

Abstract
Background
Major basic protein released from eosinophils to airway parasympathetic nerves blocks inhibitory M2 muscarinic receptors on the parasympathetic nerves, increasing acetylcholine release and potentiating reflex bronchoconstriction. Recruitment of eosinophils to airway parasympathetic neurons requires neural expression of both intercellular adhesion molecular-1 (ICAM-1) and eotaxin. We have shown that inflammatory cytokines induce eotaxin and ICAM-1 expression in parasympathetic neurons.
Objective
To test whether the β2 agonist albuterol, which is used to treat asthma, changes TNF-alpha-induced eotaxin and ICAM-1 expression in human parasympathetic neurons.
Methods
Parasympathetic neurons were isolated from human tracheas and grown in serum-free medium for one week. Cells were incubated with either (R)-albuterol (the active isomer), (S)-albuterol (the inactive isomer) or (R,S)-albuterol for 90 minutes before adding 2 ng/ml TNF-alpha for another 4 hours (for mRNA) or 24 hours (for protein).
Results and Conclusions
Baseline expression of eotaxin and ICAM-1 were not changed by any isomer of albuterol as measured by real time RT-PCR. TNF-alpha induced ICAM-1 expression was significantly inhibited by (R)-albuterol in a dose dependent manner, but not by (S) or (R,S)-albuterol. Eotaxin expression was not changed by TNF-alpha or by any isomer of albuterol. The β-receptor antagonist propranolol blocked the inhibitory effect of (R)-albuterol on TNF-alpha-induced ICAM-1 expression.
Clinical Implication
The suppressive effect of (R)-albuterol on neural ICAM-1 expression may be an additional mechanism for decreasing bronchoconstriction, since it would decrease eosinophil recruitment to the airway nerves.
Introduction
Eosinophils are in contact with airway nerves in patients with asthma and in antigen challenged animals [1], [2]. Migration and binding of eosinophils to the nerve are mediated by chemotactic factors and adhesion molecules [3], [4], [5], [6], [7], [8], [9], including eotaxin and intercellular adhesion molecule 1 (ICAM-1). Eotaxin selectively recruits eosinophils via CCR3 (C–C chemokine receptor 3) expressed on eosinophils. ICAM-1 is important for eosinophil adhesion via LFA-1, a receptor found on eosinophils. Both eotaxin and ICAM-1 are present on airway nerves in antigen-challenged guinea pigs and on cultured airway parasympathetic neurons [5], [7]. Both can be induced by inflammatory cytokines [5], [7], [10], [11]. Reducing ICAM-1 or blocking eotaxin expression on parasympathetic nerves relates to reduced parasympathetic nerves associated eosinophils, and reduced airway hyperreactivity [5], [7], [8]. Thus, controlling expression of eotaxin and ICAM-1 on airway parasympathetic nerves is critical for reducing neural inflammation and preventing airway hyperreactivity.
The short-acting β2-adrenergic bronchodilator albuterol is commonly administered to patients in racemic form, containing equal parts of its active isomer (R)- and its inactive isomer (S)-albuterol. It has been argued that (R)-albuterol (often known as levalbuterol) is more effective than the racemic (R, S)-albuterol mixture. Clinical studies demonstrate that greater clinical efficacy is achieved when (R)-albuterol is given in amounts equivalent to that found in the racemic albuterol and that (R)-albuterol is also associated with fewer side effects [12], [13]. The mechanism underlying the difference between (R)- and (R, S)- albuterol remains unclear. Since the expression of eotaxin and ICAM-1 on airway parasympathetic nerves are critical for neural inflammation, we tested the effect of (R,S)-albuterol, (R)-albuterol and (S)-albuterol on TNFα-induced eotaxin and ICAM-1 expression on human parasympathetic neurons in primary culture.
Results
β2 Receptors are Expressed on Human Parasympathetic Neurons
β2 receptor expression was shown by staining with anti-β2 receptor antibody (red, Figure 1 A and C–D). Parasympathetic neurons were identified in primary culture using antibodies to non-phosphorylated neurophilaments (green, Figure 1 B–C). Parasympathetic neurons expressed β2 receptors as shown by positive co-localization (yellow, Figure 1C) of anti-β2 receptor (red) and anti-neurophilament (green) antibodies staining. β2 receptors were expressed on the cell body (Figure 1 A,C–D) and neurites (Figure 1 D). There was no fluorescent signal in negative controls (insert of D) that were treated with normal serum in place of primary antibodies. Cell nuclei were stained blue with DAPI. (Figure 1 A–C and insert of D).
Figure 1. β2 receptors are identified by anti-β2 receptors antibody on human trachea parasympathetic neurons (red, A, B–D) under high (A,C) and low (D) power.

Neurons are labeled with anti-neurofilament antibodis (B, green) and the merged image (for neuronal and β2 receptor staining) is shown in C. Nuclei stain blue with DAPI. The insert of D is the absence of primary antibody. Magnification bars: 50 µm.
Different Effects on TNF-α Induced ICAM-1 and Eotaxin Expression by Different Albuterol Isomers
The anti-inflammatory effect of albuterol was tested by investigating the effect of albuterol on TNF-α-induced ICAM-1 and eotaxin mRNA expression (Figure 2). TNF-α significantly induced ICAM-1 mRNA expression on human parasympathetic neurons (Figure 2A) as compared to control.
Figure 2. Pretreatment with (R)-albuterol before TNF-α significantly inhibits TNF-α-induced ICAM-1 mRNA expression in human parasympathetic neurons as detected by real-time qPCR (A).
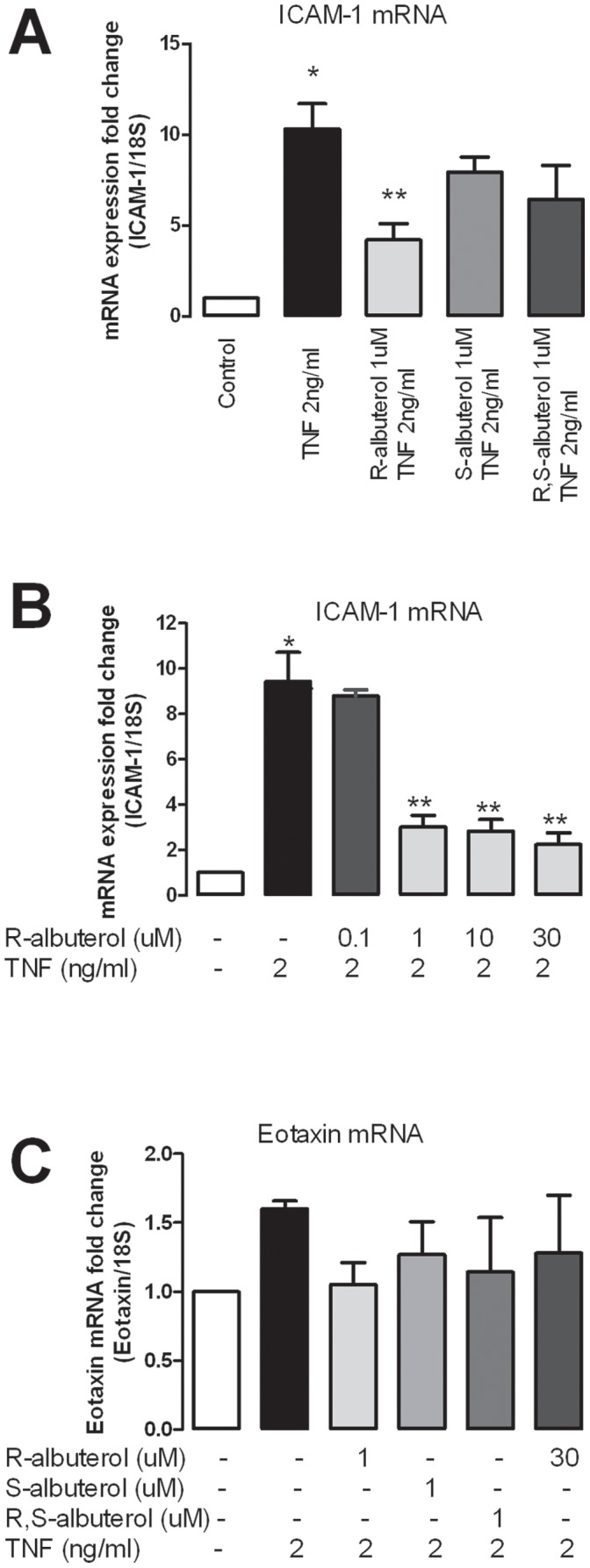
(S)-or (R,S)-albuterol does not inhibit TNF-α induced ICAM-1 (A). The inhibitory effect of (R)-albuterol on TNF-α-induced ICAM-1 mRNA expression is dose dependent (B). Neither TNF-α nor any albuterol isomer changes eotaxin expression (C). *indicates significantly different from control. **indicates significantly different from TNF-α treatment, as analyzed by one way ANOVA.
(S)-, and (R,S)-albuterol (1 uM) caused a small but not statistically significant decrease in TNF-α-induced ICAM-1 expression (P>0.05) (Figure 2A). In contrast, (R)-albuterol (1 uM) significantly inhibited TNF-α-induced ICAM-1 mRNA expression by more than 50% (P<0.05, Figure 2A). The inhibitory effect of (R)-albuterol on TNF-α-induced ICAM-1 expression was dose dependent (Figure 2B). None of the isomers of albuterol changed basal mRNA expression of ICAM-1 in human parasympathetic neurons (data not shown). Neither albuterol nor TNF-α changed the expression of eotaxin (P>0.05, Figure 2C).
The effect of albuterol on TNF-α-induced ICAM-1 protein expression was tested by measuring the fluorescence intensity of immunohistochemical staining (Figure 3). Consistent with our previous finding [5], [7], TNF-α significantly increased ICAM-1 protein expression in human parasympathetic neurons (Figure 3). (R)-albuterol significantly inhibited TNF-α-induced ICAM-1 protein expression (Figure 3A; p<0.05). In contrast, neither (S) nor (R,S)-albuterol affected TNF-α-induced ICAM-1 protein expression (Figure 3 B and C).
Figure 3. ICAM-1 protein expression is measured by fluorescence intensity of a labeled anti-ICAM-1 antibody.
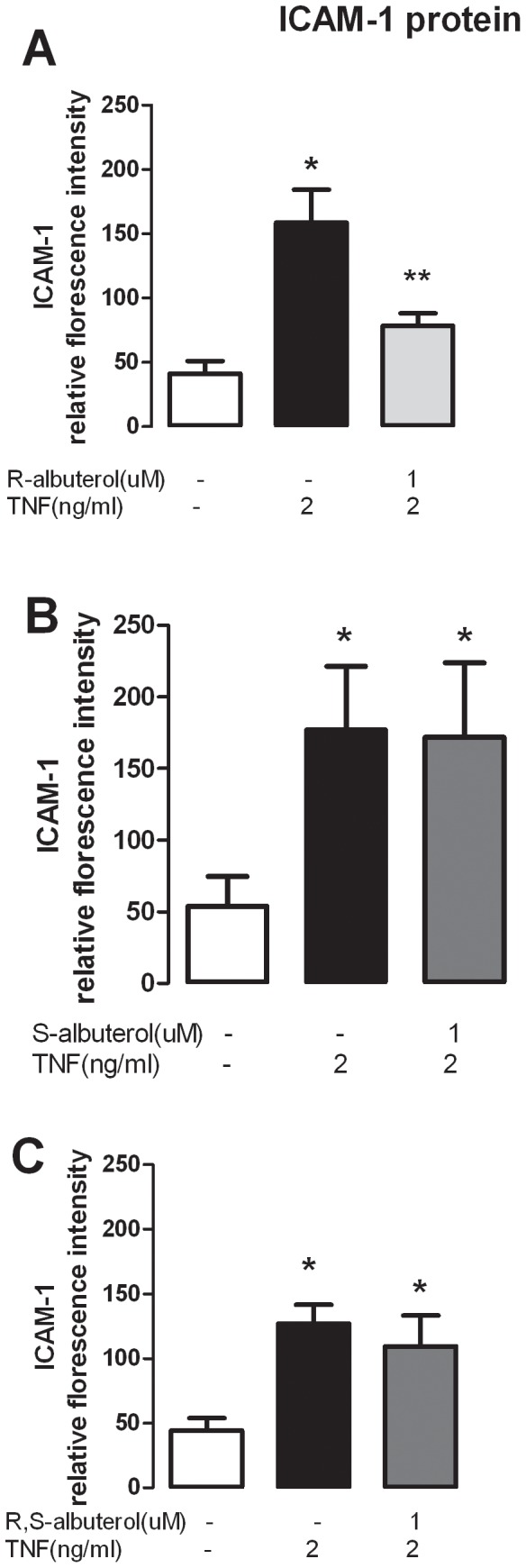
(R)-albuterol (A) but not S- or (R-,S)-albuterol (B and C) significantly inhibits TNF-α-induced ICAM-1 protein (p<0.005). *indicates significantly different from control and **indicates significantly different from TNF- treatment, as analyzed by paired T-test.
In order to test whether the inhibitory effect by R-albuterol was via β-receptors, human parasympathetic neurons were pre-incubated with and without the β-receptor antagonist propranolol before (R)-albuterol and TNF-α were applied to the cell culture (Figure 4). In neurons pre-incubated with propranolol and (R)-albuterol, ICAM-1 protein expression induced by TNF-α was significantly (6 times) higher than those with (R)-albuterol but without propranolol pre-incubation (p<0.05) (Figure 4). Additionally, ICAM-1 expression in the propranolol and (R)-albuterol pre-incubation group was not significantly different from that treated with TNF-α and propranolol (Figure 4). Thus, the suppression effect of albuterol was blocked by pre-incubation with propranolol, indicating that (R)-albuterol inhibits TNF-α induced ICAM-1 expression on human parasympathetic neurons via β-receptors.
Figure 4. Pretreatment with β-receptor antagonist propranolol completely prevents the suppressive effect of R-albuterol on TNF-α-induced ICAM-1 protein expression that is identified by fluorescence intensity of anti-ICAM-1 antibody staining in human parasympathetic nerves.
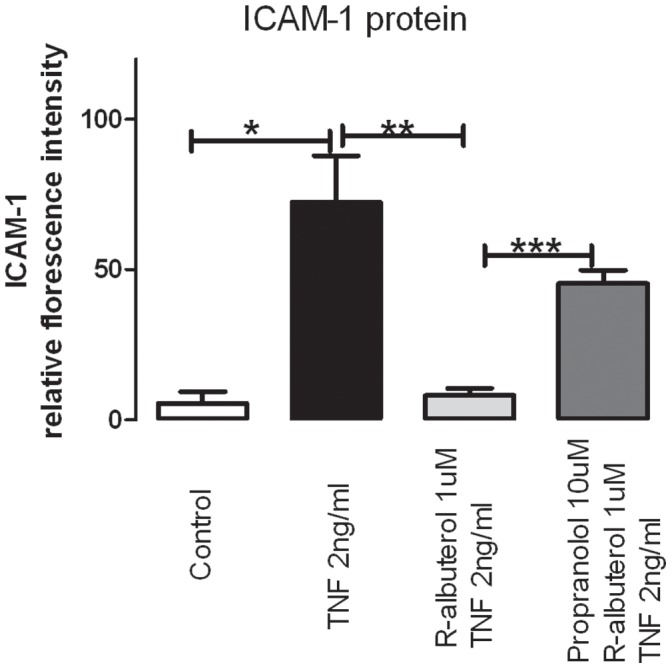
*indicates significant difference from control, ** indicates significant difference from TNF-α treatment and *** indicates significant difference from (R)-albuterol, as analyzed by one way ANOVA.
Discussion
The data presented here are the first to directly show expression of β2-receptors on parasympathetic neurons (Figure 1), although it has been investigated indirectly in physiology and pharmacology studies [14], [15], [16]. Our data show that activation of β2-receptors by (R)-albuterol, but not (S)- or (R,S)- albuterol, can inhibit TNF-α-induced ICAM-1 expression on parasympathetic neurons. Reduced ICAM-1 expression on parasympathetic nerves is directly related to reduced interaction between parasympathetic nerves and inflammatory cells, including eosinophils [7], [8]. It is known that inflammatory cells can affect the release of neurotransmitters from nerves. For example, eosinophils increase ACh release, leading to airway hyperreactivity [2], [5], [7], [8], [17], [18]. Thus, the inhibitory effect of R-albuterol on TNF-α-induced ICAM-1 expression can reduce recruitment of inflammatory cells to the parasympathetic nerve, inhibiting airway hyperreactivity.
β2-agonists have several non-bronchodilator actions that may contribute to their clinical efficacy. For example, β2-agonists have inhibitory effects on inflammatory cells, including neutrophils [19], T lymphocytes [20], and eosinophils [21]. β2-agonists can also down regulate eotaxin production in airway smooth muscle cells [22]. The observation presented here provides another piece of evidence supporting the anti-inflammatory effect of β2-adrenergic agonists (Figure 2–4). Because airway parasympathetic nerve associated eosinophils play a key role in antigen induced hyperreactivity and the migration of eosinophils is mediated by ICAM-1 [5], [7], [8], [23], [24], [25], the suppressing effect of (R)-albuterol on neuronal ICAM-1 expression may be an important anti-inflammatory effect of this drug which contribute to its clinical efficacy.
(R)-albuterol is more potent in inhibiting TNF-α-induced ICAM-1 expression than (S)-albuterol or (R,S)-albuterol. (S)-albuterol does not contribute to the anti-inflammatory effects (Figure 2 and 3), but may compete with (R)-albuterol for binding to β2-receptor in the racemic albuterol ((R,S)-albuterol) as (S)-albuterol is a partial agonist at the β2-receptor [26]. Moreover, at the same concentration of albuterol, racemic albuterol ((R,S)-albuterol) contains only half of the albuterol that can efficiently bind to β2-receptors and inhibit TNF-α-induced ICAM-1 expression. Therefore, it is not surprising that (R)-albuterol is more potent compared to (R,S)-albuterol (Figure 2 and 3) in inhibiting ICAM-1 expression on parasympathetic neurons.
Our previous study also shows that the NF-κB inhibitor blocks TNF-α-induced ICAM-1 expression, indicating that TNF-α induces ICAM-1 expression on parasympathetic neuron via a NF-κB dependent mechanism [7]. This is consistent with results from studies in other type of cells, such as airway epithelial cells [27], that NF-κB is a key player in ICAM-1 expression induced by TNF-α. Therefore, factors that influence the translocation and activation of NF-κB proteins may change TNF-α-induced ICAM-1 expression. An inhibitory effect of the β2-receptor agonist on NF-κB activity has been reported [28]. β2-receptor agonists regulate NF-κB activity via elevated cAMP and activation of PKA [28], [29], [30], [31]. This suggests that R-albuterol, a β2-receptor agonist, suppresses the activity of NF-κB via elevated cAMP and activation of PKA, inhibiting TNF-α-induced expression of ICAM-1 mRNA.
In summary, we have shown that β2-adrenergic receptors are expressed on human parasympathetic neurons and activation of β2-adrenergic receptors by (R)-albuterol can suppress TNF-α-induced ICAM-1 expression. As this effect is not seen with racemic albuterol, we speculate that (R)-albuterol may have a better therapeutic effect than racemic albuterol in asthma treatment, not only because of its bronchodilator properties, but also because of its anti-inflammatory effect.
Materials and Methods
Cell culture: Airway parasympathetic neurons were isolated from human tracheas which were donated by organ donors. The Oregon Health & Science University Research Integrity Office waived the need for ethical approval on this culture of human airway cells prior to the onset of the study. Airway parasympathetic neurons were grown in serum-free medium for 1 week as previously described [7]. Cells were incubated in fresh medium with or without albuterol isoforms (Sepracor) for 90 minutes, followed by additional incubation with recombinant human TNF-α (2 ng/ml, T0157 Sigma) for either 4 hours for real time RT-PCR, or 24 hours for immunohistochemistry. In experiments with propranolol (10 uM, Sigma P0884), the β-blocker was added to the culture medium 30 minutes before (R)-albuterol was applied.
Immunohistochemistry staining: Parasympathetic neurons were identified by staining with antibodies to non-phosphorylated neurophilament (SMI-311, 1∶10000, Covance). β2 receptors were detected using rabbit anti-β2 receptor antibody (1∶200, sc-569, Santa Cruz Biotechnology, Inc., Santa Cruz, CA). ICAM-1 expression in parasympathetic nerves from human tracheas was identified using rabbit anti-human ICAM-1 antibodies (1∶50, sc-7891, Santa Cruz Biotechnology, Inc., Santa Cruz, CA). All primary antibodies were incubated overnight at 4°C, followed by incubation with corresponding secondary antibodies, either labeled with Alexa fluor 555 (red) or Alexa fluor 488 (green) (Molecular Probes, Invitrogen Corp., Carlsbad, CA ) for 60 minutes at 37°C. Negative controls were incubated with normal serum (Vector) in place of the primary antibodies. All slides were mounted in aqueous medium with 4′-6′diamino-2-phenylindole (DAPI, Vector) to stain nuclei.
Florescence intensity of ICAM-1 staining was quantified using Metamorph. Cells, which were cultured on Lab Tek IV chamber slides (cat#154461, Nalge Nunc International), were divided into groups as follows: control, TNF-α treated only, albuterol isomer plus TNF-α treated, and propranolol plus R-albuterol plus TNF-α treated. The labeling of each slide was covered in order to eliminate investigator bias. All chamber slides were treated the same way in each step during the fixation and immunohistochemical staining and were photographed using a fluorescence microscope with the same exposure time. ICAM-1 labeled neurites from different treatment groups were selected at random and outlined. Nerve cell bodies were difficult to distinguish individually and therefore were not included in the analysis. The lower threshold, set as zero, was determined in empty areas using Metamorph, and was then used to measure the intensity in neurites. Average intensity was collected from twenty to thirty separate neurites in each treatment group and the experiment was repeated three or more times. The mean ± standard error was calculated from collected data and one way ANOVA test was used to determine the statistical significance. P values of less than 0.05 were accepted as statistically significant.
Real time RT-PCR: Cell RNA was isolated using the RNeasy Mini Kit (74106; QIAGEN), according to the manufacturer’s recommendations. The RNA was reverse transcribed using SuperScript III (18080–051; Invitrogen Corp.) with random hexamer primers. Quantitative PCR was carried out in triplicate at 60°C over 45 cycles using the Quantitect SYBR Green PCR kit (204143; QIAGEN). The amounts of PCR product were quantified using the Mx3000P real-time PCR system (Stratagene). Oligonucleotide PCR primer pairs were designed from published human sequences as follows: ICAM-1∶5′-GGCTGGAGCTGTTTGAGAAC-3′ and 5′-ACTGTGGGGTTCAACCTCTG-3′. eotaxin-1: sense, AACCACCTGCTGCTTTAACC, antisense, TCCTGCACCCACTTCTTCTT. 18S ribosomal RNA was used as an internal control, and the primer pairs were as follows: 5′-GTAACCCGTTGAACCCCATT-3′ and 5′-CCATCCAATCGGTAGTAGCG-3′. The threshold cycle number was measured, and the relative expression of ICAM-1 was adjusted for the threshold cycle for detection of 18S. Results were presented as the mean ± SE of the 3 experiments from human parasympathetic neurons isolated from the trachea. A one way ANOVA was used to determine the statistical significance of differences. P values of less than 0.05 were accepted as statistically significant.
Acknowledgments
We thank the members of the Pacific Northwest Transplant Bank, Portland, Oregon, for procuring and supplying tracheas from organ donors.
Funding Statement
The funders (grants from National Institutes of Health: HL55543 (ADF), HL54659 (DBJ), HL071795 (DBJ), ES014601 (DBJ)) had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Sepracor Inc provided albuterol and funding to support part of experiment supplies in this study. But Sepracor Inc also had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Costello RW, Schofield BH, Kephart GM, Gleich GJ, Jacoby DB, et al. (1997) Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol 273: L93–103. [DOI] [PubMed] [Google Scholar]
- 2. Elbon CL, Jacoby DB, Fryer AD (1995) Pretreatment with an antibody to interleukin-5 prevents loss of pulmonary M2 muscarinic receptor function in antigen-challenged guinea pigs. Am J Respir Cell Mol Biol 12: 320–328. [DOI] [PubMed] [Google Scholar]
- 3. Walsh MT, Curran DR, Kingham PJ, Morgan RK, Durcan N, et al. (2004) Effect of eosinophil adhesion on intracellular signaling in cholinergic nerve cells. American Journal of Respiratory Cell & Molecular Biology 30: 333–341. [DOI] [PubMed] [Google Scholar]
- 4. Kikuchi I, Kikuchi S, Kobayashi T, Hagiwara K, Sakamoto Y, et al. (2006) Eosinophil trans-basement membrane migration induced by interleukin-8 and neutrophils. Am J Respir Cell Mol Biol 34: 760–765. [DOI] [PubMed] [Google Scholar]
- 5. Fryer AD, Stein LH, Nie Z, Curtis DE, Evans CM, et al. (2006) Neuronal eotaxin and the effects of ccr3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest 116: 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nie Z, Jacoby DB, Fryer AD (2009) Etanercept Prevents Airway Hyperresponsiveness by Protecting Neuronal M2 Muscarinic Receptors in Antigen Challenged Guinea Pigs British Journal of Pharmacology. 156: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nie Z, Nelson CS, Jacoby DB, Fryer AD (2007) Expression and regulation of intercellular adhesion molecule-1 on airway parasympathetic nerves. J Allergy Clin Immunol. [DOI] [PubMed]
- 8. Sawatzky DA, Kingham PJ, Court E, Kumaravel B, Fryer AD, et al. (2002) Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am J Physiol Lung Cell Mol Physiol 282: L1279–1288. [DOI] [PubMed] [Google Scholar]
- 9. Wegner CD, Rothlein R, Gundel RH (1991) Adhesion molecules in the pathogenesis of asthma. Agents & Actions - Supplements 34: 529–544. [PubMed] [Google Scholar]
- 10. Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ (2002) Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. Journal of Clinical Investigation 110: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hakonarson H, Maskeri N, Carter C, Hodinka RL, Campbell D, et al. (1998) Mechanism of rhinovirus-induced changes in airway smooth muscle responsiveness. Journal of Clinical Investigation 102: 1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Handley DA, Tinkelman D, Noonan M, Rollins TE, Snider ME, et al. (2000) Dose-response evaluation of levalbuterol versus racemic albuterol in patients with asthma. J Asthma 37: 319–327. [DOI] [PubMed] [Google Scholar]
- 13. Nelson HS, Bensch G, Pleskow WW, DiSantostefano R, DeGraw S, et al. (1998) Improved bronchodilation with levalbuterol compared with racemic albuterol in patients with asthma. J Allergy Clin Immunol 102: 943–952. [DOI] [PubMed] [Google Scholar]
- 14. Zhang XY, Olszewski MA, Robinson NE (1995) Beta 2-adrenoceptor activation augments acetylcholine release from tracheal parasympathetic nerves. Am J Physiol 268: L950–956. [DOI] [PubMed] [Google Scholar]
- 15. Zhang XY, Zhu FX, Robinson NE (1996) Excitatory prejunctional beta 2-adrenoceptor distribution within equine airway cholinergic nerves. Respir Physiol 106: 81–90. [DOI] [PubMed] [Google Scholar]
- 16. Belvisi MG, Patel HJ, Takahashi T, Barnes PJ, Giembycz MA (1996) Paradoxical facilitation of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by isoprenaline. Br J Pharmacol 117: 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacoby DB, Gleich GJ, Fryer AD (1993) Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest 91: 1314–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Evans CM, Fryer AD, Jacoby DB, Gleich GJ, Costello RW (1997) Pretreatment with antibody to eosinophil major basic protein prevents hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. J Clin Invest 100: 2254–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reid DW, Ward C, Wang N, Zheng L, Bish R, et al. (2003) Possible anti-inflammatory effect of salmeterol against interleukin-8 and neutrophil activation in asthma in vivo. Eur Respir J 21: 994–999. [DOI] [PubMed] [Google Scholar]
- 20. Baramki D, Koester J, Anderson AJ, Borish L (2002) Modulation of T-cell function by (R)- and (S)-isomers of albuterol: anti-inflammatory influences of (R)-isomers are negated in the presence of the (S)-isomer. J Allergy Clin Immunol 109: 449–454. [DOI] [PubMed] [Google Scholar]
- 21. Volcheck GW, Kelkar P, Bartemes KR, Gleich GJ, Kita H (2005) Effects of (R)- and (S)-isomers of beta-adrenergic agonists on eosinophil response to interleukin-5. Clin Exp Allergy 35: 1341–1346. [DOI] [PubMed] [Google Scholar]
- 22. Ameredes BT, Calhoun WJ (2005) Modulation of GM-CSF release by enantiomers of beta-agonists in human airway smooth muscle. J Allergy Clin Immunol 116: 65–72. [DOI] [PubMed] [Google Scholar]
- 23. Marlin SD, Springer TA (1987) Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51: 813–819. [DOI] [PubMed] [Google Scholar]
- 24. van de Stolpe A, van der Saag PT (1996) Intercellular adhesion molecule-1. Journal of Molecular Medicine 74: 13–33. [DOI] [PubMed] [Google Scholar]
- 25. Stanciu LA, Djukanovic R (1998) The role of ICAM-1 on T-cells in the pathogenesis of asthma. European Respiratory Journal 11: 949–957. [DOI] [PubMed] [Google Scholar]
- 26. Penn RB, Frielle T, McCullough JR, Aberg G, Benovic JL (1996) Comparison of R-, S-, and RS-albuterol interaction with human beta 1- and beta 2-adrenergic receptors. Clin Rev Allergy Immunol 14: 37–45. [DOI] [PubMed] [Google Scholar]
- 27. Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, et al. (2000) Effects of TNF-alpha on expression of ICAM-1 in human airway epithelial cells in vitro. Signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol 22: 685–692. [DOI] [PubMed] [Google Scholar]
- 28. Farmer P, Pugin J (2000) beta-adrenergic agonists exert their “anti-inflammatory” effects in monocytic cells through the IkappaB/NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol 279: L675–682. [DOI] [PubMed] [Google Scholar]
- 29. Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N (1996) Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 271: 20828–20835. [DOI] [PubMed] [Google Scholar]
- 30. Parry GC, Mackman N (1997) Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol 159: 5450–5456. [PubMed] [Google Scholar]
- 31. Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, et al. (2005) Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur J Immunol 35: 31–41. [DOI] [PubMed] [Google Scholar]