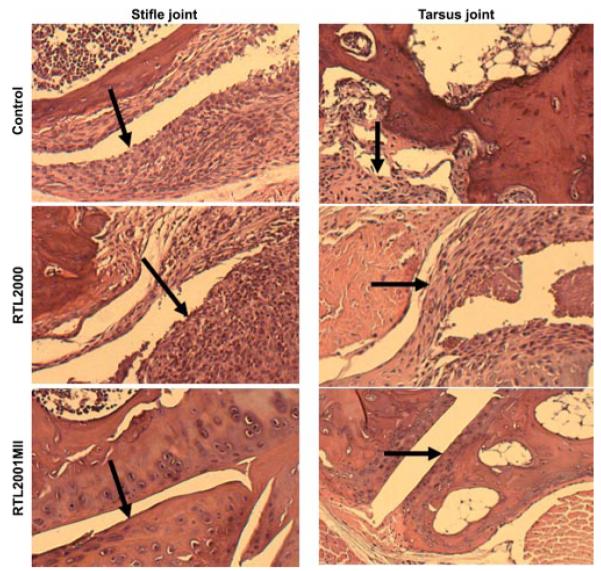
RTL2001MII treatment reduced histological signs of CIA in DBA/1LacJ mice. Hind limbs obtained from control, RTL2000 and RTL2001MII treated mice at day 70 post immunization were fixed in formalin. Limbs were decalcified in HCl and sectioned in half longitudinally through the stifle and tibio-tarsal joints. Five γm sections were processed and stained with H&E.
Table I.
The histological arthritis score among treatment groups in DBA1/LacJ mice
| Molecule | Stifle Joint |
Tibiotarsal Joint and Tarsal Joints |
||
|---|---|---|---|---|
| Synovial Proliferation Score |
Articular Inflammation Score |
Synovial Proliferation Score |
Articular Inflammation Score |
|
| Control | 3 | 2 | 3 | 2 |
| RTL2000 | 3 | 1 | 3 | 2 |
| RTL2001MII | 1 | 1 | 1 | 1 |
RTL treatment led to a systemic cytokine switch from proinflammatory to anti-inflammatory responses
We previously reported (40) that in SJL/J mice, a relapsing-remitting EAE animal disease model, murine I-As/PLP139–151 construct (termed RTL401) prevented relapses and significantly reduced clinical severity of the disease induced by PLP139–151peptide. We found that RTL treatment in SJL/J mice led to a remarkable reduction of proinflammatory cytokines and chemokines, and significantly enhanced anti-inflammatory cytokines and chemokines (40, 46). In this study, the systemic cytokine response to the RTL treatment was examined. Splenocytes were collected from each treatment group at the end of the experiment. RT-PCR was performed for the expression of various proinflammatory and anti-inflammatory cytokines using the L32 housekeeping gene as the control. Cytokine secretion was also examined at the same time using Bio-Plex cytokine kits. Our results showed that RTL2001MII treatment significantly reduced splenocyte secretion of the proinflammatory cytokines IL-1β, IL-6, IL-17 and IFN-γ compared with buffer-treated control mice (Fig. 4, A–D), but significantly increased the expression of the anti-inflammatory cytokines, IL-10 and IL-13 (Fig. 5), suggesting a cytokine “switch”. However, treatment with RTL2000 also reduced production of IL-1β and IL-6, indicating that alteration of these two cytokines was not selective for RTL2001MII.
FIGURE 4.

RTL2001MII treatment significantly reduced production of proinflammatory cytokines. In each of three separate experiments, splenocytes pooled from two individual mice from the indicated treatment groups were cultured at 4 × 106 cells/well in a 24-well flat-bottom culture plate with medium alone or 25 μg/ml bCII257–274 peptide for 48 h. Supernatants were then collected and analyzed using a Bio-Plex cytokine kit with Abs specific for IL-1β (A), IL-6 (B), IL-17 (C), and IFN-γ (D). Representative data are from one of the three experiments. α, Significance between the control and each of the treatment groups was determined using Student’s t test (p < 0.05).
FIGURE 5.

RTL2001MII treatment significantly increased the expression of anti-inflammatory cytokines. Total RNA was isolated from the splenocytes of two treated mice from each of the control, RTL2000 and RTL2001MII groups at the end of the experiment. cDNA was synthesized and real-time PCR was performed using primers specific for IL-10 (A) and IL-13 (B). Expression of each gene was calculated relative to the expression of the housekeeping gene, L32. α, Significance between the control and experimental groups was determined using Student’s t test (p < 0.05). ND, Not detectable. Representative data are from one of two separate experiments.
RTL2001MII treatment may up-regulate the T regulatory cell (Treg) population
Our previous study (47) suggested that the RTLs provided a suboptimal ligation through Ag-specific TCR to alter the T cell signaling. A previous in vitro study in human CD4+CD25− T cells (48) showed that a suboptimal signal induced by the engagement of anti-CD3/CD28 Abs could activate the expression of the FoxP3 gene. Thus, the potential effect of the RTLs on the expression of FoxP3 in the splenocytes was examined. The splenocytes were collected from each treatment group at the end of the experiment and RT-PCR was performed for the expression of the FoxP3 gene using the L32 housekeeping gene as the control. The FoxP3 gene expression in splenocytes was significantly increased in the RTL2001MII treated group as compared with the control and the “empty” the RTL2000 treated group (Fig. 6). This finding suggests up-regulation of the Treg population in RTL treated animals that conceivably could mediate bystander suppression during the course of the disease.
FIGURE 6.
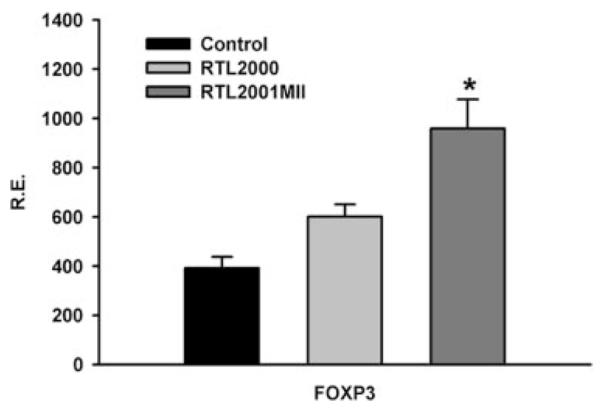
RTL2001MII treatment up-regulated the expression of the FoxP3 gene in splenocytes. Total RNA was isolated from splenocytes harvested from two treated mice from each of the control, RTL2000 and RTL2001MII groups at the end of experiment. cDNA was synthesized and real-time PCR was performed using primers specific for FoxP3. Gene expression was calculated relative to the expression of the housekeeping gene, L32. α, Significant between the control and experimental groups was determined using Student’s t test (p < 0.05). Representative data are from one of two separate experiments.
RTL2001MII treatment reduced proinflammatory cytokines in the joint tissue
At the end of the experiment, the joint tissues were collected from the animals of each treatment group. Total RNA was isolated from the synovial joint tissue and RT-PCR was performed to evaluate the effect of the RTL treatment on the expression of various cytokines in the local joint tissues. The results showed that RTL2001MII treatment significantly reduced the expression of IL-1β, IL-6, and IL-23 in synovial joint tissue (Fig. 7), with a reduction in TNF-α expression induced by both RTL2000 and RTL2001MII. The down-regulation of these cytokines in synovial joint tissue could result from less infiltrating cells and/or suppression of synovial inflammation after RTL treatment, which is in agreement with histopathological data shown in Fig. 3.
FIGURE 7.

RTL2001MII treatment down-regulated the expression of proinflammatory cytokine genes in joint tissue. Total RNA was isolated at the end of the experiment from whole frozen joint tissue harvested from two treated mice from each of the control, RTL2000, and RTL2001MII groups. cDNA was synthesized and real-time PCR was performed using primers specific for IL-1β (A), IL-6 (B), TNF-α (C), and IL-23 (D). Expression of each gene was calculated relative to the expression of the housekeeping gene, L32. α, Significance between the control and experimental groups was determined using Student’s t test (p < 0.05). Representative data are from one of two separate experiments.
Discussion
In our previous studies, we demonstrated that both monomeric DR2-derived and I-As-derived RTLs could protect and treat the clinical and histological signs of EAE in DR2 Tg mice (39, 41) and SJL mice (40), respectively. In the current study, we used a similar approach, constructing monomeric I-Aq-derived RTLs covalently linked with bCII257–270 peptide (RTL2001) (US patent no. 6,270,772). Biochemical and biophysical characterization indicated the recombinant molecule retained structural integrity. In our initial animal study, RTL2001 did not appear to protect the animals from CIA (data not shown). Previous studies (49, 50) suggested that the side chains of isoleucine at position 260 and phenylalanine at position 263 of the bCII257–270 peptide are the important P1 and P4 anchor residues for binding to the I-Aq molecule. Lysine 264 (K264) appears to be the major T cell recognition site of CII257–273 and CIA is in particular associated with recognition of K264 after posttranslational hydroxylation and subsequent attachment of a β-D-galactopyranosyl moiety (31, 32). A number of publications indicated that the posttranslational modification of arthritogenic peptide bCII257–270 may be required to induce a protective or suppressive effect. Glycosylation of the key TCR contacting residue K264 within the bCII257–270 peptide resulted in a reduction of Ab response to arthritogenic peptide and a decrease in T cell response to the glycopeptides (45, 51). We have not yet overcome the technical hurdles required for engineering a recombinant molecule with a glycosylation at only this single amino acid residue. A previous study showed that substituting a few amino acid residues on both the amino terminus (position 257) and carboxy terminus (positions 268 and 269) of the determinant core peptide (bCII260–267) could significantly alter Ag-specific T cell responses (49). Moreover, a recent study involved in the TCR specificity suggested that certain amino acid side chains on the peptide/MHC surface can actually ‘disrupt’ the TCR-peptide-MHC interaction, even though the side chains are not involved in binding to the TCR (52). Thus, we modified RTL2001 by substituting four amino acid residues (E257A, G265A, G268A, and P269A) within the arthritogenic bCII257–270 peptide (termed RTL2001MII). Our results showed that the modified murine I-Aq-derived RTL, RTL2001MII, could protect the animals from CIA. The results strongly support our hypothesis that regulating the context in which the complex of peptide/MHC interacts with the TCR could control the fate of the pathogenic T cells.
In the current study, the pretreatment with murine versions of RTLs reduced the incidence of the disease, suppressed the clinical and histological signs of CIA and induced long-term immunoregulation against arthritogenic Ags without need for further boosting. The ability of RTL2001MII to suppress arthritis appears to be correlated with a change in the cytokine response both systemically (Figs. 4 and 5) and locally (Fig. 7) and a reduction in infiltrating pathogenic cells in synovial joint tissue (Fig. 3). The dramatic reduction of infiltrating pathogenic cells in the local joint tissue at day 70 post immunization is the most significant change resulting from RTL treatment, and may explain the corresponding reduction in expression of proinflammatory cytokines. Our previous EAE study in SJL mice showed that treatment with I-As-derived RTL covalently linked to PLP139–151 peptide reduced the expression of adhesion molecules including the very late activation Ag-4 (VLA-4; CD49d) and lymphocyte function-associated Ag-1 (LFA-1; CD11a) in the CNS (40). These results suggest that the RTL treatment affected the disease systemically and also at the local level. Long-term protection against arthritogenic Ags supports the clinical application of this novel class of peptide/MHC class II construct in patients with RA.
In the CIA model, CD4+ T cells are initially activated by presentation of disease-associated CII Ags by I-Aq class II molecules, with concomitant activation of B cells that later will play an important role in the course of the disease by producing CII-specific autoantibodies. We thus examined the effect of the RTLs on the bCII-specific Ab response. Our results demonstrated that the RTL2001MII treatment significantly increased the bCII-specific IgG1 isotype and reduced the IgG2a isotype that is usually associated with a pathogenic response. However, the reduction of IgG2a was minimal, which may in part explain the residual disease score and 22% incidence of CIA in the RTL2001MII treated group. An approach that could specifically target Ag-specific B cells or plasma cells may be needed in combination with the RTLs for successful treatment of RA.
Using peptide/MHC complex as a direct approach toward Ag-driven immunosuppression has been studied previously. It has been demonstrated that partial activation of pathogenic T cells through the Ag-specific TCR without costimulators could induce anergy (34) or apoptosis (33, 35). In the current study, we produced monomeric murine I-Aq-derived RTLs and our results suggested that treatment of CIA with I-Aq-derived RTL linked to the CII peptide not only down-regulated two key proinflammatory cytokines, IL-17 and IFN-γ, but also up-regulated anti-inflammatory cytokines, IL-10 and IL-13, in the spleens of DBA/1LacJ mice. Interestingly, we previously reported that the treatment of EAE with monomeric I-As-derived RTL covalently linked to PLP139–151 peptide also resulted in a switch from proinflammatory to anti-inflammatory cytokines and reduced expression of the adhesion molecules, VLA-4 and LFA-1, in the CNS (40). In both studies, these two different murine monomeric RTLs did not significantly reduce T cell proliferation responses to bCII257–273 or PLP139–151, respectively (data not shown). These data suggest that systemic shifting the cytokine profile may be an important regulatory mechanism of these monomeric RTLs. It may be that reduction in IL-17 and IFN-γ and induction of anti-inflammatory cytokines, IL-10 and IL-13, are of particular importance because these changes were observed only after pre-treatment with the protective RTL2001MII. In contrast, both RTL2001MII and the “empty” RTL2000 reduced expression of the proinflammatory cytokines IL-1β and IL-6, thus suggesting that these changes were not directly related to protection against CIA.
In the joint tissue, treatment with RTL2001MII but not RTL2000 resulted in a significant reduction in the expression of three key proinflammatory cytokines, IL-1β, IL-6, and IL-23. These selective changes thus may have contributed to reduced disease scores and pathology in the joints. In contrast, both RTL2000 and RTL2001MII induced lower expression of TNF-α, although the inhibition of TNF-α by RTL2001MII was much more pronounced than for RTL2000. It is noteworthy that the pattern of inhibition mediated by treatment with RTL2001MII differed systemically vs locally in the joint. This discrepancy could be due to regional differences or perhaps might reflect a disparity between protein secretion (measured in spleen cell cultures stimulated with CII peptide) vs mRNA expression in whole joint tissue.
Recently, numerous publications suggested that the IL-23/IL-17 axis is “evil” in organ-specific autoimmune diseases (17, 53–56). It has been demonstrated that IL-12 primarily acts on naive T cells and plays a critical role in determining the differentiation and generation of Th1 cell populations, whereas IL-23 preferentially acts on memory T cells and is an essential cytokine for the development of organ specific inflammatory autoimmune disease such as EAE (54, 57) and CIA (55, 56). Production of IL-23 is an important pathway that drives a subset of the highly pathogenic CD4+ T cell population (termed the Th17 subset) to produce IL-17A, IL-17F, and TNF-α, but not IFN-γ or IL-4. Both IL-17A and IL-17F are members of the IL-17 family, which are homodimeric cytokines and play an important role in the regulation of autoimmunity (53, 58–60). A recent study showed that IL-17 enhanced osteoclastogenesis by inducing the receptor activator of NF-κB (RANKL) on mesenchymal cells (17). Moreover, both IL-1β and IL-6 have been suggested to promote the development of the Th17 subset (61, 62). Clearly, the reduction in IL-1β, IL-6, and IL-23 could have profound effects on joint inflammation and bone destruction in CIA. These results, in combination with RTL-induced systemic inhibition of IL-17 and IFN-γ and up-regulation of IL-10 and IL-13 suggest that long-term tolerance against arthritogenic Ags may be mediated through a “cytokine switch” mechanism.
Expression of the FoxP3 gene in splenocytes was significantly increased in the RTL2001MII treated group vs the controls or “empty” RTL2000 treated mice. These data suggest that RTL treatment may up-regulate the Treg population, although we were unable to detect an increase in the number of FOXP3+ Treg cells in spleen. Tregs are critical regulators of immune tolerance (63), and their suppressive control of effector T cells was observed in both experimental systems (63) and humans (64, 65). Tregs are defined by their function, and express the transcription factor FoxP3 and/or suppressive cytokines (IL-10 and TGF-β) (66–69). The process for induction of FoxP3 gene expression is still unclear. In vitro studies suggested that the expression of FoxP3 was inducible, and a suboptimal stimulation by anti-CD3/CD28 Abs could activate the expression of FoxP3 (48). Moreover, a recent study demonstrated that de novo generation of Ag-specific CD4+CD25+ regulatory T cells from human CD4+CD25− cells could be achieved through an Ag specific TCR with a cognate peptide/MHC complex (70). We believe that the RTLs may be capable of providing such a suboptimal signal as suggested by our previous study in which the RTLs led to a partial calcium signal and transient T cell signaling (47). We postulate that when a peptide/MHC class II complex interacts with cognate TCR in the absence of costimulators, such engagement could result in a suboptimal signal in the T cell that could trigger FoxP3 expression.
In conclusion, the current study demonstrates that the murine I-Aq-derived RTLs could reduce the incidence of the disease, suppress the clinical and histological signs of CIA and induce long-term clinical benefits. These effects on CIA appeared to be mediated by a cytokine switch mechanism involving a systemic increase of anti-inflammatory factors, IL-10, IL-13, and FoxP3, coupled with both systemic and local reduction of proinflammatory factors. Additionally, RTL treatment resulted in increased IgG1 and decreased IgG2a specific for bCII. This is the first report demonstrating effective treatment of joint inflammation and clinical signs of CIA with I-Aq-derived RTLs, thus supporting its possible clinical use for treating rheumatoid arthritis in humans. We believe that the general strategy developed in this study may have application to other organ specific autoimmune diseases with risk associated MHC class II alleles for which potential T cell target Ags are known or suspected.
Acknowledgments
We thank Eva Niehaus for assistance in preparation of the manuscript.
Footnotes
Disclosures Arthur A. Vandenbark, Halina Offner, Gregory G. Burrows, and Oregon Health and Science University have a significant financial interest in Artielle ImmunoTherapeutics, a company that may have a commercial interest in the result of this research and technology. This potential conflict of interest has been reviewed and managed by the Oregon Health and Science University and Veterans Affairs Medical Center Conflict of Interest in Research Committees.
This work was supported by National Institutes of Health Grants MD001833, AI43960, NS41965, and DK068881, Grant 10255 from the Northwest Health Foundation, and Biomedical Laboratory Research and Development Service, Department of Veterans Affairs.
Abbreviations used in this paper: RA, rheumatoid arthritis; RTL, recombinant T cell receptor ligands; CIA, collagen-induced arthritis; CII, type II collagen; Th17, Th type 17; Tg, transgenic; Treg, T regulatory cell.
References
- 1.Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat. Rev. Drug Discov. 2003;2:473–488. doi: 10.1038/nrd1109. [DOI] [PubMed] [Google Scholar]
- 2.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol. Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 4.Nepom GT. Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv. Immunol. 1998;68:315–332. doi: 10.1016/s0065-2776(08)60563-5. [DOI] [PubMed] [Google Scholar]
- 5.Nepom GT. The role of the DR4 shared epitope in selection and commitment of autoreactive T cells in rheumatoid arthritis. Rheum. Dis. Clin North Am. 2001;27:305–315. doi: 10.1016/s0889-857x(05)70203-9. [DOI] [PubMed] [Google Scholar]
- 6.Roudier J. Association of MHC and rheumatoid arthritis: association of RA with HLA-DR4: the role of repertoire selection. Arthritis Res. 2000;2:217–220. doi: 10.1186/ar91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firestein GS. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J. Clin. Invest. 2004;114:471–474. doi: 10.1172/JCI22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- 9.Kim WU, Cho ML, Jung YO, Min SY, Park SW, Min DJ, Yoon JH, Kim HY. Type II collagen autoimmunity in rheumatoid arthritis. Am. J. Med. Sci. 2004;327:202–211. doi: 10.1097/00000441-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Trentham DE, Dynesius RA, David JR. Passive transfer by cells of type II collagen-induced arthritis in rats. J. Clin. Invest. 1978;62:359–366. doi: 10.1172/JCI109136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmdahl R, Klareskog L, Rubin K, Larsson E, Wigzell H. T lymphocytes in collagen II-induced arthritis in mice: characterization of arthritogenic collagen II-specific T-cell lines and clones. Scand. J. Immunol. 1985;22:295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x. [DOI] [PubMed] [Google Scholar]
- 12.Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J. Exp. Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmdahl R, Jansson L, Larsson A, Jonsson R. Arthritis in DBA/1 mice induced with passively transferred type II collagen immune serum: immunohistopathology and serum levels of anti-type II collagen auto-antibodies. Scand. J. Immunol. 1990;31:147–157. doi: 10.1111/j.1365-3083.1990.tb02754.x. [DOI] [PubMed] [Google Scholar]
- 14.Wooley PH, Luthra HS, Singh SK, Huse AR, Stuart JM, David CS. Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clin. Proc. 1984;59:737–743. doi: 10.1016/s0025-6196(12)65583-9. [DOI] [PubMed] [Google Scholar]
- 15.Thomas R, McIlraith M, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in CD45RBdim mature memory T cells that are potent helpers for B cell differentiation. Arthritis Rheum. 1992;35:1455–1465. doi: 10.1002/art.1780351209. [DOI] [PubMed] [Google Scholar]
- 16.Panayi GS, Corrigall VM, Pitzalis C. Pathogenesis of rheumatoid arthritis: the role of T cells and other beasts. Rheum. Dis. Clin. North Am. 2001;27:317–334. doi: 10.1016/s0889-857x(05)70204-0. [DOI] [PubMed] [Google Scholar]
- 17.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosloniec EF, Whittington KB, He X, Stuart JM, Kang AH. Collagen-induced arthritis mediated by HLA-DR1 (*0101) and HLA-DR4 (*0401) Am J. Med. Sci. 2004;327:169–179. doi: 10.1097/00000441-200404000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin. Immunopathol. 2003;25:3–18. doi: 10.1007/s00281-003-0127-1. [DOI] [PubMed] [Google Scholar]
- 20.Wooley PH. Immunotherapy in collagen-induced arthritis: past, present, and future. Am. J. Med. Sci. 2004;327:217–226. doi: 10.1097/00000441-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Holmdahl R, Bockermann R, Backlund J, Yamada H. The molecular pathogenesis of collagen-induced arthritis in mice: a model for rheumatoid arthritis. Ageing Res. Rev. 2002;1:135–147. doi: 10.1016/s0047-6374(01)00371-2. [DOI] [PubMed] [Google Scholar]
- 22.Myers LK, Miyahara H, Terato K, Seyer JM, Stuart JM, Kang AH. Collagen-induced arthritis in B10.RIII mice (H-2r): identification of an arthritogenic T-cell determinant. Immunology. 1995;84:509–513. [PMC free article] [PubMed] [Google Scholar]
- 23.Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen-induced arthritis in mice, I: major histocompatibility complex (I region) linkage and antibody correlates. J. Exp. Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 25.Winchester R, Dwyer E, Rose S. The genetic basis of rheumatoid arthritis: the shared epitope hypothesis. Rheum. Dis. Clin. North Am. 1992;18:761–783. [PubMed] [Google Scholar]
- 26.Michaelsson E, Andersson M, Engstrom A, Holmdahl R. Identification of an immunodominant type-II collagen peptide recognized by T cells in H-2q mice: self tolerance at the level of determinant selection. Eur. J. Immunol. 1992;22:1819–1825. doi: 10.1002/eji.1830220722. [DOI] [PubMed] [Google Scholar]
- 27.Brand DD, Myers LK, Terato K, Whittington KB, Stuart JM, Kang AH, Rosloniec EF. Characterization of the T cell determinants in the induction of autoimmune arthritis by bovine α 1(II)-CB11 in H-2q mice. J. Immunol. 1994;152:3088–3097. [PubMed] [Google Scholar]
- 28.Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, Woods A, Altmann DM, Stuart JM, Kang AH. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosloniec EF, Brand DD, Myers LK, Esaki Y, Whittington KB, Zaller DM, Woods A, Stuart JM, Kang AH. Induction of autoimmune arthritis in HLA-DR4 (DRB1*0401) transgenic mice by immunization with human and bovine type II collagen. J. Immunol. 1998;160:2573–2578. [PubMed] [Google Scholar]
- 30.Rosloniec EF, Whittington KB, Zaller DM, Kang AH. HLA-DR1 (DRB1*0101) and DR4 (DRB1*0401) use the same anchor residues for binding an immunodominant peptide derived from human type II collagen. J. Immunol. 2002;168:253–259. doi: 10.4049/jimmunol.168.1.253. [DOI] [PubMed] [Google Scholar]
- 31.Malmstrom V, Backlund J, Jansson L, Kihlberg J, Holmdahl R. T cells that are naturally tolerant to cartilage-derived type II collagen are involved in the development of collagen-induced arthritis. Arthritis Res. 2000;2:315–326. doi: 10.1186/ar106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corthay A, Backlund J, Holmdahl R. Role of glycopeptide-specific T cells in collagen-induced arthritis: an example how post-translational modification of proteins may be involved in autoimmune disease. Ann. Med. 2001;33:456–465. doi: 10.3109/07853890109002094. [DOI] [PubMed] [Google Scholar]
- 33.Rhode PR, Burkhardt M, Jiao J, Siddiqui AH, Huang GP, Wong HC. Single-chain MHC class II molecules induce T cell activation and apoptosis. J. Immunol. 1996;157:4885–4891. [PubMed] [Google Scholar]
- 34.Appel H, Seth NP, Gauthier L, Wucherpfennig KW. Anergy induction by dimeric TCR ligands. J. Immunol. 2001;166:5279–5285. doi: 10.4049/jimmunol.166.8.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nag B, Kendrick T, Arimilli S, Yu SC, Sriram S. Soluble MHC II-peptide complexes induce antigen-specific apoptosis in T cells. Cell. Immunol. 1996;170:25–33. doi: 10.1006/cimm.1996.0130. [DOI] [PubMed] [Google Scholar]
- 36.Burrows GG, Bebo BF, Jr., Adlard KL, Vandenbark AA, Offner H. Two-domain MHC class II molecules form stable complexes with myelin basic protein 69–89 peptide that detect and inhibit rat encephalitogenic T cells and treat experimental autoimmune encephalomyelitis. J. Immunol. 1998;161:5987–5996. [PubMed] [Google Scholar]
- 37.Burrows GG, Chang JW, Bachinger HP, Bourdette DN, Offner H, Vandenbark AA. Design, engineering, and production of functional single-chain T cell receptor ligands. Protein Eng. 1999;12:771–778. doi: 10.1093/protein/12.9.771. [DOI] [PubMed] [Google Scholar]
- 38.Chang JW, Mechling DE, Bachinger HP, Burrows GG. Design, engineering, and production of human recombinant T cell receptor ligands derived from human leukocyte antigen DR2. J. Biol. Chem. 2001;276:24170–24176. doi: 10.1074/jbc.M101808200. [DOI] [PubMed] [Google Scholar]
- 39.Huan J, Meza-Romero R, Mooney JL, Chou Y, Edwards DM, Rich C, Link JM, Vandenbark AA, Bourdette DN, Bachinger H-P, Burrows GG. Rationally designed mutations convert complexes of human recombinant T cell receptor ligands into monomers that retain biological activity. J. Chem. Tech. Biotech. 2005;80:2–12. doi: 10.1002/jctb.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huan J, Subramanian S, Jones R, Rich C, Link J, Mooney J, Bourdette DN, Vandenbark AA, Burrows GG, Offner H. Monomeric recombinant TCR ligand reduces relapse rate and severity of experimental autoimmune encephalomyelitis in SJL/J mice through cytokine switch. J. Immunol. 2004;172:4556–4566. doi: 10.4049/jimmunol.172.7.4556. [DOI] [PubMed] [Google Scholar]
- 41.Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, Fugger L, Offner H, Jones R, Burrows GG. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35–55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J. Immunol. 2003;171:127–133. doi: 10.4049/jimmunol.171.1.127. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian S, Tovey M, Afentoulis M, Krogstad A, Vandenbark AA, Offner H. Ethinyl estradiol treats collagen-induced arthritis in DBA/1LacJ mice by inhibiting the production of TNF-α and IL-1β. Clin. Immunol. 2005;115:162–172. doi: 10.1016/j.clim.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Leung BP, Sattar N, Crilly A, Prach M, McCarey DW, Payne H, Madhok R, Campbell C, Gracie JA, Liew FY, McInnes IB. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J. Immunol. 2003;170:1524–1530. doi: 10.4049/jimmunol.170.3.1524. [DOI] [PubMed] [Google Scholar]
- 44.Burrows GG, Chou YK, Wang C, Chang JW, Finn TP, Culbertson NE, Kim J, Bourdette DN, Lewinsohn DA, Lewinsohn DM, et al. Rudimentary TCR signaling triggers default IL-10 secretion by human Th1 cells. J. Immunol. 2001;167:4386–4395. doi: 10.4049/jimmunol.167.8.4386. [DOI] [PubMed] [Google Scholar]
- 45.Dzhambazov B, Nandakumar KS, Kihlberg J, Fugger L, Holmdahl R, Vestberg M. Therapeutic vaccination of active arthritis with a glycosylated collagen type II peptide in complex with MHC class II molecules. J. Immunol. 2006;176:1525–1533. doi: 10.4049/jimmunol.176.3.1525. [DOI] [PubMed] [Google Scholar]
- 46.Offner H, Subramanian S, Wang C, Afentoulis M, Vandenbark AA, Huan J, Burrows GG. Treatment of passive experimental autoimmune encephalomyelitis in SJL mice with a recombinant TCR ligand induces IL-13 and prevents axonal injury. J. Immunol. 2005;175:4103–4111. doi: 10.4049/jimmunol.175.6.4103. [DOI] [PubMed] [Google Scholar]
- 47.Wang C, Mooney JL, Meza-Romero R, Chou YK, Huan J, Vandenbark AA, Offner H, Burrows GG. Recombinant TCR ligand induces early TCR signaling and a unique pattern of downstream activation. J. Immunol. 2003;171:1934–1940. doi: 10.4049/jimmunol.171.4.1934. [DOI] [PubMed] [Google Scholar]
- 48.Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. J. Clin. Invest. 2003;112:1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosloniec EF, Whittington KB, Brand DD, Myers LK, Stuart JM. Identification of MHC class II and TCR binding residues in the type II collagen immunodominant determinant mediating collagen-induced arthritis. Cell. Immunol. 1996;172:21–28. doi: 10.1006/cimm.1996.0210. [DOI] [PubMed] [Google Scholar]
- 50.Kjellen P, Brunsberg U, Broddefalk J, Hansen B, Vestberg M, Ivarsson I, Engstrom A, Svejgaard A, Kihlberg J, Fugger L, Holmdahl R. The structural basis of MHC control of collagen-induced arthritis: binding of the immunodominant type II collagen 256–270 glycopeptide to H-2Aq and H-2Ap molecules. Eur. J. Immunol. 1998;28:755–767. doi: 10.1002/(SICI)1521-4141(199802)28:02<755::AID-IMMU755>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 51.Backlund J, Treschow A, Bockermann R, Holm B, Holm L, Issazadeh-Navikas S, Kihlberg J, Holmdahl R. Glycosylation of type II collagen is of major importance for T cell tolerance and pathology in collagen-induced arthritis. Eur. J. Immunol. 2002;32:3776–3784. doi: 10.1002/1521-4141(200212)32:12<3776::AID-IMMU3776>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 52.Huseby ES, Crawford F, White J, Marrack P, Kappler JW. Interface-disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat. Immunol. 2006;7:1191–1199. doi: 10.1038/ni1401. [DOI] [PubMed] [Google Scholar]
- 53.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J. Clin. Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 55.Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, Jin HT, Min SY, Ju JH, Park KS, et al. STAT3 and NF-κB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 2006;176:5652–5661. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 56.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 60.Hoeve MA, Savage ND, de Boer T, Langenberg DM, de Waal Malefyt R, Ottenhoff TH, Verreck FA. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur. J. Immunol. 2006;36:661–670. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 61.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 63.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset, I: evidence for the active participation of T cells in natural self-tolerance, deficit of a T cell subset as a possible cause of autoimmune disease. J. Exp. Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shevach EM. Certified professionals: CD4+CD25+ suppressor T cells. J. Exp. Med. 2001;193:F41–F46. doi: 10.1084/jem.193.11.f41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, Thunberg S, Deniz G, Valenta R, Fiebig H, et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J. Exp. Med. 2004;199:1567–1575. doi: 10.1084/jem.20032058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 67.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 69.Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, et al. Functional impairment of CD8+ T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20:293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- 70.Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH. De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+CD25− cells. Proc. Natl. Acad. Sci. USA. 2005;102:4103–4108. doi: 10.1073/pnas.0407691102. [DOI] [PMC free article] [PubMed] [Google Scholar]