

Abstract
Gap junctions (GJs) are composed of tens to many thousands of double-membrane spanning GJ channels that cluster together to form densely packed channel arrays (termed GJ plaques) in apposing plasma membranes of neighboring cells. In addition to providing direct intercellular communication (GJIC, their hallmark function), GJs, based on their characteristic double-membrane-spanning configuration, likely also significantly contribute to physical cell-to-cell adhesion. Clearly, modulation (up-/down-regulation) of GJIC and of physical cell-to-cell adhesion is as vitally important as the basic ability of GJ formation itself. Others and we have previously described that GJs can be removed from the plasma membrane via the internalization of entire GJ plaques (or portions thereof) in a cellular process that resembles clathrin-mediated endocytosis. GJ endocytosis results in the formation of double-membrane vesicles (termed annular gap junctions [AGJs] or connexosomes) in the cytoplasm of one of the coupled cells. Four recent independent studies, consistent with earlier ultrastructural analyses, demonstrate the degradation of endocytosed AGJ vesicles via autophagy. However, in TPA-treated cells others report degradation of AGJs via the endo-/lysosomal degradation pathway. Here we summarize evidence that supports the concept that autophagy serves as the cellular default pathway for the degradation of internalized GJs. Furthermore, we highlight and discuss structural criteria that seem required for an alternate degradation via the endo-/lysosomal pathway.
Keywords: Autophagy, Cell-Cell Junctions, Connexin 43, Gap Junctions, Protein Degradation, Ubiquitin
Introduction
Direct cell-to-cell communication is a pivotal cellular function of multi-cellular organisms. It is established by gap junction (GJ) channels, that bridge apposing plasma membranes of neighboring cells. Typically, tens to thousands of GJ channels cluster into densely packed two-dimensional arrays, termed GJ plaques that can reach several micrometers in diameter. GJ channels are assembled from a ubiquitously expressed class of four-pass trans-membrane proteins, termed connexins, with connexin 43 (Cx43) being the most abundantly expressed connexin type. Six connexin polypeptides oligomerize into a ring to form a hexameric trans-membrane structure with a central hydrophilic pore, called a hemi-channel or connexon. Once trafficked to the plasma membrane, two connexons, one provided by each of two neighboring cells, dock head-on in the extra-cellular space to form the complete double-membrane spanning GJ channel that is completely sealed-off to the extracellular space. Recruitment of additional GJ channels along the outer edge enlarges the GJ plaques, while simultaneous removal of older channels from plaque centers balances GJ channel turnover (Falk et al. 2009; Gaietta et al. 2002; Lauf et al. 2002). In addition to providing intercellular communication (their hallmark function), GJs, based on their characteristic double-membrane-spanning configuration and their tight extracellular head-to-head docking, are also likely to contribute significantly to physical cell-to-cell adhesion that generally is attributed to the adhesive force exerted by adherens- and tight junctions. Down-regulation of cell-cell adhesion contributes to many physiological and pathological conditions in which cells physically uncouple from their neighbors. It is for example seen during cell migration in development and wound healing, mitosis, apoptosis, leukocyte extravasation, ischemia, hemorrhage, edema, and cancer metastasis. Thus, to effectively reduce or abolish cell-to-cell adhesion under such conditions, all major types of cell-cell adhesion structures (including desomosomes, adherens-, tight-, and gap junctions) will either have to reduce their adhesive force, or will have to be removed from the plasma membrane.
GJ channels are known to gate such that physiological parameters including intracellular pH, Ca2+ concentration and Cx phosphorylation can modulate the status of GJ channels (opened, closed), and the extent of GJ-mediated intercellular communication (GJIC) (Delmar et al. 2004; Laird 2005; Lampe and Lau 2004; Moreno 2005; Pahujaa et al. 2007; Solan and Lampe 2009; Warn-Cramer and Lau 2004). Importantly however, GJ channel gating (channel closure, not channel removal) unlikely will significantly affect the level of GJ-mediated cell-to-cell adhesion. Alternatively, the extend of GJIC could also be regulated by splitting GJ channels (similar to adherens junctions), or by reducing the number of plasma membrane-located GJs. Both mechanisms also would contribute to a reduction in physical cell-to-cell adhesion. Goodenough and Gilula, and Ghoshroy et al. reported that connexons, once docked, appear inseparable under physiological conditions (Ghoshroy et al. 1995; Goodenough and Gilula 1974), suggesting that GJs, especially under conditions where physical cell-cell adhesion needs to be reduced/abolished, will be removed from the plasma membrane. Here, we summarize recent findings that support the view that GJ removal from the plasma membrane is achieved by the internalization/endocytosis of GJs. Next, we will address two potential cellular degradation pathways, autophagososmal versus endo-/lysosomal, that both have been described recently to degrade endocytosed GJs (Figure 1). We provide evidence that supports the concept that autophagy appears as the most likely cellular degradation pathway for removing internalized GJs form the cytoplasm. Finally, we discuss important structural criteria that may allow an alternate endo-/lysosomal degradation of internalized GJs.
Figure 1.

Schematic representation of proposed steps that lead to GJ internalization (steps 1–3), cytoplasmic AGJ vesicle formation and fragmentation (steps 4, 5), and AGJ vesicle degradation by phago-/lysosomal (steps 6–10) and endo-/lysosomal pathways (steps 11–15) based on the previous work by others and us (see text for details). Note the proposed non-junctional membrane domains missing the green GJ label (shown in steps 4, 5, 11, 12), and the increased phosphorylation and ubiquitination on AGJ vesicles that fuse with endosomes (steps 11, 12 versus 6, 7). (Adapted from Falk et al. 2009 and Fong et al. 2012.)
Plasma membrane-located GJs are endocytosed resulting in the formation of cytoplasmic double-membrane GJ vesicles termed AGJs or connexosomes
We reported previously that cells continuously, -and effectively after treatment with natural inflammatory mediators-, internalize and turn over their GJs via a combined endo-/exocytic process (Baker et al. 2008; Falk et al. 2009; Gilleron et al. 2008; Gumpert et al. 2008; Piehl et al. 2007) (Figure 1, steps 1–5). Internalization was found to occur preferentially into one of two coupled cells, indicating a highly regulated process (Falk et al. 2009; Piehl et al. 2007). The scaffolding protein, ZO-1, a well-known binding partner of Cx43 (Giepmans and Moolenaar 1998; Hunter et al. 2005; Toyofuku et al. 1998), was found to be displaced from GJ plaques on the side of plaque invagination (Baker et al. 2008; Gilleron et al. 2008). This observation suggests that ZO-1, not surprising for a scaffolding protein, may play multiple roles in GJ biosynthesis and function, including regulation of GJ plaque size (Hunter et al. 2005), accrual of channels to GJ plaques (Rhett and Gourdie 2011; Rhett et al. 2011), and defining directionality of GJ plaque internalization (Baker et al. 2008; Gilleron et al. 2008). Further analyses indicated that GJ internalization utilizes well-known components of the clathrin-mediated endocytosis (CME) machinery, including the classical endocytic coat protein clathrin, the clathrin-adaptors AP-2 and Dab2, the GTPase dynamin2, the retrograde actin motor myosin VI (myo6), as well as the process of actin polymerization (Gumpert et al. 2008; Piehl et al. 2007) (Figure 1, steps 1–4). GJ internalization generates characteristic cytoplasmic double-membrane GJ vesicles, termed annular GJs (AGJs) or connexosomes. Note that the outer membrane of the generated AGJ vesicles corresponds to the plasma membrane of the host-cell, while the inner membrane and the vesicle lumen correspond to plasma membrane and cytoplasm of the neighboring donor cell (Figure 1, steps 1–5). Others and we further found that internalization is highly regulated and can occur very efficiently. For example, acute GJ internalization can be seen in primary porcine pulmonary artery endothelial cells (PAECs) exposed to natural inflammatory mediators such as thrombin and endothelin (Baker et al. 2008), well-known inhibitors of GJIC (Blomstrand et al. 2004; Postma et al. 1998; Spinella et al. 2003; van Zeijl et al. 2007), and in Cx43-GFP expressing NRK cells (Jordan et al. 2001). Efficient internalization of cell-cell junctions reduces/abolishes cell-cell adhesion most likely allowing the endothelial cells to migrate to the inflamed tissue regions in order to aid in the healing process and to modulate endothelial barrier function (Baldwin and Thurston 2001; Falk 2010; Lum and Malik 1994; Wong et al. 2004). Efficient GJ internalization is also seen in Sertoli cells in response to treatment with the non-genomic carcinogen lindane (Gilleron et al. 2008), and it is seen under pathological conditions such as in the failing canine ventricular myocardium (Hesketh et al. 2010).
Continuous, as well as induced endocytosis of GJ plaques (consisting of double-membrane spanning GJ channels) is supported (I) by numerous earlier ultrastructural and live-cell based analyses that detected AGJ vesicles in the cytoplasm of cells in situ, especially occurring in differentiating tissues and sometimes in association with lysosomes (Ginzberg and Gilula 1979; Hesketh et al. 2010; Jordan et al. 2001; Larsen et al. 1979; Leach and Oliphant 1984; Mazet et al. 1985; Pfeifer 1980; Severs et al. 1989); (II) the fundamental observation that connexons, once docked, are inseparable under physiological conditions (Ghoshroy et al. 1995; Goodenough and Gilula 1974); and (III) by the typical short half-life of connexins and GJ channels of only 1–5 hours (Beardslee et al. 1998; Berthoud et al. 2004; Falk et al. 2009; Fallon and Goodenough 1981; Gaietta et al. 2002).
What happens to cytoplasmic AGJ vesicles after their generation?
Four recent studies, Hesketh et al. (2010), Lichtenstein et al. (2011), Fong et al. (2012), and Bejarano et al. (2012) report the degradation of endocytosed AGJ vesicles via autophagy (Figure 1, steps 6–10), while Leithe et al. (Leithe et al. 2009; Leithe and Rivedal 2004b; Fykerud et al. 2012) report degradation of endocytosed AGJ vesicles via the cellular endo-/lysosomal degradation pathway in cells that were treated with the phorbol ester, TPA (12-O-tetradecanoylphorbol 13-acetate), an analog of the secondary messenger DAG (diacylglycerol) (Figure 1, steps 11–15). Hesketh et al. (2010) report loss of GJs from the plasma membrane, GJ endocytosis, and AGJ degradation by autophagy in pacing-induced failing canine ventricular myocardium. Lichtenstein et al. (2011) report that autophagy contributes to the degradation of endogenously (NRK cells and mouse embryonic fibroblasts) and exogenously (HeLa cells) expressed wild type Cx43 protein, and of wild type and cataract-associated mutant Cx50 proteins in both un-induced cells and in cells in which autophagy was induced by starvation (Lichtenstein et al. 2011). Fong et al. (2012) report the autophagic degradation of AGJ vesicles in HeLa cells expressing exogenous fluorescently tagged Cx43, and in primary porcine pulmonary artery endothelial cells (PAECs) endogenously expressing Cx43. Bejarano et al. (2012) report the Nedd4-mediated ubiquitin-dependent autophagic degradation of internalized GJs in situ (mouse liver) as well as in starved and fed cultured cells expressing Cx43 endogenously and exogenously (mouse embryonic fibroblasts, NIH3T3, COS7, and NRK cells). In all four studies, cytoplasmic AGJ vesicles were detected inside phagophores by ultrastructural analyses, and AGJs were observed to colocalize with the most commonly used autophagy marker protein, LC3-II/Atg8 (Figure 2). Autophagosomes exhibit a highly characteristic, clearly recognizable double-membrane structure on ultra-thin sections, and thus conventional electron microscopy, as performed by Hesketh et al., Lichtenstein et al., Fong et al., and Bejarano et al. is still one of the best techniques for the characterization of autophagosomes (Mizushima 2004) (Figure 2D–H). Microtubule-associated protein light chain 3 (LC3, the mammalian homolog of the yeast autophagic protein Atg8) is an abundant soluble cytoplasmic protein. It is proteolytically processed by the removal of a few N-terminal amino acid residues shortly after translation that generates LC3-I. LC3-I is recruited to developing phagophores, is covalently conjugated to phosphatidyl-ethanolamine (PE) of the phagophore membrane (termed LC3-II), and remains on autophagosomes for most of their lifetime (Kabeya et al. 2000; Mizushima 2004). Thus LC3 protein that was used by all four laboratories in colocalization studies is one of the most useful generic marker-proteins for the characterization of autophagosomes (Kabeya et al. 2000) (Figure 2A).
Figure 2.

Evidence for autophagic AGJ vesicle degradation. (A) HeLa cells were cotransfected with Cx43-mApple and the mammalian autophagy marker protein GFP-LC3 (left panel), or the activation-deficient LC3-mutant GFP-LC3(G120A) (right panel). In cells, a fraction of cytoplasmic LC3 (LC3-I) is covalently conjugated to phagophore-membranes (LC3-II) that localizes to autophagosomes; LC3(G120A) cannot be conjugated and remains cytoplasmic. Representative merged fluorescence images acquired 24 hours post transfections are shown. Individual and merged fluorescence signals of the boxed areas are shown below at higher magnification. Robust colocalization of cytoplasmic AGJ vesicles present in Cx43-mApple expressing cells (red puncta) with GFP-LC3-II (green puncta) was observed in GFP-LC3 expressing cells, but not in GFP-LC3(G120A) expressing cells. Representative colocalizing AGJ vesicles are marked with arrows; GJs are marked with arrowheads. Scale bars = 10 m.
(B) Western blot analyses of total Cx43-GFP, or Cx43-YFP protein in transiently and stably expressing HeLa cells 72 hours after Beclin-1, a crucial autophagy-protein was depleted by RNAi-oligonucleotide transfection. SI-control cells were transfected with a scrambled non-targeting RISC-activating control oligonucleotide. Cx43-GFP/YFP was detected by probing with polyclonal anti-Cx43 antibodies. Normalized quantitative analyses revealed a two to three-fold accumulation of Cx43-GFP/YFP protein in Beclin-1-KD over RNAi-control cells.
(C) Quantitative AGJ vesicle analyses performed 72 hours post RNA oligonucleotide-transfection indicated a significant cytoplasmic AGJ accumulation in Beclin-1/(Atg6), LAMP-2, and p62/SQSTM1 (all autophagy-related proteins) depleted cells (≥ 50%, marked with asterisks) compared to RNAi-control cells (panel 1, left). Less pronounced AGJ vesicle-accumulation was observed in LC3-depleted cells (32%), and this was attributed to sufficient inactive LC3-I that may have remained in the LC3-KD cells and may have been converted into active LC3-II. In addition, quantitative analyses of AGJ vesicles (panel 2, center left), LC3-positive autophagosomes (panel 3, center right), and colocalizing AGJ/autophagosomes (panel 4, right) in Beclin-1 and p62/SQSTM1 knockdown cells revealed a significant increase of AGJs, a significant decrease of autophagosomes, and significantly reduced AGJ/autophagosome colocalization in three independent experiments (** = p<0.01; *** = p<0.001).
(D) Multiple stages characteristic of progressive autophagosome formation and maturation that formed around AGJ vesicles revealed by ultrastructural analyses of Cx43-GFP expressing HeLa cell preparations. Double-membrane cisternae (presumably isolation membranes, marked with arrows) progressively encircled AGJ vesicles (panels a–c), coalesced into phagophores (panels c–e) and fused with lysosomes (L, panels d, e), resulting in AGJ degradation inside the phagosome (panel f). (Figure portions A to D are reproduced with permission from Fong et al. 2012.)
(E–H) Autophagosomes presumably degrading endogenous AGJs in vivo, identified in the equine stratum spinosum (E, reproduced with permission from Leach and Oliphant 1984); rat liver (F, reproduced with permission from Pfeifer 1980); mouse embryonic fibroblasts (G, reproduced with permission from Lichtenstein et al. 2011); and mammalian cardiomyocytes (H, reproduced with permission from Severs et al. 1989). Scale bars = 100 nm
While the Lichtenstein et al. and Bejarano et al. studies were aimed more broadly at a potential role of autophagy contributing to connexin and GJ degradation in general, the Fong et al., and the Hesketh et al. studies were aimed specifically at investigating the fate of internalized AGJ vesicles that others and we had characterized previously (Baker et al. 2008; Gumpert et al. 2008; Jordan et al. 2001; Piehl et al. 2007). To support their findings, Lichtenstein et al. and Bejarano et al., besides using other approaches, knocked down the autophagy-related proteins Atg5 and Atg7 in cells expressing either endogenous or exogenous Cx43, and used the drugs chloroquine and 3MA to inhibit autophagy. In contrast, Fong et al. knocked down expression of the autophagy related proteins, Beclin-1 (Atg6), LC3 (Atg8), LAMP-2, and p62/sequestosome 1 (SQSTM1) (Figure 2B, C), and used the drugs 3MA, Wortmannin, and Bafilomycin A1 in Cx43-GFP expressing HeLa cells. Taken together, all four complementary studies (Bejarano et al. 2012; Fong et al. 2012; Hesketh et al. 2010; Lichtenstein et al. 2011) provide convincing evidence that under physiological, as well as pathological conditions, GJ plaques are endocytosed from the plasma membrane, and the resulting AGJ vesicles are degraded by autophagy (Figures 1, 2). As mentioned above, in the Lichtenstein et al., Fong et al., and Bejarano et al. studies the ubiquitin-binding protein p62/SQSTM1 was identified as a protein that targets internalized GJs to autophagic degradation. Knocking down p62/SQSTM1 protein levels as performed by Fong et al. resulted in a significantly increased accumulation of cytoplasmic AGJs (av. 55%, n=4) and a significantly reduced colocalization (av. 69.5%, n=3) of AGJs with autophagosomes (Figure 2C). Remarkably, although autophagic degradation of GJs had been described in several classical ultrastructural analyses of various cells and tissues in situ including heart, dermis, and liver (Leach and Oliphant 1984; Mazet et al. 1985; Pfeifer 1980; Severs et al. 1989) (see Figure 2E, F, H), until recently, not much attention was attributed to this fundamental GJ degradation pathway. Autophagic degradation of GJs plays a significant role in the regulation of GJ function, as inhibition of cellular autophagy increases GJIC, prevents internalization of GJs, slows down the degradation of connexins, and causes cytoplasmic accumulation of internalized GJ vesicles in situ, as well as in cultured cells expressing either endogenously or exogenously connexin proteins (Bejarano et al. 2012; Fong et al. 2012; Lichtenstein et al. 2011). Some characteristics that aid in a better understanding of the autophagic degradation pathway will be described next.
(Macro)Autophagy
Cells have developed three principal degradation pathways: the proteasomal, the endo-/lysosomal, and the phago-/lysosomal system (termed macroautophagy or simply autophagy), and all three have been implicated previously at various steps in the regulation of GJ stability and connexin degradation (Hesketh et al. 2010; Laing et al. 1997; Leach and Oliphant 1984; Leithe and Rivedal 2004a; Musil et al. 2000; Pfeifer 1980; Qin et al. 2003). While the two latter ones utilize the lysosome for final degradation and are designed for the degradation of protein aggregates, multi-protein complexes and cytoplasmic organelles, the proteasomal system is designed for the degradation of single polypeptide chains that require unfolding to be inserted into the tubular core of the cytoplasmically located proteasome. Since AGJ vesicles are highly oligomeric multi-subunit protein assemblies, their degradation by the proteasome degrading single, unfolded polypeptides appears unlikely, and to our knowledge, no evidence exists that would suggest proteasome-mediated degradation of assembled GJ plaques or of AGJ vesicles. Also, it should be noted here that lysosomal inhibitors such as leupeptin, chloroquine, NH4Cl, and E-64, that previously have been used to gain evidence for endo-/lysosomal degradation of GJs (Berthoud et al. 2004; Laing et al. 1997; Musil et al. 2000; Qin et al. 2003), will also inhibit autophagic GJ degradation, and thus obtained results may not have been accurately interpreted. Additional, future experiments that specifically target one or the other cellular degradation pathway may be required to clarify specific roles of both pathways in GJ degradation. GJ endocytosis, post-endocytic sorting, and the role of Cx43 ubiquitination for proteasomal and endo-/lysosomal degradation of connexin polypeptides, connexons, non-aggregated GJ channels, and of GJ plaques has recently been extensively reviewed (Kjenseth et al. 2010; Leithe et al. 2011; Su and Lau 2012). Here, we therefore concentrate on the autophagosomal degradation of AGJs, and on addressing a few prerequisites and factors that appear crucial for allowing a potential alternate endo-/lysosomal degradation of AGJ vesicles (Figure 1).
Historically, autophagy has been known as a lysosomal degradation pathway that is essential for cell survival following nutrient depletion. However, substantial research over the past decade has indicated that autophagy, beside its well-known function in organelle degradation during starvation, represents a much more common and highly-conserved lysosome-based cellular degradation pathway that is specifically designed to remove and degrade protein aggregates, multi-protein complexes, organelles, and invading pathogens from the cytoplasm (Bjorkoy et al. 2005; Hung et al. 2009; Pohl and Jentsch 2009; Ravikumar et al. 2008). Recent studies have further shown that protein aggregates, such as the ones formed by huntingtin and β-amyloid protein, and cellular structures such as the midbody ring, a mitotic cytokinesis left-over multi-protein complex, are all degraded by autophagy (Bjorkoy et al. 2005; Hung et al. 2009; Pohl and Jentsch 2009; Ravikumar et al. 2008). Clearly, these cellular structures are degraded by autophagy independent of starvation. In addition, autophagosomal degradation of membranous/vesicular organelles, as for example, malfunctioning mitochondria, is common. Since the catabolic activity of lysosomes is used in this process, degradation-prone structures first need to be separated from the cytoplasm. This is due to the destructive activity of lysosomal enzymes, which cannot be released directly into the cytoplasm. Indeed, cytoplasmic structures targeted for degradation are engulfed in double-membraned vesicles called autophagosomes that allow lysosomal fusion, degradation, and subsequent recycling of the phagosome cargo and the phagosome membrane (Figure 1, steps 6–8).
Autophagosomes are formed by the elongation and fusion of phagophore membrane cisternae, which are derived from pre-autophagosomal structures. The membrane origin of phagophores is still unclear and likely involves multiple sources, such as the plasma membrane (Ravikumar et al. 2010), outer mitochondrial membranes (Hailey et al. 2010), the endoplasmic reticulum (Matsunaga et al. 2010), and the Golgi apparatus (Yen et al. 2010); reviewed recently in Mari et al. 2011). A large number of proteins essential or relevant for autophagosome formation and autophagic degradation (termed Atg-proteins) have been characterized and can be used as reliable markers for autophagic degradation. Of these, the ubiquitin-like proteins Atg12 (that is conjugated to Atg5) and LC3/Atg8, Atg7, and the PI3-kinase complex-component Beclin-1 (BECN1)/Atg6 (essential for phagophore nucleation, as described above) are especially important for autophagosome formation and autophagic degradation. All of them have been targeted in the four above-mentioned GJ/autophagy studies (Bejarano et al. 2012; Fong et al. 2012; Hesketh et al. 2010; Lichtenstein et al. 2011) (also see Figure 2A–C).
Autophagosomal degradation of AGJs
Since cytoplasmic vesicles in general can fuse with endosomes, at first glance, autophagic degradation of AGJ vesicles might not appear intuitive. However, considering the structural organization of AGJ vesicles, including the dense packing of channels that occupy most or even the entire surface of AGJs in a double-membrane arrangement and their cytoplasmic location, autophagic degradation of AGJ vesicles appears much more plausible. In addition, the two tightly bound membranes of AGJ vesicles may not easily allow their fusion with single-membraned endosomes. Since, as explained above, lysosomal enzymes cannot be released into the cytosol, it is likely that the isolation membrane (the phagophore) formed during the initial steps of autophagy is required to sequester and separate the AGJ vesicle from the cytoplasm, thus providing the sealed membrane container that is required for subsequent lysosome fusion and lysosomal-based AGJ degradation. Finally, the unique structural composition of AGJ vesicles with lumen and inner membrane derived from the neighboring cell (being foreign to the AGJ-receiving host cell) may further direct AGJs to autophagic degradation. Also, while cell-cell communication provided by GJs prior to endocytosis can only accommodate the diffusion of small molecules, ions, and secondary messengers across the connected plasma membranes, endocytosis of GJs and formation of AGJ vesicles could in addition allow the transfer of potentially harmful substances from cell to cell, as for example unwanted regulatory proteins, other larger molecules (e.g. micro RNAs), or even cytoplasmic pathogens that could get entrapped in the AGJ vesicle lumen, further requiring efficient AGJ degradation. Taken together, these structural and functional characteristics, together with the fact that autophagy serves as the generic pathway for cytoplasmically localized degradation products (organelles and protein-aggregates), renders autophagic degradation the most likely cellular pathway for AGJ degradation.
Signals that may direct AGJs for degradation
Post-translational modification of proteins is a widespread mechanism to fine-tune the structure, function, and localization of proteins. One of the most versatile and intriguing protein-modifications is the covalent attachment of ubiquitin (Ub) or Ub-like modifications to target proteins. Ub is a 76-amino acid long protein, and either single or multiple Ub moieties can be conjugated to lysine amino acid residues of target proteins. An incredible diversity of mono- and poly-Ub chains (in which Ub moieties can be linked to each other via the Ub residues Met1-, Lys6-, Lys11-, Lys27-, Lys29-, Lys33-, Lys48-, and Lys63-) conjugated to target proteins have been characterized that can range in function from protein activation to protein degradation (see Fushman and Wilkinson 2011 for a recent review). Multiple mono-Ubs, and Lys48- and Lys63-linked poly-Ubs have been recognized as important signals for protein degradation. For example, conjugation of Ub moieties to proteins has been recognized as a signal for both proteasomal targeting (addition of Lys48-linked poly-Ub chains) and more recently as a sorting signal for internalized vesicles of the late endocytic pathway. This is achieved through the addition of multiple mono-Ub moieties or of Lys63-linked poly-Ub chains which ultimately lead to degradation by lysosomes (Hicke 2001; Hicke and Dunn 2003; Schnell and Hebert 2003; Shih et al. 2002; Stahl and Barbieri 2002). In addition, Lys-63-linked poly-ubiquitination can act as an internalization signal for clathrin-mediated endocytosis (CME) (Belouzard and Rouille 2006; Geetha et al. 2005). In this process, multiple mono-Ub moieties are attached to the target protein and are recognized by specific CME-machinery protein-components that associate with a subset of Ub-binding proteins, specifically Epsin1 and Eps15 (Barriere et al. 2006; Hawryluk et al. 2006; Madshus 2006). Further work has shown that the Ub-binding protein, p62/SQSTM1, recognizes and interacts via its UBA-domain with poly-ubiquitinated proteins (Ciani et al. 2003; Seibenhener et al. 2004; Wilkinson et al. 2001) and delivers poly-ubiquitinated (Lys63-linked) oligomeric protein complexes to the autophagic degradation pathway (Bjorkoy et al. 2005; Pankiv et al. 2007). Ubiquitination of Cx43-based GJs has been described previously (Catarino et al. 2011; Girao et al. 2009; Leithe et al. 2009; Leithe and Rivedal 2004b; reviewed in Kjenseth et al. 2010; Leithe et al. 2011). The findings that Cx43-based GJs can become ubiquitinated, the known affinity of p62/SQSTM1 for ubiquitinated protein complexes, its colocalization with plasma membrane GJs in HeLa, COS7, and PAE cells (Bejarano et al. 2012; Fong et al. 2012; Lichtenstein et al. 2011), and its apparent involvement in targeting AGJ vesicles to autophagic degradation (Fong et al. 2012) suggest that ubiquitination of Cx43 (and at least Cx50), besides serving as a likely signal for GJ internalization, may also serve as the signal for targeting AGJ vesicles to autophagic degradation. Future research will be required to determine the potentially numerous types (multiple mono-Ubs, Lys48- and Lys63-linked poly-Ubs, etc.) and functions of connexin ubiquitination (see Kjenseth et al. 2010; Leithe et al. 2011; Su and Lau 2012 for recent reviews that discuss Cx-ubiqitination). Very recently, Kjenseth et al. (2012) described an additional, Ub-like posttranslational modification of Cx43, SUMOylation (SUMO, small ubiquitin-like modifier) that appears to be involved in regulating GJ stability and turnover. The small Ub-like protein SUMO was found to be conjugated to lysines 144 and 237 of the Cx43-C-terminal domain further complicating the role of Ub and Ub-like signals in the maintenance and degradation of GJs.
Alternative, endo-/lysosomal degradation of AGJs
Interestingly, a recent paper by Leithe et al. (2009) reports that in TPA-treated cells (a structural analog of the secondary messenger molecule diacylglycerol [DAG]), internalized GJs appear to be degraded by the endo-/lysosomal and not the autophagosomal pathway (Figure 1, steps 11–15). Very recently, the Leithe lab identified the protein Smurf2 (the HECT E3 ubiquitin ligase smad ubiqitination regulatory factor-2) as a critical factor that regulates GJ internalization and endo-/lysosomal targeting in TPA-treated cells (Fykerud et al. 2012). DAG is a known potent activator of protein kinase C (PKC), and PKC is known to phosphorylate and promote ubiquitination of Cx43 (Leithe et al. 2009; Leithe and Rivedal, 2004b; Postma et al. 1998). Based on these and our own results, it is tempting to speculate that cells might be able to regulate by which pathway (endo-/lysosomal versus phago-/lysosomal) specific cargo is sequestered and processed (e.g. endo-/lysosomal and phago-/lysosomal pathways might process internalized GJs in different ways). Furthermore, the level of cargo-phosphorylation and/or ubiquitination might determine which of these pathways is ultimately chosen (basic phosphorylation/ubiquitination signaling autophagic AGJ vesicle degradation; elevated phosphorylation/ubiquitination signaling endo-/lysosomal AGJ vesicle degradation) (see Figure 1, steps 6–10 versus 11–15).
Endo-/lysosomal degradation of AGJs as observed in TPA-treated cells by Leithe et al. (Leithe et al. 2009) of course raises an important question: How is it structurally possible for a double-membrane vesicle that consists of tightly bonded membrane layers and densely packed GJ channels to fuse with a single-membrane endosome? The Rivedal and Leithe laboratories suggest that subsequent to GJ internalization and AGJ formation, the inner AGJ membrane splits and peels away from the outer AGJ membrane, generating a single-membraned cytoplasmic AGJ vesicle that then can fuse with a single-membraned endosome (Kjenseth et al. 2010; Kjenseth et al. 2012; Leithe et al. 2009; Leithe et al. 2011). However, since docked GJ channels can not split into undocked connexons under physiological conditions (Ghoshroy et al. 1995; Goodenough and Gilula 1974), -which appears to be the apparent reason for double-membrane GJ endocytosis-, it is not clear how membrane separation could be initiated in the AGJ vesicles shortly after their generation. Clearly low pH, a characteristic of late endosomes and lysosomes, and a potential initiator of GJ splitting can be excluded because AGJ vesicle membrane-separation needs to occur before AGJ/endosome fusion. Su and Lau also do not speculate on how cytoplasmic AGJs may end up inside endosomes, as is suggested in their recent review article (Su and Lau 2012).
Interestingly, we found that AGJ vesicles examined by electron microscopy may include a small area where the two membranes are void of GJ channels and are not docked or linked to each other (Piehl et al. 2007) (Figure 3A, and shown schematically in Figure 1, steps 4, 5, 11 and 12). Similar small AGJ membrane separations were also observed in classical ultrastructural analyses of GJs and AGJ vesicles (see e.g. Mazet et al. 1985). Possibly, these non-junctional membrane domains consist of plasma membrane that is derived from both neighboring cells, and we postulate that these areas might originate from plasma membrane regions that were located immediately adjacent to the GJ plaques and as well were internalized. To gain further support for this hypothesis, we incubated inducible stably Cx43-YFP expressing HeLa cells (described in Lauf et al. 2002) for 2–4 hours with a fluorescently-tagged lectin, Alexa594-wheat germ agglutinin (WGA), and examined AGJ vesicles by high-resolution fluorescence microscopy. WGA binds specifically to sialic acid and N-acetylglucosaminyl carbohydrate moieties commonly found on extracellular-exposed carbohydrate side-chains of plasma membrane proteins. Due to its relatively large size (~38 kDa), WGA is not able to traverse the plasma membrane in living cells. However, WGA will label the extracellular surface of plasma membranes and only subsequently will be endocytosed to also label intracellular membrane compartments (Wright 1984). Interestingly we found that a portion of cytoplasmic AGJ vesicles that were likely generated during the WGA-incubation period, exhibited red-fluorescent WGA-puncta (labeled with arrows in Figure 3B). These results support our hypothesis that the un-docked membrane domains we detected by EM on AGJ vesicles indeed represent plasma membrane that was located in the immediate vicinity of GJ plaques and was concomitantly internalized in the AGJ endocytosis process. For us, it is tempting to speculate that this non-junctional membrane domain could provide the single membrane area that would allow double-membrane AGJ vesicles to fuse with single-membrane endosomes.
Figure 3.
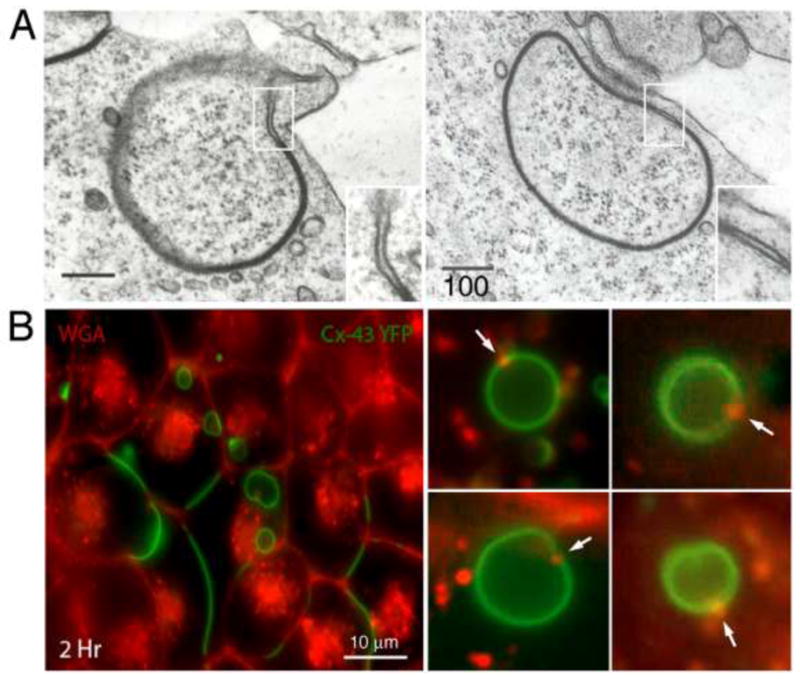
Fine structure and composition of AGJ vesicles. (A) Double-membrane AGJ vesicles can contain a patch of non-junctional membrane where the two membrane layers are separated (enlarged in inserts). These non-junctional membrane patches appear to be derived from plasma membrane that was located immediately adjacent to the GJ plaque and was concomitantly endocytosed. (B) These non-junctional AGJ membrane domains label with extracellularly applied, fluorescence-labeled wheat germ agglutinin (WGA) (red puncta on green AGJ vesicles marked with arrows) in Cx43-YFP expressing cells. Stable Cx43-YFP expressing HeLa cells were incubated for 2–4 h with Alexa594-labelled WGA and examined by fluorescence microscopy. Low magnification survey image (panel 1, left) and high-resolution images of internalized AGJ vesicles (panels 2–5, right) are shown (scale bars = nm in [A], and m in [B]). (Figure 3A is adapted from (Piehl et al. 2007).)
Concluding remarks
A number of recent and classic manuscripts describe the endocytosis of GJ plaques as double-membrane spanning structures (Baker et al. 2008; Falk et al. 2009; Ginzberg and Gilula 1978; Gumpert et al. 2008; Jordan et al. 2001; Larsen et al. 1979; Mazet et al. 1985; Piehl et al. 2007) and the autophagosomal, and potentially endo-/lysosomal degradation of the resulting cytoplasmic double-membrane AGJ vesicles (Bejarano et al. 2012; Fong et al. 2012; Hesketh et al. 2010; Leach and Oliphant 1984; Leithe et al. 2009; Lichtenstein et al. 2011; Pfeifer 1980; Severs et al. 1989). Based on the recent and classic observations described above, we hypothesize that double-membrane GJ endocytosis is the only practical means of how cells can efficiently remove plasma membrane-localized GJ plaques. Potential GJ splitting, or GJ dispersion (Lane and Swales 1980) have not been observed by us in any of our multiple life-cell recordings and therefore are not considered efficient mechanisms of GJ removal. Furthermore, based on the recently published evidence suggesting autophagosomal degradation of AGJ vesicles (Fong et al. 2012; Hesketh et al. 2010; Lichtenstein et al. 2011), the knowledge that autophagy serves as the generic pathway for the degradation of cytoplasmically localized organelles and protein-aggregates, and based on the known structural composition of AGJ vesicles consisting of tightly sealed-together membrane layers, we further hypothesize that the default degradation pathway for these cytoplasmic structures is (macro)autophagy. Alternatively, endo-/lysosomal AGJ degradation described to occur in TPA-treated cells may be achieved based on the presence of AGJ membrane-domains where the two membrane-layers are not linked to each other (such as the ones we have characterized and described above, see Figure 3), providing the structural prerequisite to allow single-membrane endosomes to fuse with double-membrane AGJ vesicles.
Finally, we speculate that the level of connexin phosphorylation and/or ubiquitination might allow cells to regulate by which pathway endo-/lysosomal versus phago-/lysosomal, endocytosed AGJ vesicles are sequestered and processed; an interesting hypothesis considering that autophagosomal and endo-/lysosomal processing may render different (degradation) products. Future research will elucidate whether our hypotheses are correct, and if there are additional cellular pathways that may dictate the fate of endocytosed GJs.
Acknowledgments
Work in the laboratory of M.M.F. is supported by NIHs NIGMS (grant GM55725) and by Lehigh University. We thank additional members of the Falk laboratory for critical comments. We wish to extend our sincere appreciation to Ross Johnson who’s dedication to gap junctions has revealed so many exciting aspects of this truly amazing cellular structure. His excitement and passion has fueled this research, and will fuel the passion of many generations of researchers to come. Although we know so much about gap junctions, so much more still needs to be discovered! His contributions to this field have been significant.
Abbreviations used
- AGJ
annular gap junction
- CME
clathrin mediated endocytosis
- Cx
connexin
- DAG
diacylglycerol
- GFP
green fluorescent protein
- GJ
gap junction
- GJIC
gap junction mediated intercellular communication
- LAMP
lysosomal associated membrane protein
- LC3
microtubule-associated protein light chain 3
- PAEC
pulmonary artery endothelial cell
- PE
phosphatidyl-ethanolamine
- PKC
protein kinase C
- RNAi
RNA interference
- SQSTM1
sequestosome 1
- SUMO
small Ub-like modifier
- TPA
12-O-tetradecanoylphorbol 13-acetate
- Ub
ubiquitin
- WGA
wheat germ agglutinin
- YFP
yellow fluorescent protein
References
- Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008;582:4039–4046. doi: 10.1016/j.febslet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AL, Thurston G. Mechanics of endothelial cell architecture and vascular permeability. Crit Rev Biomed Eng. 2001;29:247–278. doi: 10.1615/critrevbiomedeng.v29.i2.20. [DOI] [PubMed] [Google Scholar]
- Barriere H, Nemes C, Lechardeur D, Khan-Mohammad M, Fruh K, Lukacs GL. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in Mammalian cells. Traffic. 2006;7:282–297. doi: 10.1111/j.1600-0854.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012;23:2156–2169. doi: 10.1091/mbc.E11-10-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S, Rouille Y. Ubiquitylation of leptin receptor OB-Ra regulates its clathrin-mediated endocytosis. EMBO J. 2006;25:932–942. doi: 10.1038/sj.emboj.7600989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62:256–267. doi: 10.1016/j.cardiores.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomstrand F, Venance L, Siren AL, Ezan P, Hanse E, Glowinski J, Ehrenreich H, Giaume C. Endothelins regulate astrocyte gap junctions in rat hippocampal slices. Eur J Neurosci. 2004;19:1005–1015. doi: 10.1111/j.0953-816x.2004.03197.x. [DOI] [PubMed] [Google Scholar]
- Catarino S, Ramalho JS, Marques C, Pereira P, Girao H. Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem J. 2011;437:255–267. doi: 10.1042/BJ20102059. [DOI] [PubMed] [Google Scholar]
- Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget’s disease of bone. J Biol Chem. 2003;278:37409–3712. doi: 10.1074/jbc.M307416200. [DOI] [PubMed] [Google Scholar]
- Delmar M, Coombs W, Sorgen P, Duffy HS, Taffet SM. Structural bases for the chemical regulation of Connexin43 channels. Cardiovasc Res. 2004;62:268–275. doi: 10.1016/j.cardiores.2003.12.030. [DOI] [PubMed] [Google Scholar]
- Falk MM. Adherens junctions remain dynamic. BMC Biol. 2010;8:34. doi: 10.1186/1741-7007-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW., 3rd Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20:3342–3352. doi: 10.1091/mbc.E09-04-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell Biol. 1981;90:521–526. doi: 10.1083/jcb.90.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JT, Kells RM, Gumpert AM, Marzillier JY, Davidson MW, Falk MM. Internalized gap junctions are degraded by autophagy. Autophagy. 2012 May 1;8(5) doi: 10.4161/auto.19390. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 2011;3:26. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fykerud TA, Kjenseth A, Schink KO, Sirnes S, Bruun J, Omori Y, Brech A, Rivedal E, Leithe E. Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J Cell Sci. 2012 May 23; doi: 10.1242/jcs.093500. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell. 2005;20:301–312. doi: 10.1016/j.molcel.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Ghoshroy S, Goodenough DA, Sosinsky GE. Preparation, characterization, and structure of half gap junctional layers split with urea and EGTA. J Membr Biol. 1995;146:15–28. doi: 10.1007/BF00232677. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- Gilleron J, Fiorini C, Carette D, Avondet C, Falk MM, Segretain D, Pointis G. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008;121:4069–4078. doi: 10.1242/jcs.033373. [DOI] [PubMed] [Google Scholar]
- Ginzberg RD, Gilula NB. Modulation of cell junctions during differentiation of the chicken otocyst sensory epithelium. Dev Biol. 1979;68:110–129. doi: 10.1016/0012-1606(79)90247-1. [DOI] [PubMed] [Google Scholar]
- Girao H, Catarino S, Pereira P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res. 2009;315:3587–3597. doi: 10.1016/j.yexcr.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Gilula NB. The splitting of hepatocyte gap junctions and zonulae occludentes with hypertonic disaccharides. J Cell Biol. 1974;61:575–590. doi: 10.1083/jcb.61.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008;582:2887–2892. doi: 10.1016/j.febslet.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk MJ, Keyel PA, Mishra SK, Watkins SC, Heuser JE, Traub LM. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic. 2006;7:262–281. doi: 10.1111/j.1600-0854.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, Kass DA, Machamer CE, Van Eyk JE, Tomaselli GF. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res. 2010;106:1153–1163. doi: 10.1161/CIRCRESAHA.108.182147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009;5:502–510. doi: 10.4161/auto.5.4.8096. [DOI] [PubMed] [Google Scholar]
- Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114:763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjenseth A, Fykerud T, Rivedal E, Leithe E. Regulation of gap junction intercellular communication by the ubiquitin system. Cell Signal. 2010;22:1267–1273. doi: 10.1016/j.cellsig.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Kjenseth A, Fykerud TA, Sirnes S, Bruun J, Kolberg M, Yohannes Z, Omori Y, Rivedal E, Leithe E. The gap junction channel protein connexin43 is covalently modified and regulated by SUMOylation. J Biol Chem. 2012 Mar 12; doi: 10.1074/jbc.M111.281832. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing JG, Tadros PN, Westphale EM, Beyer EC. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane NJ, Swales LS. Dispersal of junctional particles, not internalization, during the in vivo disappearance of gap junctions. Cell. 1980;19:579–586. doi: 10.1016/s0092-8674(80)80034-1. [DOI] [PubMed] [Google Scholar]
- Larsen WJ, Tung HN, Murray SA, Swenson CA. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol. 1979;83:576–587. doi: 10.1083/jcb.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DH, Oliphant LW. Degradation of annular gap junctions of the equine hoof wall. Acta Anat (Basel) 1984;120:214–219. doi: 10.1159/000145923. [DOI] [PubMed] [Google Scholar]
- Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci. 2009;122:3883–3893. doi: 10.1242/jcs.053801. [DOI] [PubMed] [Google Scholar]
- Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004a;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem. 2004b;279:50089–50096. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- Leithe E, Sirnes S, Fykerud T, Kjenseth A, Rivedal E. Endocytosis and post-endocytic sorting of connexins. Biochim Biophys Acta. 2011;1818:1870–1879. doi: 10.1016/j.bbamem.2011.09.029. [DOI] [PubMed] [Google Scholar]
- Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124:910–920. doi: 10.1242/jcs.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- Madshus IH. Ubiquitin binding in endocytosis--how tight should it be and where does it happen? Traffic. 2006;7:258–261. doi: 10.1111/j.1600-0854.2006.00393.x. [DOI] [PubMed] [Google Scholar]
- Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25. doi: 10.3410/B3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazet F, Wittenberg BA, Spray DC. Fate of intercellular junctions in isolated adult rat cardiac cells. Circ Res. 1985;56:195–204. doi: 10.1161/01.res.56.2.195. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Moreno AP. Connexin phosphorylation as a regulatory event linked to channel gating. Biochim Biophys Acta. 2005;1711:164–171. doi: 10.1016/j.bbamem.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- Pahujaa M, Anikin M, Goldberg GS. Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp Cell Res. 2007;313:4083–4090. doi: 10.1016/j.yexcr.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pfeifer U. Autophagic sequestration of internalized gap junctions in rat liver. Eur J Cell Biol. 1980;21:244–246. [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol. 2009;11:65–70. doi: 10.1038/ncb1813. [DOI] [PubMed] [Google Scholar]
- Postma FR, Hengeveld T, Alblas J, Giepmans BN, Zondag GC, Jalink K, Moolenaar WH. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Shao Q, Igdoura SA, Alaoui-Jamali MA, Laird DW. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J Biol Chem. 2003;278:30005–30014. doi: 10.1074/jbc.M300614200. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121:1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett JM, Gourdie RG. The perinexus: A new feature of Cx43 gap junction organization. Heart Rhythm. 2011;9:619–623. doi: 10.1016/j.hrthm.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell DJ, Hebert DN. Protein translocons: multifunctional mediators of protein translocation across membranes. Cell. 2003;112:491–505. doi: 10.1016/s0092-8674(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severs NJ, Shovel KS, Slade AM, Powell T, Twist VW, Green CR. Fate of gap junctions in isolated adult mammalian cardiomyocytes. Circ Res. 1989;65:22–42. doi: 10.1161/01.res.65.1.22. [DOI] [PubMed] [Google Scholar]
- Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol. 2002;4:389–393. doi: 10.1038/ncb790. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinella F, Rosano L, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 decreases gap junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem. 2003;278:41294–41301. doi: 10.1074/jbc.M304785200. [DOI] [PubMed] [Google Scholar]
- Stahl PD, Barbieri MA. Multivesicular bodies and multivesicular endosomes: the “ins and outs” of endosomal traffic. Sci STKE. 2002;2002:pe32. doi: 10.1126/stke.2002.141.pe32. [DOI] [PubMed] [Google Scholar]
- Su V, Lau AF. Ubiquitination, intracellular trafficking, and degradation of connexins. Arch Biochem Biophys. 2012 Jan 3; doi: 10.1016/j.abb.2011.12.027. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- van Zeijl L, Ponsioen B, Giepmans BN, Ariaens A, Postma FR, Varnai P, Balla T, Divecha N, Jalink K, Moolenaar WH. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2007;177:881–891. doi: 10.1083/jcb.200610144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lau AF. Regulation of gap junctions by tyrosine protein kinases. Biochim Biophys Acta. 2004;1662:81–95. doi: 10.1016/j.bbamem.2003.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson CR, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, Gordon C. Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol. 2001;3:939–943. doi: 10.1038/ncb1001-939. [DOI] [PubMed] [Google Scholar]
- Wong CW, Christen T, Kwak BR. Connexins in leukocytes: shuttling messages? Cardiovasc Res. 2004;62:357–367. doi: 10.1016/j.cardiores.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Wright CS. Structural comparison of the two distinct sugar binding sites in wheat germ agglutinin isolectin II. J Mol Biol. 1984;178:91–104. doi: 10.1016/0022-2836(84)90232-8. [DOI] [PubMed] [Google Scholar]
- Yen WL, Shintani T, Nair U, Cao Y, Richardson BC, Li Z, Hughson FM, Baba M, Klionsky DJ. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol. 2010;188:101–114. doi: 10.1083/jcb.200904075. [DOI] [PMC free article] [PubMed] [Google Scholar]