

During our studies on aromatic borylation,[1] we considered the combination of a highly electrophilic R2BNTf2 reagent with a base that would neutralize the HNTf2 byproduct of borylation without deactivating the electrophile. In principle, these requirements might be satisfied by 1,8-bis(dimethylamino)naphthalene (1), a hindered and exceptionally basic aniline that finds numerous applications as a basic catalyst or reagent due to its legendary lack of nucleophilicity.[2, 3] Strong electrophiles interact weakly, if at all, with the amine nitrogens, and very few examples are known where stable bonds to nitrogen can be formed between 1 and electrophilic groups larger than hydrogen.[2, 4-7] Among these exceptional cases, cyclic boronium structures 2 and 3 are relatively stable because the subunits BH2 and BF2 have minimal steric requirements.[5] However, the more hindered BMe2 derivative 4 has not been detected and no analogous BR2 structures are known.[5a, 8] In view of this long history, we were somewhat surprised to find that an adduct is readily formed simply upon mixing 1 with the 9-BBN bistriflimide reagent 5a despite the transannular steric demands of the 9-BBN core and the need to form adjacent quaternary bonds to boron as well as nitrogen.[9, 10] The remarkable structural features and unusual reactivity of this adduct are the subject of the following communication.
The previously unreported 5a was easily prepared from the commercially available 9-BBN dimer and bis(trifluoromethanesulfonyl)imide upon heating in toluene. Combination of the bulky boron reagent 5a with a stoichiometric amount of 1 in CD2Cl2 at room temperature formed a deep red solution that turned colorless within seconds of mixing the reagents. Analysis of the resulting solution by 11B NMR spectroscopy revealed a signal at δ 16.2 ppm, suggesting that a single tetracoordinate[11] boron atom is present in the product. The 19F NMR spectrum showed a single peak at δ -79.4 ppm, which is characteristic of bistriflimide anion,[12] so the boron-containing fragment was thus identified to be a cation. The 1H NMR spectrum suggested that the solution structure of the cation is highly symmetrical on the NMR timescale at room temperature. Only four groups of protons corresponding to the diamine subunit 1 were observed, including one sharp singlet for all four methyl groups, and a well-resolved (at 500 MHz) AMX system for the aromatic protons. Other peaks in the 1H and 13C NMR spectra were also consistent with a symmetrical time-averaged structure for the cation (for example, a single 13C methyl peak at δ 57.1 ppm, and only 3 peaks for the 9-BBN cage carbons). Since the covalent adduct 6 (X = NTf2) is ruled by observation of the bistriflimide anion,[12b] and the tricoordinate cationic borenium structure 7 (X = NTf2) is not consistent with the observed 11B NMR chemical shift, the most plausible structure for the species formed from 1 and 5a is the exceptionally hindered boronium salt 8a. Formation of 8a may follow the logical sequence in Scheme 1, but alternative mechanisms have not been excluded.[13] Formation of >95% 8a depends on the low nucleophilicity of the counterion, and only partial conversion to the boronium ion was observed when 5b was used instead of 5a. Thus, when equimolar 1 and the triflate reagent 5b were mixed in CD2Cl2, the resulting solution showed both the starting 1 and the product 8b (ca. 1.2:1 ratio by 1H NMR assay).
Scheme 1.
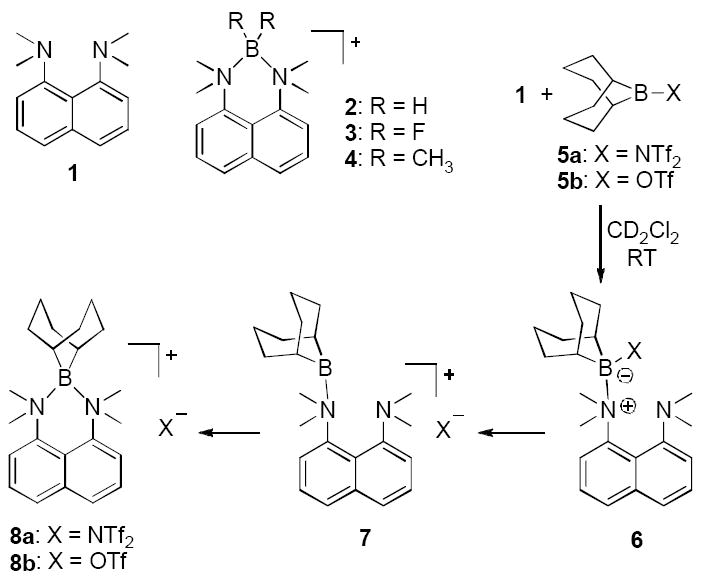
The low temperature 1H NMR behavior of 8a in CD2Cl2 is complex and indicates the presence of unsymmetrical species. Decreasing the temperature broadens the 1H NMR singlet corresponding to the N-methyl groups, until it turns into a broad set of at least three maxima that are not fully resolved even at -80°C. Additional information about the solution structure is provided by the very different shielding of the two bridgehead hydrogen atoms of the 9-BBN cage observed in the low temperature 1H NMR spectra. While the spectrum taken at room temperature shows only one peak for both bridgehead protons, two distinct resonances are observed at -80 °C. One of the bridgehead hydrogens gives rise to a peak at an unremarkable δ 1.57 ppm, while the other hydrogen appears at δ 0.33 ppm. Such prominent shielding by the aromatic ring is consistent with the 9-BBN cage being tilted toward one side of the naphthalene plane to place the shielded proton above the aromatic π-system.
Slow cooling of the solution of 8a in a mixture of CH2Cl2 and hexanes produced large platelike crystals, suitable for X-ray diffraction studies.[14] Due to strain imposed by the hindered environment, the structure of 8a is non-symmetrical, the B-N bonds are very long, and both the 9-BBN cage and the bis(dimethylamino)naphthalene unit are severely twisted. While the C15-B1-C19 angle (106°) and C-B bond lengths (1.63 Å, B1-C15; 1.61 Å, B1-C19) are within the expected range for 9-BBN derivatives,[9a, 15] the C20-C19-C15-C22 and C18-C19-C15-C16 dihedral angles are in excess of 10°. Furthermore, strong distortion of the diamine base is evidenced by the N2-C9-C1-N1 dihedral angle of 28.9°, compared to 20.3° in 1.[16] On the other hand, the aniline N⋯N distance in 8a (2.65 Å) is substantially shorter than that reported for 1 (2.79 Å), and only slightly exceeds that in salts of protonated 1, such as the sulfonimide salt 1·HNMs2 (2.60 Å).[17]
The arrangement of the 9-BBN cage in crystals of 8a is also noteworthy. The bridgehead carbons C15 and C19 are quite distinct, and C15 is pseudo-axial with respect to the distorted half-chair boron heterocycle. This places C15-H above the aromatic π-system, consistent with the low temperature 1H NMR result indicating substantial shielding of one of the bridgehead protons. Another prominent structural detail is the length of the B-N bonds (B1-N1 1.72 Å; B1-N2 1.73 Å), compared to values of 1.58-1.60 Å in simpler boronium cations such as [H2B(NMe3)(MeIm)]+ or [H2B(NH2Me)(MeIm)]+ (MeIm = 1-methylimidazole).[18,19] Since the 1.72-1.73 Å distance greatly exceeds the sum of covalent radii for B and N atoms (1.55 Å),[20] the calculated Pauling bond order for both B-N bonds in 8a is only ca. 0.55.[21]
The unusual structural features prompted computational modeling of the cation 8a.[22a] Gas phase geometry optimization at the M06-2X/6-31G(d,p) level produced a structure that is in close agreement with the X-ray data (for example, B-N bond lengths are within 0.01 Å of the experimental values).[22b] NBO analysis[22c] performed on the optimized structure indicates that the boron atom carries the bulk of the positive charge (NBO charge 0.97), and Wiberg bond orders of the two B-N bonds are 0.52 and 0.53.
It was also of interest to compare the interactions of other amines with the potent Lewis acid 5a in solution. When triethylamine was combined with 5a in CD2Cl2, clean formation of the borenium ion 10 was observed (Scheme 2). No evidence for a boronium structure 11 was detected, even when excess triethylamine was used. The borenium character of 10 is substantiated by a strongly deshielded 11B NMR peak at δ 85.1 ppm, as well as the bistriflimide anion peak at δ -79.5 ppm in the 19F NMR spectrum,[12a] evidence that rules out the alternative structure 9. For simplicity, 9 is tentatively shown as a precursor of 10, although direct conversion from 5a is not ruled out.
Scheme 2.
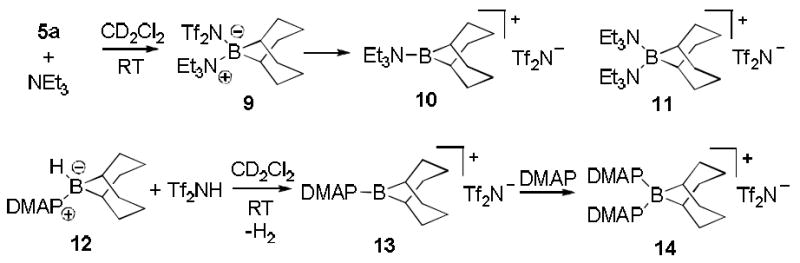
In contrast to triethylamine, 4-(dimethylamino)pyridine (DMAP) reacted with a stoichiometric amount of 5a to afford mostly the isolable boronium cation 14 according to the 11B NMR shift of δ 3.0 ppm (CD2Cl2, rt) along with traces of the borenium cation 13 (δ 66.5 ppm).[23] A much better way to generate 13 in situ was to protonate the amine borane complex 12 with Tf2NH. This method confirmed the chemical shift of 13 and afforded solutions also containing relatively minor amounts of the boronium salt 14 (ca. 7-11:1 13:14). However, the more hindered 2,6-di-tert-butyl-4-methylpyridine did not interact with 5a at room temperature according to 1H and 11B NMR assay.
The most remarkable feature of the borenium salt 10 is the absence of any stabilizing π-donor or n-donor substituents at boron, in contrast to 13 and to all previously reported persistent borenium ions generated in the condensed phase.[24, 25] A comparison of 11B NMR shifts for 10 (δ 85.1 ppm) and 13 (δ 66.5 ppm) indicates extensive cation stabilization by π delocalization between DMAP and the boron atom. Other π-stabilized borenium cations have been observed in the 11B chemical shift range of δ 58.2 to 66 ppm,[1, 24, 25] suggesting that 10 may be an exceptionally electrophilic member of the borenium family of structures.
High electrophilicity of boron cations is crucial for potential applications in electrophilic aromatic borylation.[1, 26] Thus, different combinations of the bistriflimide 5a with basic amines generated reagents that react with electron-rich heterocycles to provide B-heteroaryl 9-BBN derivatives along with the protonated amines. The reagent consisting of 5a and the non-complexing 2,6-di-tert-butyl-4-methylpyridine was the most reactive, and borylated N-methylindole in seconds at room temperature to afford 16a (>95% conversion by NMR spectroscopy), while the cationic reagents 8a and 10 required several hours at 50 °C for similar conversion. No added base was needed with 8a or 10 because both reagents already contain a “built-in” base (proton sponge 1 and triethylamine, respectively) to neutralize the HNTf2 that forms during borylation. On the other hand, neither 13 nor 14 reacted with N-methylindole under these conditions. [27]
While the boronium salt 8a is less potent than the reagent from 5a and 2,6-di-tert-butyl-4-methylpyridine, 8a is a far more convenient borylating agent. Practical access to 8a on gram scale is possible using a one-pot procedure from 9-BBN, 1, and HNTf2 without having to isolate the highly sensitive 5a (see Supporting Information). Crystallized 8a is much easier to handle compared to 5a, and even survives up to a month of exposure to dry air (dessicator over Drierite), in contrast to 5a or 10. Furthermore, the aromatic borylation products obtained using 8a are easy to isolate (Table 1). Crystalline products 16a-d were obtained in high purity simply by extracting the reaction mixtures with hexanes, where neither the unreacted 8a nor the byproduct 1·HNTf2 is soluble, followed by solvent evaporation. This procedure minimizes the risk of competing protodeboronation using 8a, but it is not feasible with the reagent from 5a and 2,6-di-tert-butyl-4-methylpyridine due to differences in reagent solubility.
Table 1.
Borylation of nitrogen heterocycles using 8a.[a]
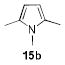
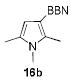
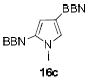
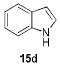
1.05 equiv of 8a; CH2Cl2; 50 °C.
2.10 equiv of 8a
The structures of boranes 16a-d were established by multinuclear NMR spectroscopy, as well as X-ray crystallography in the case of 16a.[28] Borylation of N-methylindole afforded exclusively the 3-substituted regioisomer, in sharp contrast to the previously reported reaction with B(C6F5)3, which produces the 2-borylated N-methylindole.[29] Pyrrole 15c gave a mixture of mono-borylated regioisomers along with some of the diborylated 16c using one equivalent of 8a (ca. 80% conversion of 15c), but two equivalents of the borylating agent cleanly produced the diborylated pyrrole 16c. In the reaction with unsubstituted indole, the known N-borylation product was produced first,[30] followed by much slower C3-borylation to afford 16d.
Several reactions of 8a suggest that it is in equilibrium with the starting 1 and 5a. Thus, equimolar 8a and Tf2NH produced the protonated diamine (1·HNTf2) and released 5a (NMR assay). Furthermore, reaction of 8a with triethylammonium bistriflimide (Et3NH+ Tf2N−) yielded 1·HNTf2 and the tricoordinate cation 10, representing an unusual route from boronium to borenium ions involving the formal migration of the 9-BBN fragment to a different amine. These events can be understood if dissociation of 8a to 1 + 5a is the first step. The same dissociative mechanism may also help explain the borylations of Table 1, although the identity of the key boron electrophile is not clear. Depending on the timing of bond dissociation and borylation events, a role for the tricoordinate borenium ion 7[1] or even a dicoordinate borinium ion[26a] cannot be ruled out at this point. The equilibrium between 8a and 1 + 5a is not directly observable by 1H NMR spectroscopy, but the analogous process does occur in the related system 8b and 1 + 5b, containing the more nucleophilic triflate anion (vide supra).
To summarize, the covalent boron bistriflimide 5a was used to access the unusual boron salts 8a and 10 by exploiting the excellent leaving group ability of bistriflimide anion. The triethylamine-derived 10 expands the range of borenium salts observed in the condensed phase, proving that resonance delocalization of the positive charge is not required for a persistent borenium cation. The hindered boronium salt 8a cautions against interpreting the name “proton sponge” too literally: in fact, this study proves that 1 can act as a chelating ligand for species much larger than a proton. Also, the unusual structure of 8a raises a rhetorical question: is there a distinct boundary between the cationic species called “boronium” (tetra-substituted B), “borenium” (tri-substituted B), and “borinium” (di-substituted B), according to Nöth’s terminology?[24] While 8a should be most appropriately called a boronium salt, the long B-N distances increase the “borinium-like” character, and the unusual reactivity adds a small hint of a borenium ion (7). Aside from the structural features of 8a, the chemoselectivity of its formation also deserves attention. In view of earlier reports that strong electrophiles attack the aromatic system of 1[3] or abstract hydride from one of the N-methyl groups,[3a, 31] it is quite intriguing that the reaction between 1 and 5a proceeds to form 8a, the most hindered of all plausible products. As evidenced by the electrophilic borylations (Table 1), the steric hindrance of 8a is responsible for extraordinary reactivity compared to less hindered boronium salts such as 14.[32]
Supplementary Material
Figure 1.
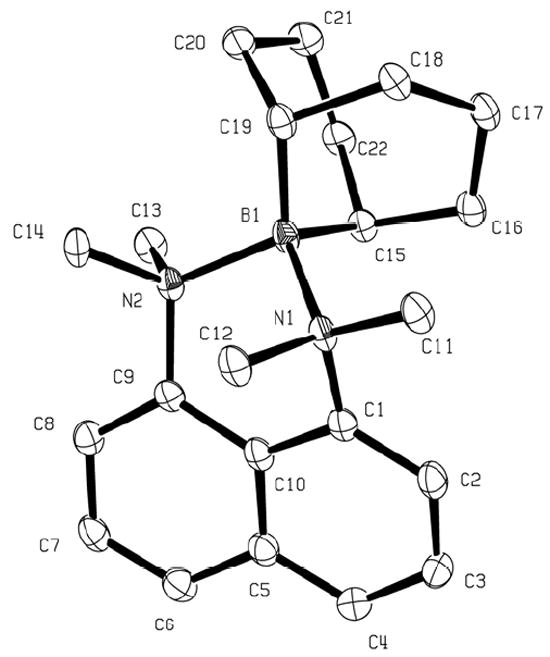
ORTEP drawing of 8a (50% probability thermal ellipsoids). The counterion and hydrogen atoms are omitted for clarity.
Footnotes
This work was supported by the Institute of General Medical Sciences, NIH (GM067146)
Supporting information for this article is available on the WWW under http://www.angewandte.org.
References
- 1.De Vries TS, Prokofjevs A, Harvey JN, Vedejs E. J Am Chem Soc. 2009;131:14679–14687. doi: 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alder RW, Bowman PS, Steele WRS, Winterman DR. Chem Commun (London) 1968:723–724. [Google Scholar]
- 3.Reviews: Barner BA, Faler CA, Joullié MM. 1,8-Bis(dimethylamino)naphthalene e-EROS Encyclopedia of Reagents for Organic Synthesis. doi: 10.1002/047084289X.rb144.pub2., and references therein; Pozharskii AF, Ozeryanskii VA. In: The Chemistry of Anilines, Part 1. Rappoport Z, editor. John Wiley & Sons Ltd.; Chichester: 2007. pp. 931–1026.
- 4.N-Monomethylation of 1,8-bis(dimethylamino)naphthalene: Alder RW, Goode NC. J Chem Soc Chem Commun. 1976:108–109.
- 5.a) Axtell DD, Cambell AC, Keller PC, Rund JV. J Coord Chem. 1976;5:129–134. [Google Scholar]; b) Onak T, Rosendo H, Siwapinyoyos G, Kubo R, Liauw L. Inorg Chem. 1979;18:2943–2945. [Google Scholar]; c) Keller PC, Rund JV. Inorg Chem. 1979;18:3197–3199. [Google Scholar]; d) Hartman JS, Shoemaker JAW. Polyhedron. 2000;19:165–176. [Google Scholar]
- 6.Only one transition metal complex of 1,8-bis(dimethylamino)-naphthalene has been structurally characterized: Yamasaki T, Ozaki N, Saika Y, Ohta K, Goboh K, Nakamura F, Hashimoto M, Okeya S. Chem Lett. 2004;33:928–929.
- 7.1,8-Bis(dimethylamino)naphthalene as a proposed ligand for metals: Lucht BL, Bernstein MP, Remenar JF, Collum DB. J Am Chem Soc. 1996;118:10707–10718.; Farzaneh F, Majidian M, Ghandi M. J Mol Catal A: Chem. 1999;148:227–233.; Collman JP, Zhong M, Zhang C, Costanzo S. J Org Chem. 2001;66:7892–7897. doi: 10.1021/jo010615u.
- 8.The BCl2 analog has been claimed, but definitive evidence has not been reported (see reference 5a).
- 9.For 9-BBN-derived boronium salts containing relatively non-hindered pyridine ligands, see: Ma K, Bats JW, Wagner M. Acta Crystallogr Sect E: Struct Rep Online. 2001;E57:o846–o848.; Köster R, Grassberger MA. Justus Liebigs Ann Chem. 1968;719:169–186.; Hünig S, Wehner I. Heterocycles. 1989;28:359–363.
- 10.Cyclic aminoboranes derived from 1,8-diaminonaphthalene and 9-BBN: Bar-Haim G, Kol M. J Org Chem. 1997;62:6682–6683.; Bar-Haim G, Shach R, Kol M. Chem Commun. 1997:229–230.
- 11.Nöth H, Wrackmeyer B. Nuclear Magnetic Resonance Spectroscopy of Boron Compounds. Springer; Berlin: 1978. [Google Scholar]
- 12.Tf2N− salts: Arvai R, Toulgoat F, Langlois BR, Sanchez J-Y, Médebielle M. Tetrahedron. 2009;65:5361–5368.; b) A single 19F peak at δ -79.8 ppm is observed at -80 °C vs. δ -79.4 ppm at rt, typical of Tf2N− anion. Covalent bistriflimide 5a gives a signal at δ -70.0 ppm.
- 13.Alternatives to consider include an electron transfer process, a process involving borinium intermediates, or a direct displacement mechanism from 5a and 1 to 8a via a transition state that resembles 7.
- 14.See Supporting Information for X-ray crystallography details. CCDC-791459 contains the supplementary crystallographic data for 8a. The cif file can be obtained free of charge from Cambridge Crystallographic Data Center (http://www.ccdc.cam.ac.uk); b) The counterion displays a number of close contacts to the cation, the shortest being the distance between a bistriflimide oxygen and an N-methyl hydrogen (2.49Å).
- 15.Yalpani M, Boese R, Köster R. Chem Ber. 1990;123:1275–1283. [Google Scholar]; Brock CP, Fu Y, Niedenzu K, Nöth H. Main Group Met Chem. 1992;15:53–60. [Google Scholar]; Wrackmeyer B, Schwarze B, Milius W. J Organomet Chem. 1995;489:201–205. [Google Scholar]
- 16.Einspahr H, Robert J-B, Marsh RE, Roberts JD. Acta Crystallogr Sect B: Struct Crystallogr Cryst Chem. 1973;29:1611–1617. [Google Scholar]
- 17.Henschel D, Moers O, Lange I, Blaschette A, Jones PG. Z Naturforsch, B: J Chem Sci. 2002;57b:777–790. [Google Scholar]
- 18.Fox PA, Griffin ST, Reichert WM, Salter EA, Smith AB, Tickell MD, Wicker BF, Cioffi EA, Davis JH, Jr, Rogers RD, Wierzbicki A. Chem Commun. 2005:3679–3681. doi: 10.1039/b504631a. [DOI] [PubMed] [Google Scholar]; Soutullo MD, Odom CI, Smith AB, McCreary DR, Sykora RE, Salter EA, Wierzbicki A, Davis JH., Jr Inorg Chim Acta. 2007;360:3099–3102. [Google Scholar]
- 19.A B-N bond distance of 1.61 Å has been reported for a 9-BBN-derived boronium ion with bipyridyl as the nitrogen ligand (ref. 9a).
- 20.Cordero B, Gómez V, Platero-Prats AE, Revés M, Echeverría J, Cremades E, Barragán F, Alvarez S. Dalton Trans. 2008:2832–2838. doi: 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- 21.Pauling L. J Am Chem Soc. 1947;69:542–553. [Google Scholar]
- 22.a) Gaussian 09, Revision A.02 suite of computational programs was used. See Supporting Information for full details; Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241.; NBO Version 3.1, Glendening ED, Reed AE, Carpenter JE, Weinhold F.
- 23.Narula CK, Nöth H. Inorg Chem. 1985;24:2532–2539. [Google Scholar]
- 24.a) Piers WE, Bourke SC, Conroy KD. Angew Chem Int Ed. 2005;44:5016–5036. doi: 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]; b) Kölle P, Nöth H. Chem Rev. 1985;85:399–418. [Google Scholar]
- 25.Reports of π-stabilized borenium cations published after ref. 24a: Uddin MK, Nagano Y, Fujiyama R, Kiyooka S, Fujio M, Tsuno Y. Tetrahedron Lett. 2005;46:627–630.; Chiu C, Gabbaï FP. Organometallics. 2008;27:1657–1659.; Matsumoto T, Gabbaï FP. Organometallics. 2009;28:4252–4253.; Other recently reported borenium ions: Kato T, Tham FS, Boyd PDW, Reed CA. Heteroatom Chem. 2006;17:209–216.; Bonnier C, Piers WE, Parvez M, Sorensen TS. Chem Commun. 2008:4593–4595. doi: 10.1039/b808739c.; Dureen MA, Lough A, Gilbert TM, Stephan DW. Chem Commun. 2008:4303–4305. doi: 10.1039/b808348g.; Ott H, Matthes C, Ringe A, Magull J, Stalke D, Klingebiel U. Chem Eur J. 2009;15:4602–4609. doi: 10.1002/chem.200802669.; see also ref. 26a.
- 26.Del Grosso A, Pritchard RG, Muryn CA, Ingleson MJ. Organometallics. 2010;29:241–249.. For a review of transition metal catalyzed borylation, see I A, Mkhalid I, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p.
- 27.The reagent from 5a + 2,6-di-tert-butyl-4-methylpyridine (DTBMP) did not borylate toluene or benzofuran at rt, but N,N-diethylaniline reacted slowly with 5a + DTBMP at rt, or with 8a at 50 °C.
- 28.See Supporting Information for X-ray crystallography details. CCDC-791460 contains the supplementary crystallographic data for 16a. The cif file can be obtained free of charge from Cambridge Crystallographic Data Center (http://www.ccdc.cam.ac.uk).
- 29.Focante F, Camurati I, Nanni D, Leardini R, Resconi L. Organometallics. 2004;23:5135–5141. [Google Scholar]
- 30.Wrackmeyer B, Maisel HE, Schwarze B, Milius W, Köster R. J Organomet Chem. 1997;541:97–107. [Google Scholar]; Wrackmeyer B, Schwarze B. J Organomet Chem. 1997;534:207–211. [Google Scholar]
-
31.A cationic product i has been reported from treatment of 1 with the potent electrophile B(C6F5)3: Di Saverio A, Focante F, Camurati I, Resconi L, Beringhelli T, D’Alfonso G, Donghi D, Maggioni D, Mercandelli P, Sironi A. Inorg Chem. 2005;44:5030–5041. doi: 10.1021/ic0502168..
- 32.Boronium salt 14 is more stable than 8a, but more reactive than unhindered analogues (ref. 24b): methanolysis of 14 or 8a occurs to ca. 90% conversion within ca. 14 h at 50 °C or 10 min at rt, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.