

Abstract
Vitamin D3, and its most active form, 1,25(OH)2D3, are well known to stimulate osteoclastogenesis through stromal cell induction of the receptor activator of nuclear factor-κB ligand (RANKL). MAPK phosphatase-1 (MKP-1) is a phosphatase classically known to negatively regulate the innate immune response through dephosphorylation of p38, ERK, and c-Jun N-terminal kinase activity. This paper describes a new function of MKP-1 in permitting genomic 1,25(OH)2D3 signaling and downstream osteoclastogenesis through RANKL. Initially, quantitative RT-PCR (qRT-PCR) and immunoblot analysis comparing bone marrow stromal cells (BMSC) revealed that 1,25(OH)2D3-induced vitamin D receptor (VDR), cytochrome P 45024a1, and RANKL mRNA expression and protein were significantly attenuated or absent in MKP-1−/− BMSC. Immunoblot analysis from cellular fractions of wild type and MKP-1−/− BMSC stimulated with 10−7 m 1,25(OH)2D3 revealed retinoid X receptor (RXR)α nuclear import was impaired in MKP-1−/− BMSC, whereas VDR import was not. Proximity ligation assays revealed that baseline VDR-RXRα heterodimer translocation was unchanged, yet 1,25(OH)2D3-induced nuclear translocation of VDR-RXRα heterodimers was reduced in MKP-1−/− BMSC. A functional consequence was observed as BMSC from MKP-1−/− mice treated with 1,25(OH)2D3 and cocultured with RAW 264.7 cells had a 91% decrease in osteoclastogenesis and a 94.5% decrease in mineralized matrix resorption compared with wild-type cocultures (P < 0.01). These results reveal an unexpected, permissive role for MKP-1 in canonical 1,25(OH)2D3 signaling via VDR-RXRα heterodimer nuclear import and downstream osteoclastogenesis through stromal cell RANKL expression.
Since its discovery as an antirachitic factor, vitamin D has been widely studied for its role in skeletal homeostasis (1). The active form of this steroid hormone, 1,25-(OH)2D3, and the vitamin D receptor (VDR) have been well studied for their roles in bone mineralization and turnover. It is widely accepted that the downstream genomic events of 1,25-(OH)2D3-bound VDR are cell specific and can significantly alter the formation and activity of osteoblasts and osteoclasts. A human deficiency in this hormone can result in rickets in children and osteomalacia in adults (2, 3). The importance of the VDR as a 1,25-(OH)2D3 receptor is highlighted by type II vitamin D-dependent rickets (OMIM 277440) whereby mutations in the DNA-binding domain of the VDR result in a rickettsial phenotype that is clinically similar to vitamin D deficiency (4).
The classic genomic mechanism by which 1,25-(OH)2D3-ligated VDR regulates gene expression in osteoblasts is through ligand-initiated heterodimerization with retinoid X receptor (RXR)-α, translocation to the nucleus, and binding of specific DNA regions, namely vitamin D-responsive elements (VDRE) and coactivator recruitment (5, 6). RXRα is a nuclear receptor family member that regulates the action of many endocrine processes through heterodimerization with a variety of hormone receptors, including thyroid receptor, peroxisome proliferator-activated receptor, VDR, and other retinoic acid receptors (7). RXRα has been shown to have a profound role in VDR genomic signaling, in part by regulating subcellular localization of the RXRα-VDR heterodimer (8). The mouse VDR gene has two primary intronic VDR-RXR binding VDRE at +20 and +29 kb and therefore induces its own expression in the presence of 1,25-(OH)2D3 (9, 10). VDRE have been found on the promoter regions of many genes involved in calcium homeostasis including receptor activator of nuclear factor-κB ligand (RANKL), an essential cytokine in the bone-resorbing process of osteoclastogenesis (11–13). Five mouse and human VDRE have been identified upstream of the transcriptional start site of the RANKL gene (TNFS11), with the most distal and potent murine VDRE (mRLD5) occurring at −76 kb (11, 14). Along with PTH as one of the primary endocrine inducers of RANKL in active osteoblasts, vitamin D and its antagonists have been widely studied for their therapeutic potential in bone pathologies (15–17). Recently, both 1,25-(OH)2D3 and exogenous RANKL stimulation of primary bone marrow-derived macrophage cells have been shown to induce the immune regulatory protein MAPK phosphatase-1 (18, 19).
The MAPK phosphatase (MKP) family of proteins has the ability to negatively regulate MAPK activity by dephosphorylating MAPK proteins. MAPK phosphatase-1 (MKP-1) is a phosphatase that negatively regulates the innate immune response through dephosphorylation of p38, ERK, and c-Jun N-terminal kinase (JNK) activity in response to various stimuli (20–23). This effect has been demonstrated by our laboratory where lipopolysaccharide-challenged MKP-1−/− mice exhibit a significant increase in osteoclastogenesis and subsequent alveolar bone loss compared with wild-type controls (24). Recently, MKP-1 has gained acceptance as a potential pharmacological target for the treatment of obesity and metabolic disorders (25).
Although MAPK activity has also been shown to inhibit genomic vitamin D activity through RXRα modification (26, 27), the role of MKP in vitamin D signaling and steroid receptor translocation is currently unknown. Limited studies on MKP-1's effect on skeletal homeostasis reveal that female, but not male, MKP-1−/− mouse femurs have decreased trabecular density at 8 wk, with a decrease in mineral apposition rate and bone formation rate (18). Although these studies strongly suggest a role for MKP-1 in skeletal homeostasis and maintenance, there has yet to be a clearly defined mechanism for its involvement in these processes. This paper presents a new, highly novel role for MKP-1 whereby it is required in both the genomic 1,25-(OH)2D3-induced VDRE binding of the RXRα-VDR heterodimer and downstream target gene expression.
Materials and Methods
Reagents
General biochemicals were obtained from Sigma Chemical Co. (St. Louis, MO). 1,25-(OH)2D3 was obtained from Enzo Life Sciences (Farmingdale, NY). Life Technologies MEM, α-MEM, and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA), as were oligonucleotides used in EMSA and chromatin immunoprecipitation (ChIP) analysis. ON-TARGETplus pooled MKP-1 small interfering RNA (siRNA) was obtained from Dharmacon (Lafayette, CO). Anti-RXRα [sc-553(x)] and anti-VDR [sc13133(x)] were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). [α-32P]dCTP (BLU513Z250UC) (6000 Ci/mmol) was obtained from PerkinElmer Corp. (Wellesley, MA).
Animals
Wild-type (WT) and MKP-1-knockout mice were purchased from Bristol-Myers Squibb Pharmaceutical Research Institute and bred at the Medical University of South Carolina and were treated in accordance with the standards of humane animal care. These mice are maintained on a mixed C57/129 background and housed under specific pathogen-free conditions with food and tap water ad libitum.
Cell culture
Primary bone marrow cells were harvested by flushing the femurs and tibia of 5- to 10-wk-old mice with α-MEM supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin and 10% heat-inactivated fetal bovine serum. Cells were passed through a 40-μm filter into a 100-mm cell culture plate. After 24 h of incubation, nonadherent cells were removed, and the plate was washed vigorously with PBS. Cells were washed again at 6 d and at 10 d the cells were scraped and plated at a density of 5 × 105 cells per well in a 12-well plate. Before 1,25-(OH)2D3 or vehicle (EtOH) treatment, bone marrow stromal cells (BMSC) and UAMS-32 cells were deinduced for 6 h with 0.3% fetal bovine serum.
RT-quantitative PCR (qPCR)
Whole-cell BMSC were harvested in TRIzol, and RNA was harvested according to protocol. Total RNA was quantified and 500 ng were reverse-transcribed into cDNA, which was used in qPCR performed in a TaqMan system using an Applied Biosystems StepOnePlus RT-PCR Real-Time Thermal Cycler, optimized Assays-On-Demand primers for the target genes MKP-1, RANKL, VDR, cytochrome P450 (CYP)24a1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for normalization.
siRNA studies
All siRNA duplexes from purchased from Dharmacon RNA Technologies (Lafayette, CO) and electroporation-based transfection was performed using the Neon Transfection System obtained from Invitrogen.
Western blot analyses
Whole-cell samples were harvested in RIPA buffer, and 20 μg total protein were loaded onto 10% SDS-PAGE gels and transferred to nitrocellulose for immunoblotting to detect RXRα and VDR protein expression. Nuclear and cytoplasmic fractions were isolated using the NE-PER kit from Thermo Scientific (Rockford, IL) according to manufacturer's instructions. Digital images were acquired and quantified using Quantity One software from Bio-Rad Laboratories, Inc. (Hercules, CA).
EMSA
WT and MKP-1−/− nuclear extracts (6 μg) were isolated (as described above) and were pretreated on ice with 10−7 m 1,25-(OH)2D3 and 1 μg polydeoxyinosinic deoxycytidylic acid for 20 min in a binding buffer [25 mm Tris-HCl (pH 8.0), 5% glycerol, 0.5 mm dithiothreitol, 100 mm KCl]. α-32P-labeled mouse osteopontin (mOP) VDRE oligonucleotide (mOP VDRE; 100 fmol) was added and incubated for 20 min at room temperature (forward, 5′–3′ GGGTACAAGGTTCACGAGGTTCACGTCTTA). Indicated lanes were also incubated with anti-RXRα antibody (sc-553x), recognizing the NH2-terminal domain of RXRα, anti-VDR antibody (sc13133x), specific to VDR amino acids 344–424, or control rabbit and mouse IgG.
ChIP assays
Primary BMSC were isolated and cultured as described above and plated on 150-mm cell culture plates and treated with or without 10−7 m 1,25-(OH)2D3 for 6 h before cells were cross-linked with 1% formaldehyde α-MEM. ChIP experiments were performed using reagents supplied following manufacturer's instructions supplied with the ChIP-IT Express kit from Active Motif (Carlsbad, CA). Chromatin pellets were sonicated to an average DNA size of 250-1000 bp (determined by gel electrophoresis) using a Fisher Model 100 Sonic Dismembranator on ice (Fisher Scientific, Pittsburgh, PA). Incubation with the indicated antibodies was performed overnight at 4 C. DNA fragments were subjected to PCR using primers designed to amplify the murine RANKL D5 region located −76 kb from the RANKL transcription start site (TSS) (forward, 5′-AAGGGTCAAGAGGGGCCTGACT; reverse, 5′-TGTCCCGGTCTCTGTCCCTGG) and a negative control region at −50 kb from the TSS designated IS4 (forward, 5′-CCTCCTATCTGTTTTACTGACGTT; reverse, 5′-CACATAGGCAGAAAGTTGAAAAGC) (11). Input DNA was acquired before immunoprecipitation and was loaded at 10% to determine the presence of gene fragments for PCR evaluation. PCR for both primer sets were run for 35 cycles and resolved on 2% agarose gels.
Immunofluorescence and in situ proximity ligation assay (PLA)
WT and MKP-1−/− BMSC were seeded at 3 × 105 cells per well in eight-well chamber slides (Labtek, Scotts Valley, CA). After a 24-h attachment period and 6 h in 0.5% α-MEM, cells were treated with vehicle control (ethanol) or 10−7 m 1,25-(OH)2D3 for 1 h. Medium was removed, and intact cells were fixed and permeabilized with −20 C MeOH for 8 min. Anti-RXRα (sc-553) and anti-VDR (sc13133) antibodies were used in immunofluorescence detection of nuclear/extranuclear RXRα and VDR native proteins, and in the PLA assay at a 1:500 dilution to detect the nuclear/extranuclear localization of the native VDR-RXRα heterodimer. Incubation with appropriate PLA probe (antimouse or antirabbit antibody), antibody blocking, ligation of probes via DNA ligase, DNA polymerase amplification, and slide mounting were performed using the proprietary solutions supplied in the Duolink kit from Olink Biosciences (Uppsala, Sweden) following manufacturer's instructions and supplied reagents (28). Negative control experiments (either anti-RXRα or anti-VDR were replaced with same species control IgG) were performed in parallel. Both immunofluorescence and PLA slides were analyzed using a Leica SP5 inverted confocal laser microscope (Leica Microsystems SA, Deerfield, IL) with a ×40 oil immersion objective. Z-stacks were taken at a density of one photo/0.5 μm, and the z-range was limited to the dimension of nuclear 4′,6-diamidino-2-phenylindole staining. Z-stacks were overlaid, and image processing and analysis were done using the Leica confocal software. Punctate PLA signals were quantified by a blinded observer.
Osteoclast coculture analysis
WT and MKP-1−/− BMSC were depleted of CD11b+ cells using a Miltenyi autoMACS Pro separator magnetic cell sorter (Miltenyi Biotec, Bergisch Gladbach, Germany) to reduce the RANKL-induced osteoclastogenesis in uncommitted primary macrophage populations (29), and plated (5 × 104 cells per well) in four-well chamber slides or Osteologic discs (Becton Dickinson, Franklin Lakes, NJ). CD11b− BMSC were pretreated with 10−8 m 1,25-(OH)2D3 for 2 d before 1 × 104 RAW 264.7 cells were added to each well. (10−8 m) 1,25-(OH)2D3 and half medium was changed every other day for 7 d [tartrate-resistant acid phosphatase (TRAP) staining] or 10 d (pit assay). Images of cultured cells were digitally captured using a Nikon TS100 inverted microscope and a Nikon 5.1 megapixel camera (Nikon, Melville, NY). Osteoclast or pit measurements were enumerated by blinded technicians. Pit areas were quantified with the assistance of NIH ImageJ version 1.44.
TRAP staining
Osteoclast cocultures were fixed with 10% glutaraldehyde and stained with TRAP as described using an Acid Phosphatase, Leukocyte TRAP Kit (Sigma).
Statistical analysis
All values presented in the figures are expressed as the mean ± sem unless otherwise stated. All statistical analyses were performed with GraphPad PRISM version 4 (GraphPad Software Inc., San Diego, CA). Differences between groups were evaluated by two-way ANOVA or Student's two-tailed t test. Significance was determined at P < 0.05.
Results
MKP-1 is required for RANKL expression in BMSC
Previously published reports from our laboratory indicate that MKP-1−/− mice have increased inflammatory bone loss in response to subgingival lipopolysaccharide (LPS) injections due to an elevation in cytokine-induced osteoclastogenesis (24). We hypothesized that this was due to an increase in RANKL expression and accordingly tested this hypothesis by treating primary WT and MKP-1−/− BMSC with the inflammatory cytokines IL-1β and TNF-α, as well as LPS from oral microbes Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis, and 1,25-(OH)2D3. Although we observed a 2-fold elevation in baseline RANKL mRNA expression, the only treatment in MKP-1−/− BMSC that was significantly different from WT was the 1,25-(OH)2D3 signaling. Surprisingly, we observed a 12-fold decrease in 1,25-(OH)2D3-stimulated RANKL mRNA expression in MKP-1−/− BMSC (Fig. 1A). To test whether this finding correlated with soluble RANKL protein expression, primary WT and MKP-1−/− BMSC were incubated with 10−8 m 1,25-(OH)2D3 for 48 h, and an ELISA was performed for secreted/membrane cleaved RANKL in the culture medium (Fig. 1B). Consistent with the RT-qPCR findings, soluble RANKL protein expression was also decreased in MKP-1−/− BMSC in response to 1,25-(OH)2D3. Subsequent experiments sought to determine the mechanism underlying these observation and determine the role of MKP-1 signaling in 1,25-(OH)2D3-mediated stromal/osteoblast gene expression.
Fig. 1.

MKP-1 is required in BMSC for 1,25-(OH)2D3 (D3) induction of RANKL. A, RT-qPCR analysis of RANKL mRNA expression in response to 18 h treatment with inflammatory stimuli and 10−8 m 1,25-(OH)2D3. B, ELISA of soluble RANKL expression from cultured media containing indicated cytokines, LPS, or 10−8 m 1,25-(OH)2D3 for 48 h (n = 4; ***, P < 0.001). Aa, Aggregatibacter actinomycetemcomitans; Pg, Porphyromonas gingivalis.
RT-qPCR analysis of 1,25-(OH)2D3 target gene expression in MKP-1−/− BMSC
Although data from RT-qPCR and ELISA analysis showed that MKP-1 is necessary for 1,25-(OH)2D3-stimulated RANKL expression in BMSCs, we sought to determine whether the requirement of MKP-1 in 1,25-(OH)2D3 signaling was limited to RANKL induction. In addition, we wanted to understand if MKP-1-mediated RANKL inhibition was immediate or occurred over several hours (30). To address these questions, we treated WT and MKP-1−/− BMSC with 10−8 m 1,25-(OH)2D3 for 0, 2, 4, 8, 12, 18, and 24 h and then performed RT-qPCR for three well-established 1,25-(OH)2D3 target genes: RANKL, VDR, and CYP24a1 (Fig. 2A). As expected, this kinetic assay showed that all three 1,25-(OH)2D3 target genes in WT BMSC had a peak expression between 12 and 18 h. Conversely, although MKP-1−/− BMSC exhibited similar baseline expression up to 4 h, peak mRNA expression in the 12- to 18-h range did not occur, indicating a defect in genomic 1,25-(OH)2D3 signaling. Western blotting for VDR expression after 24-h 1,25-(OH)2D3 treatment indicated that MKP-1−/− BMSC had a decrease in VDR protein expression as well (Fig. 2, B and C). These data are consistent with the notion that MKP-1 was required for 1,25-(OH)2D3 genomic signaling.
Fig. 2.

MKP-1 is necessary for maximal genomic expression of 1,25-(OH)2D3 target genes RANKL, VDR, and CYP24a1. A, WT and MKP-1−/− BMSC were stimulated with 10−8 m 1,25-(OH)2D3 for 0, 2, 4, 8, 12, 18, and 24 h. RT-qPCR results above were normalized to GAPDH expression and shown as fold change relative to WT control (n = 3). B,) Western blot analysis for VDR after 24 h 10−8 m 1,25-(OH)2D3 stimulation indicates that MKP-1 is required for 1,25-(OH)2D3 induction of VDR protein expression. C, Densitometric analysis of VDR expression normalized to GAPDH (n = 3; *, P < 0.05, **, P < 0.01).
MKP-1 is required for the maximal 1,25-(OH)2D3 target gene expression and RANKL promoter activity in UAMS-32 stromal/osteoblast cells
Because primary BMSC are not a homogenous population of cells, and 1,25-(OH)2D3 target gene expression in osteoblasts has been well described (6, 31), we wanted to confirm our hypothesis that MKP-1 plays a role in stromal osteoblast development. We transfected the UAMS-32 mouse stromal/osteoblast cell line with 100 nm pooled MKP-1 siRNA using an optimized electroporation protocol and were able to knock down MKP-1 mRNA expression by 70% after an 18-h 1,25-(OH)2D3 treatment (Fig. 3A). MKP-1 knockdown had no effect on unstimulated RANKL, VDR, and CYP24a1 (data not shown), yet RT-qPCR revealed that 1,25-(OH)2D3-stimulated CYP24a1 and RANKL mRNA expression was significantly decreased, and VDR mRNA expression was actually found to be below baseline transcript levels (Fig. 3A). These findings demonstrate that MKP-1 is necessary for 1,25-(OH)2D3 target gene expression in stromal/osteoblast cells and that the decrease in their expression, as shown in Fig. 2, is due, at least in part, to the permissive role of MKP-1 in stromal/osteoblast cells.
Fig. 3.
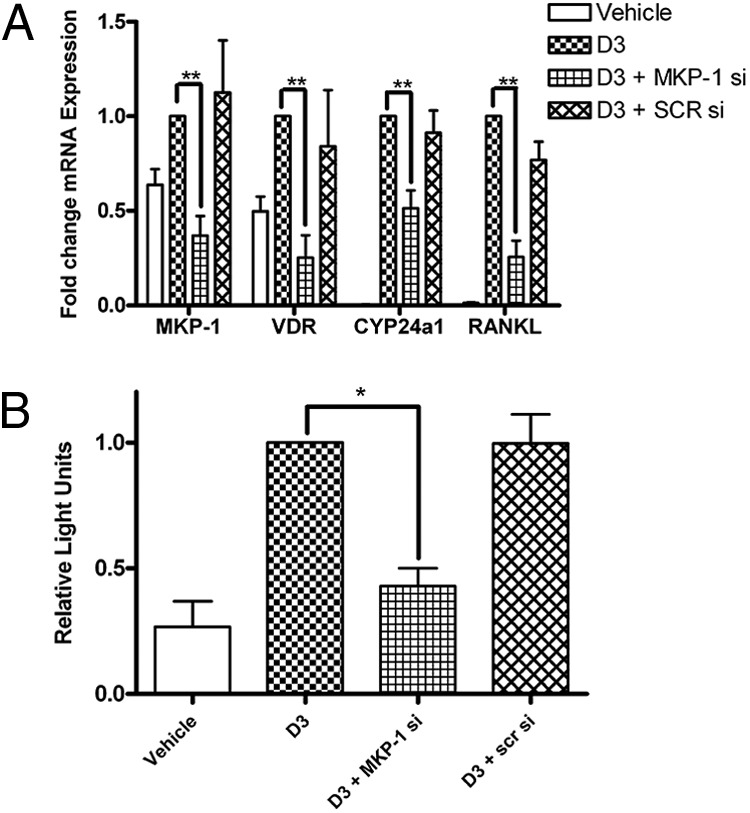
MKP-1 is needed for maximal 1,25-(OH)2D3 target mRNA expression and RANKL promoter activity in a UAMS32 Luc-reporter stromal cell line. A, UAMS-32 stromal cells were treated with 10−8 m 1,25-(OH)2D3 for 18 h followed by qPCR analysis. B, MKP-1 siRNA-transfected UAMS-32 stromal cells were stimulated with 10−8 m 1,25-(OH)2D3 for 18 h, followed by qPCR analysis. (n = 4; *, P < 0.05; **, P < 0.01). SCR, Scrambled.
Understanding that MKP-1 is required for 1,25-(OH)2D3 target mRNA expression, we examined whether MKP-1 signaling had an effect at the transcription level. We performed a similar MKP-1 siRNA experiment with UAMS-32 cells that contained a bacterial artificial chromosome-based transcriptional reporter containing the entire mouse RANKL gene with 120 kb of RANKL 5′-flanking region and 14-kb 3′-flanking region, as described previously (32). As positive controls, reporter activity of these cells was significantly increased by PTH (data not shown) and 1,25-(OH)2D3 treatment, with a 50% decreased response upon VDR siRNA treatment (data not shown). Time course experiments revealed maximal 1,25-(OH)2D3 induction of RANKL gene promoter activity at 18 h (our unpublished data). Using the same conditions as shown in Fig. 3A, we demonstrated that the MKP-1 knockdown in osteoblasts significantly decreased 1,25-(OH)2D3-induced RANKL promoter activity by 57% (P = 0.05; Fig. 3B).
Gel mobility shift and ChIP analysis reveals that VDR-RXRα heterodimer VDRE binding is attenuated in MKP-1−/− BMSC
Having determined that MKP-1 is necessary for the genomic expression of several 1,25-(OH)2D3 target genes and for RANKL gene promoter activity, we explored the possibility that MKP-1 permits the VDR-RXRα heterodimer's ability to access/bind VDRE in BMSC nuclear extracts. We treated primary WT and MKP-1−/− BMSC with 10−7 m 1,25-(OH)2D3 for 1 h and performed an EMSA with the harvested nuclear extracts (containing phosphatase inhibitors) and the mouse osteopontin (mOP) oligonucleotide containing the VDRE sequence GGTTCAxxxGGTTCA, which strongly interacts with the VDR-RXRα heterodimer (9, 33). Incubation with the α-32P-labeled mOP oligonucleotide yielded a relatively equal shift with both WT and MKP-1−/− extract that was abolished using nonradiolabeled mOP competition. Incubation with antibodies against VDR and RXRα consistently produced a strong supershift in WT extracts that diminished the original shift (Fig. 4). Using MKP-1−/− nuclear extracts, these RXRα and VDR antibodies consistently produced a visibly fainter supershift, whereas incubation with control rabbit IgG had no effect. In EMSA assays in which BMSC were only treated with vehicle control, a VDR and an RXRα supershift was observed in WT cells only (our unpublished data). ChIP analysis was used to test VDR and RXRα's ability to bind the distal (−76 kb) RANKL VDRE mRLD5 in WT and MKP−/− BMSC containing the binding sequences GAGTCAccgAGTTGA and GGTTGCctgAGTTCA (9). Although immunoprecipitates show that VDR is strongly induced to bind to the mRLD5 region by 1,25-(OH)2D3 in both genotypes, we only observe RXRα DNA binding in WT BMSC. These data provide evidence that MKP-1 is required for both maximal 1,25-(OH)2D3-induced nuclear binding of VDR and RXRα to a consensus VDRE and 1,25-(OH)2D3-induced binding of endogenous RXRα to the RANKL VDRE mRLD5.
Fig. 4.

EMSA analysis shows 1,25-(OH)2D3-stimulated MKP-1−/− BMSC have a decreased VDRE binding capacity. A, The α-32P-labeled mOP VDRE was incubated with 1,25-(OH)2D3-treated nuclear extracts from WT and MKP-1−/− BMSC and electrophoresed through a nondenaturing gel. R, RXRα supershifts; V, VDR antibody supershifts; *, the putative mOP-RXRα/VDR heterodimer complex. Results are representative of three independent experiments. B, ChIP analysis of the RANKL VDRE enhancer region located −76 kb from the RANKL TSS (mRLD5) and control non-VDRE region located −50 kb from the RANKL TSS (IS4). Results are representative of three independent experiments. Ctrl, Control.
1,25-(OH)2D3-induced nuclear import of the VDR-RXRα heterodimer is attenuated in MKP-1−/− BMSC
After the observation that there is a decreased capacity for VDRE binding in MKP-1−/− BMSC nuclear extracts, we sought to determine whether MKP-1−/− BMSC have an altered subcellular localization of RXRα and VDR. We treated WT and MKP-1−/− BMSC with 1,25-(OH)2D3 for 1 h, isolated the nuclear and cytoplasmic fractions, and subjected the protein extracts to Western blotting for RXRα and VDR. The results indicate that 1,25-(OH)2D3 induces a VDR translocation from the cytoplasmic to the nuclear fraction in both cell genotypes, although this VDR translocation may be delayed in MKP-1−/− BMSC, as evidenced by the relatively small reduction in cytoplasmic VDR upon 1,25-(OH)2D3 stimulation (Fig. 5A). The results show that upon 1,25-(OH)2D3 treatment, RXRα translocates freely into the nucleus from the cytosol in WT BMSC, presumably as a VDR-RXRα heterodimer. In MKP-1−/− BMSC, however, there is no detectable translocation, as evidenced by the lack of an increase in RXRα upon 1,25-(OH)2D3 treatment (Fig. 5, B and C). These findings were supplemented by fluorescent immunocytochemical experiments in which 1,25-(OH)2D3 stimulation caused visible VDR nuclear translocation in both genotypes, but only RXRα translocation in WT BMSC (Fig. 5D). Although MKP-1 does not appear to inhibit 1,25-(OH)2D3-induced VDR translocation, these data provide clear evidence that MKP-1 may be required for RXRα nuclear translocation.
Fig. 5.

MKP-1 is required for 1,25-(OH)2D3-induced RXRα nuclear import. A, Representative Western blot of cytoplasmic/nuclear expression of VDR and RXRα from BMSC treated with 10−7 m 1,25-(OH)2D3 for 1 h. B, Densitometric quantification of RXRα and VDR protein levels normalized to GAPDH or lamin A/C. C, Densitometric comparison of VDR and RXRα nuclear import in primary WT and MKP-1−/− BMSC and total relative VDR and RXRα present in nuclear and cytoplasmic fractions. D, Immunofluorescence cytochemistry of WT and MKP-1−/− BMSC for RXRα and VDR protein localization. White scale bar, 10 μm. (n = 3; *, P < 0.05). Ctrl, Control; Veh, vehicle.
When this 1-h 1,25-(OH)2D3 stimulation was repeated and WT and MKP-1−/− BMSC were subjected to a PLA using both anti-RXRα (rabbit) and anti-VDR (mouse) antibodies, we observed that there was little difference in unstimulated total or nuclear PLA signals and an insignificant difference in the total 10−7 m 1,25-(OH)2D3-stimulated PLA signals formed; yet the number of nuclear signals after 10−7 m 1,25-(OH)2D3 stimulation was 64.9% lower (P < 0.05) in the MKP-1−/− BMSC. Negative controls stimulated with 10−7 m 1,25-(OH)2D3 and incubated with anti-RXRα only or anti-VDR only plus species control IgG yielded few PLA signals (Fig. 6, A and B). Taken together, use of the PLA system suggests that endogenous 1,25-(OH)2D3-induced RXRa-VDR heterodimer nuclear translocation in primary BMSC is highly dependent upon MKP-1.
Fig. 6.

MKP-1 is required for 1,25-(OH)2D3-induced nuclear translocation of the VDR-RXRα heterodimer. WT and MKP-1−/− BMSC were treated with vehicle control or 10−7 m 1,25-(OH)2D3 for 1 h and were subjected to PLA with antibodies against VDR (mouse) and RXRα (rabbit); confocal images were captured for quantification. A, Representative confocal images of PLA signals in WT and MKP-1−/− BMSC. B, Quantification of total and nuclear PLA signals in the Z-range of the DAPI stained nuclei for the indicated treatments. White scale bar, 10 μm (n = 3; *, P < 0.05). Ctrl, Control.
MKP-1 is required in stromal cells for 1,25-(OH)2D3 induction of osteoclastogenesis
The findings suggest that BMSC are dependent on MKP-1 for their ability to induce osteoclastogenesis in response to a 1,25-(OH)2D3 stimulus. To test this prediction, we used an osteoclast coculture system (34, 35) in which WT and MKP-1−/− BMSC were magnetically depleted of CD11b+ cells to reduce primary macrophage contamination. They were then cultured with the RAW264.7 cells, after which they were treated with 10−8 m 1,25-(OH)2D3 ± recombinant osteoprotegerin. After 7 d of treatment, TRAP staining was performed and osteoclasts (TRAP+, ≥ 3 nuclei) were enumerated (Fig. 7A). Cocultures on hydroxyapatite discs were incubated for 10 d to allow for matrix resorption, and pit area and pit number were quantified (Fig. 7, B and C). As anticipated, MKP-1−/− BMSC and RAW264.7 cell cocultures had a significant reduction in the number of osteoclasts formed, as well as a decrease in the number of nuclei/osteoclasts (Fig. 6A). When was osteoprotegerin was added, the osteoclastogenesis was almost completely blocked in both cocultures. The area of resorbed matrix per hydroxyapatite disc was also significantly reduced in the MKP-1−/− coculture, as was the number of circumscribed areas of resorption. Taken together, these coculture data demonstrate a functional role for MKP-1 in permitting the 1,25-(OH)2D3 induction of RANKL-mediated osteoclastogenesis in BMSC that is predicted by MKP-1's role in canonical 1,25-(OH)2D3 signaling.
Fig. 7.

MKP-1 is required for maximal 1,25-(OH)2D3-induced osteoclastogenesis. WT and MKP-1−/− BMSC were cocultured with RAW264.7 cells to test the osteoclast-inducing potential of 1,25-(OH)2D3-stimulated stromal cells. A, Quantification of osteoclasts per well and nuclei per osteoclast. Coculture osteoclast-resorbing activity was quantified on osteologic discs. B, Representative pit images and C) pits per well and total pit area per well. (n = 3; P < 0.05; **, P < 0.01; ***, P < 0.001). Ctrl, Control; OPG, osteoprotegerin.
Discussion
In addition to cellular interactions via cytokines, skeletal and immune cellular processes display significant cross talk through common transcription factors, signaling molecules, and membrane receptors (39). MKP-1 is one such immune regulatory protein that has been studied recently for its additional role in osteoclastogenesis and mineralization (18, 40). Immunologically, MKP-1 is primarily known for its role in the negative regulation of inflammatory cytokine responses through dephosphorylation of p38 and JNK MAPK. For a stress-induced gene historically thought of as a key negative regulator for many cellular processes (41–43), evidence that MKP-1 actually permits RANKL expression truly represents a paradigm shift in our understanding of MKP-1 signaling.
Previous work in our laboratory focused on the role of MKP-1 in attenuating chronic inflammatory bone loss generated in experimental periodontal disease models. TRAP staining of LPS-treated maxilla indicated that MKP-1−/− mice had increased localized osteoclastogenesis, and micro-computed tomography confirmed that there was increased bone loss (24). Likewise, gain of function studies showed that adenoviral delivery of MKP-1 attenuated IL-6 expression, inflammatory infiltrate, and osteoclastogenesis (44). We followed up on this finding in cultured primary BMSC by investigating the expression of RANKL in response to a IL-1β, TNF-α, and LPS challenge from two periodontal pathogens known to stimulate RANKL along with a positive control of 1,25-(OH)2D3. Because the presence of MKP-1 has been found to attenuate the expression of inflammatory cytokines (45, 46), the finding that MKP-1 permits RANKL expression was quite surprising, and supports a novel role for MKP-1 in 1,25-(OH)2D3-induced gene expression.
Not knowing whether our initial finding was limited to RANKL only, our data indicate that 1,25-(OH)2D3-induced VDR and CYP24a1 mRNA expression [two well-established 1,25-(OH)2D3 targets] are highly dependent on the presence of MKP-1 signaling. Thus, the role of MKP-1 in 1,25-(OH)2D3 target gene expression is not restricted only to RANKL and may be common for the 1,25-(OH)2D3 genomic signaling pathway. Although we cannot completely rule out a role for MKP-1 in vitamin D's rapid, nongenomic membrane effects, no evidence for this activation exists for these genes, and similar mRNA expression levels were seen at 0, 2, and 4 h, indicating that MKP-1 plays a primary role in the genomic vitamin D pathway (30). Additionally, because all three vitamin D target genes (RANKL, VDR, and CYP24a1) are inhibited in MKP-1−/− BMSC at approximately the same time, baseline VDR protein levels are similar in both genotypes, and cycloheximide treatment did not rescue 1,25-(OH)2D3 induction (data not shown), we do not suspect that decreased target-gene expression results from a primary defect in VDR expression.
Although evidence suggests that osteoblasts are not the only cells that express RANKL (47), it has been established that cells of stromal osteoblast origin are the primary RANKL-expressing cells (37, 47, 48). To confirm that the MKP-1 has this novel role in stromal cells, we knocked down MKP-1 expression in a stromal/osteoblast cell line (UAMS-32), which revealed a significant decrease in RANKL, VDR, and CYP24a1 mRNA after 1,25-(OH)2D3 treatment. These data were very similar to our observations in primary BMSC, leading us to believe that stromal osteoblasts were a significant contributor. To test whether this consequence was due to a decrease in promoter activity, we repeated this siRNA and 1,25-(OH)2D3 treatment on the same cell line containing a bacterial artificial chromosome-based transcriptional reporter construct containing the entire mouse RANKL gene with 120 kb of RANKL 5′-flanking region and 14 kb 3′ flanking region (32). The result that MKP-1 siRNA was able to knockdown 1,25-(OH)2D3-induced RANKL gene promoter activity in UAMS-32 cells supports a role for MKP-1 in regulating both 1,25-(OH)2D3 RANKL mRNA and genomic RANKL activation in stromal osteoblasts. Similar MKP-1 silencing data showing reduced 1,25-(OH)2D3-induced VDR and CYP24a1 expression suggest that MKP-1 signaling may be required for other vitamin D target genes' expression as well. Based on these findings, we suspected that MKP-1 regulates 1,25-(OH)2D3 genomic action by permitting VDRE promoter activity, and that the results obtained using primary BMSC are due, in part, to a contribution from stromal/osteoblast cells.
This suspicion was strengthened because 1,25-(OH)2D3-stimulated MKP-1−/− BMSC nuclei had a decreased capacity for VDRE binding. The mOP oligonucleotide is an established VDRE consensus sequence, and its binding by VDR and RXRα through EMSA has been used to independently simulate genomic 1,25-(OH)2D3 genomic action (9, 33). Previous work by Kim et al. (11) showed the mRLD5 located at −76kb is the primary site of RANKL gene activation by 1,25-(OH)2D3-induced VDR-RXRα heterodimer binding in mouse osteoblast cells. Based on this work, our ChIP results suggest that MKP-1 may be required for 1,25-(OH)2D3-induced RXRα species binding at the mRLD5 region. The observed increase in 1,25-(OH)2D3-induced VDR binding at mRLD5 in MKP-1−/− cells may represent an increase in VDR-VDR homodimer binding or weaker VDR monomer the VDRE binding of which is strengthened via cross-linking. Either way, this new VDR species binding in MKP-1−/− cells likely occurs as compensation for a decrease in available nuclear VDR-RXRα heterodimer. Because coregulatory complex recruitment and transcriptional activation depends on the VDR-RXRα heterodimer (49, 50), we predict that these findings result in the observed decrease in RANKL gene activation and could cause the decrease in VDR and CYP-24a1 expression through a similar mechanism. This disruption in VDRE binding could depend on MKP-1's function to prevent a defect in VDR-RXRα heterodimer conformation, formation, or in its nuclear translocation.
The possibility that MKP-1 is involved in 1,25-(OH)2D3-induced nuclear trafficking was addressed initially by immunoblot analysis and confocal microscopy. Experimental evidence presented reveals that 1,25-(OH)2D3 induces a VDR translocation from the cytoplasmic to the nuclear fraction in both cell genotypes, although this VDR translocation may be delayed in MKP-1−/− BMSC, as evidenced by the relatively small reduction in cytoplasmic VDR upon 1,25-(OH)2D3 stimulation. The results show that RXRα translocates freely into the nucleus from the cytosol in WT BMSC, presumably as a VDR-RXRα heterodimer. However, despite slightly higher baseline RXRα protein in MKP-1−/− BMSC, there is no detectable translocation, as evidenced by the unchanging protein levels after 1,25-(OH)2D3 treatment. Therefore, given that RXRα dictates VDR-RXRα heterodimer translocation (51), and the VDR-RXRα heterodimer is known to have a significantly higher affinity for genomic VDRE than the VDR protein alone (8), this suggests that the decreased RXRα nuclear translocation contributes to the inhibition of VDR-RXRα VDRE binding observed in vitro by EMSA and endogenously by ChIP. Although RXRα is not known to translocate in response to 1,25-(OH)2D3 as a monomer, we confirm this by PLA, which tests specifically for RXRα-VDR heterodimer localization. Although there was little difference in unstimulated total or nuclear PLA signals and an insignificant difference in the total 10−7 m 1,25-(OH)2D3-stimulated PLA signals, the number of nuclear signals after 10−7 m 1,25-(OH)2D3 stimulation is significantly lower in the MKP-1−/− BMSC. These data support observations obtained from nuclear translocation studies and indicate that MKP-1 is required for 1,25-(OH)2D3-induced nuclear translocation of the VDR-RXRα heterodimer. Although not significant, the decrease in total PLA signals observed in MKP-1−/− cells leaves open the possibility that MKP-1 may also contribute to 1,25-(OH)2D3 ligand-initiated VDR-RXRα heterodimer formation. Although we cannot rule out that MKP-1 potentially preserves RXRα or VDR baseline expression or half-life, VDR and RXRα total protein levels were very similar in both genotypes; therefore, this should be considered a minor contribution to the observed phenotype, if at all. These data show that MKP-1 is necessary for the proper nuclear transport of VDR-RXRα heterodimer that is a likely contributor to the observed decreased capacity for VDR-RXRα VDRE binding in MKP-1−/− BMSC.
Having established that stromal osteoblast cells have a diminished capacity for 1,25-(OH)2D3-induced RANKL promoter activity, mRNA, and soluble protein expression, we predicted that BMSC are dependent upon MKP-1 for their ability to induce osteoclastogenesis in response to a 1,25-(OH)2D3 stimulus. The reduced osteoclastogenesis, nuclei/osteoclasts and mineralized matrix resorption in the primary BMSC and RAW264.7 coculture system is a predictable result given the insufficient stromal osteoclastogenic signal (RANKL) in response to 1,25-(OH)2D3. Because osteoclastogenesis was almost completely blocked by osteoprotegerin in both cocultures, the observed reduction in osteoclast formation/activity in MKP-1−/− cocultures is likely due to reduced 1,25-(OH)2D3-induced RANKL expression in the stromal population.
Future studies will focus on potential phosphorylation targets of MKP-1 in genomic 1,25-(OH)2D3 signaling. MKP-1 is known to dephosphorylate and deactivate the p38, JNK, and ERK pathways, and so future studies will focus on the potential that our observations stem from an increase in MAPK phosphorylation/activation in the absence of MKP-1. A potential complicating factor in MKP-1−/− BMSC is that the already reduced nuclear RXRα population may also have an altered structural conformation, due to posttranslational modification(s) that would limit its VDRE binding ability. Previous studies have shown that MAPK activation can directly inhibit genomic vitamin D activity through RXRα phosphorylation by JNK at serine 260 in human keratinocytes (26), and by ERK in the AF-1 domain in human prostate epithelial cells (27); however, the mechanism for how RXRα phosphorylation inhibits genomic vitamin D signaling was not shown. Given this knowledge and our findings that VDR-RXRα heterodimer translocation is impaired, it is possible that MKP-1 may be necessary to prevent a phosphorylation of RXRα, or phosphorylation of heterodimerized VDR, which impairs the 1,25-(OH)2D3-initiated translocation of the VDR-RXRα heterodimer, contributing to diminished VDRE binding and decreased 1,25-(OH)2D3 target gene expression.
This work provides the first evidence that MKP may regulate RXRα cellular trafficking and that these effects have functional consequences in vitamin D signaling. An additional consequence of these findings is that MKP-1 may have more global consequences in RXR heterodimer partners; therefore, the action of thyroid hormone, the prostanoids, and retinoids may also have altered signaling in the absence of MKP-1, which may contribute to the phenotype of MKP-1−/− mice. Additionally, the induction of MKP-1 mRNA by 1,25-(OH)2D3 and the need for MKP-1 in 1,25-(OH)2D3 signaling is suggestive of a positive feedback role for MKP-1.
These findings support a novel role for MKP-1 in permitting canonical 1,25-(OH)2D3 signaling via VDR-RXRα heterodimer nuclear import in BMSC. If MKP-1 in humans shows similar regulation of genomic 1,25-(OH)2D3 signaling, then these results may provide a scientific basis for future therapies whereby MKP-1 is targeted to control downstream 1,25-(OH)2D3 target gene expression. Taken together, we provide a clear link between a new permissive role for MKP-1 in 1,25-(OH)2D3 genomic signaling and its functional consequences in osteoclastogenesis via RANKL, while presenting mechanistic evidence for MKP-1 as a new player in osteoimmunology.
Acknowledgments
We thank Dr. Courtney Haycraft (Medical University of South Carolina) for her assistance with confocal microscopy and Dr. Renny Franceschi (University of Michigan) for his critical review of this manuscript.
This work was supported by National Institutes of Health grants R01 DE018290, T32 DE01755, and 2P20 RR017696.
Authors' roles are as follows: Study design: A.G. and K.K.. Study conduct: A.G. Data collection: A.G.. Data analysis: A.G., M.K., and K.K. Data interpretation: A.G. and K.K. Drafting manuscript: A.G. and K.K. Revising manuscript content: A.G. and K.K. Approving final version of manuscript: A.G., M.K., and K.K. K.K. takes responsibility for the integrity of the data analysis.
Disclosure Summary: The authors state that they have no conflicts of interest.
NURSA Molecule Pages†:
Nuclear Receptors: VDR;
Ligands: 1,25-dihydroxyvitamin D3.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- BMSC
- Bone marrow stromal cells
- ChIP
- chromatin immunoprecipitation
- CYP
- cytochrome P450
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- JNK
- c-Jun N-terminal kinase
- LPS
- lipopolysaccharide
- MKP-1
- MAPK phosphatase-1
- mOP
- mouse osteopontin
- PLA
- proximity ligation assay
- qPCR
- quantitative PCR
- RANKL
- receptor activator of nuclear factor-κB
- RXR
- retinoid X receptor
- siRNA
- small interfering RNA
- TRAP
- tartrate-resistant acid phosphatase
- TSS
- transcription start site
- VDR
- vitamin D receptor
- VDRE
- vitamin D response element
- WT
- wild type.
References
- 1. Bickle DA, J, Christakos S. 2008. Vitamin D: production, metabolism, mechanism of action, and clinical requirements. In: Rosen CJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society for Bone and Mineral Research; 141–147 [Google Scholar]
- 2. Pettifor JM. 2005. Rickets and vitamin D deficiency in children and adolescents. Endocrinol Metab Clin North Am 34:537–553, vii [DOI] [PubMed] [Google Scholar]
- 3. Bordier P, Pechet MM, Hesse R, Marie P, Rasmussen H. 1974. Response of adult patients with osteomalacia to treatment with crystalline 1α-hydroxy vitamin D3. N Engl J Med 291:866–871 [DOI] [PubMed] [Google Scholar]
- 4. Kristjansson K, Rut AR, Hewison M, O'Riordan JL, Hughes MR. 1993. Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1,25 dihydroxyvitamin D3. J Clin Invest 92:12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pike JW, Shevde NK. 2005. The Vitamin D Receptor. In: Feldman D, Pike JW, Glorieux FH, eds. Vitamin D. 2nd ed San Diego: Elsevier; 167–191 [Google Scholar]
- 6. Kim S, Shevde NK, Pike JW. 2005. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J Bone Miner Res 20:305–317 [DOI] [PubMed] [Google Scholar]
- 7. Mangelsdorf DJ, Evans RM. 1995. The RXR heterodimers and orphan receptors. Cell 83:841–850 [DOI] [PubMed] [Google Scholar]
- 8. Prüfer K, Racz A, Lin GC, Barsony J. 2000. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem 275:41114–41123 [DOI] [PubMed] [Google Scholar]
- 9. Pike JW, Meyer MB, Watanuki M, Kim S, Zella LA, Fretz JA, Yamazaki M, Shevde NK. 2007. Perspectives on mechanisms of gene regulation by 1,25-dihydroxyvitamin D3 and its receptor. J Steroid Biochem Mol Biol 103:389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zella LA, Kim S, Shevde NK, Pike JW. 2006. Enhancers located within two introns of the vitamin D receptor gene mediate transcriptional autoregulation by 1,25-dihydroxyvitamin D3. Mol Endocrinol 20:1231–1247 [DOI] [PubMed] [Google Scholar]
- 11. Kim S, Yamazaki M, Zella LA, Shevde NK, Pike JW. 2006. Activation of receptor activator of NF-κB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol 26:6469–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pike JW, Zella LA, Meyer MB, Fretz JA, Kim S. 2007. Molecular actions of 1,25-dihydroxyvitamin D3 on genes involved in calcium homeostasis. J Bone Miner Res 22(Suppl 2):V16–V19 [DOI] [PubMed] [Google Scholar]
- 13. Boyle WJ, Simonet WS, Lacey DL. 2003. Osteoclast differentiation and activation. Nature 423:337–342 [DOI] [PubMed] [Google Scholar]
- 14. Kim S, Yamazaki M, Zella LA, Meyer MB, Fretz JA, Shevde NK, Pike JW. 2007. Multiple enhancer regions located at significant distances upstream of the transcriptional start site mediate RANKL gene expression in response to 1,25-dihydroxyvitamin D3. J Steroid Biochem Mol Biol 103:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cranney A, Horsley T, O'Donnell S, Weiler H, Puil L, Ooi D, Atkinson S, Ward L, Moher D, Hanley D, Fang M, Yazdi F, Garritty C, Sampson M, Barrowman N, Tsertsvadze A, Mamaladze V. 2007. Effectiveness and safety of vitamin D in relation to bone health. Evid Rep Technol Assess (Full Rep) 158:1–235 [PMC free article] [PubMed] [Google Scholar]
- 16. Ishizuka S, Kurihara N, Reddy SV, Cornish J, Cundy T, Roodman GD. 2005. (23S)-25-Dehydro-1α-hydroxyvitamin D3-26,23-lactone, a vitamin D receptor antagonist that inhibits osteoclast formation and bone resorption in bone marrow cultures from patients with Paget's disease. Endocrinology 146:2023–2030 [DOI] [PubMed] [Google Scholar]
- 17. Kurihara N, Reddy SV, Araki N, Ishizuka S, Ozono K, Cornish J, Cundy T, Singer FR, Roodman GD. 2004. Role of TAFII-17, a VDR binding protein, in the increased osteoclast formation in Paget's disease. J Bone Miner Res 19:1154–1164 [DOI] [PubMed] [Google Scholar]
- 18. Carlson J, Cui W, Zhang Q, Xu X, Mercan F, Bennett AM, Vignery A. 2009. Role of MKP-1 in osteoclasts and bone homeostasis. Am J Pathol 175:1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Leung DY, Richers BN, Liu Y, Remigio LK, Riches DW, Goleva E. 2012. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol 188:2127–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abraham SM, Clark AR. 2006. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans 34:1018–1023 [DOI] [PubMed] [Google Scholar]
- 21. Dickinson RJ, Keyse SM. 2006. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci 119:4607–4615 [DOI] [PubMed] [Google Scholar]
- 22. Liu Y, Shepherd EG, Nelin LD. 2007. MAPK phosphatases–regulating the immune response. Nat Rev Immunol 7:202–212 [DOI] [PubMed] [Google Scholar]
- 23. Franklin CC, Kraft AS. 1997. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem 272:16917–16923 [DOI] [PubMed] [Google Scholar]
- 24. Sartori R, Li F, Kirkwood KL. 2009. MAP kinase phosphatase-1 protects against inflammatory bone loss. J Dent Res 88:1125–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roth Flach RJ, Bennett AM. 2010. Mitogen-activated protein kinase phosphatase-1—a potential therapeutic target in metabolic disease. Expert Opin Ther Targets 14:1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Solomon C, White JH, Kremer R. 1999. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor α. J Clin Invest 103:1729–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Z, Kovalenko P, Cui M, Desmet M, Clinton SK, Fleet JC. 2010. Constitutive activation of the mitogen-activated protein kinase pathway impairs vitamin D signaling in human prostate epithelial cells. J Cell Physiol 224:433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000 [DOI] [PubMed] [Google Scholar]
- 29. Hayashi H, Nakahama K, Sato T, Tuchiya T, Asakawa Y, Maemura T, Tanaka M, Morita M, Morita I. 2008. The role of Mac-1 (CD11b/CD18) in osteoclast differentiation induced by receptor activator of nuclear factor-κB ligand. FEBS Lett 582:3243–3248 [DOI] [PubMed] [Google Scholar]
- 30. Wali RK, Kong J, Sitrin MD, Bissonnette M, Li YC. 2003. Vitamin D receptor is not required for the rapid actions of 1,25-dihydroxyvitamin D3 to increase intracellular calcium and activate protein kinase C in mouse osteoblasts. J Cell Biochem 88:794–801 [DOI] [PubMed] [Google Scholar]
- 31. Atkins GJ, Anderson PH, Findlay DM, Welldon KJ, Vincent C, Zannettino AC, O'Loughlin PD, Morris HA. 2007. Metabolism of vitamin D3 in human osteoblasts: evidence for autocrine and paracrine activities of 1 α,25-dihydroxyvitamin D3. Bone 40:1517–1528 [DOI] [PubMed] [Google Scholar]
- 32. Fu Q, Manolagas SC, O'Brien CA. 2006. Parathyroid hormone controls receptor activator of NF-κB ligand gene expression via a distant transcriptional enhancer. Mol Cell Biol 26:6453–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. 1990. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci USA 87:9995–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rossa C, Jr, Liu M, Kirkwood KL. 2008. A dominant function of p38 mitogen-activated protein kinase signaling in receptor activator of nuclear factor-κB ligand expression and osteoclastogenesis induction by Aggregatibacter actinomycetemcomitans and Escherichia coli lipopolysaccharide. J Periodontal Res 43:201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patil CS, Liu M, Zhao W, Coatney DD, Li F, VanTubergen EA, D'Silva NJ, Kirkwood KL. 2008. Targeting mRNA stability arrests inflammatory bone loss. Mol Ther 16:1657–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Teitelbaum SL. 2000. Bone resorption by osteoclasts. Science 289:1504–1508 [DOI] [PubMed] [Google Scholar]
- 37. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. 1998. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176 [DOI] [PubMed] [Google Scholar]
- 38. Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, Wang L, Xia XZ, Elliott R, Chiu L, Black T, Scully S, Capparelli C, Morony S, Shimamoto G, Bass MB, Boyle WJ. 1999. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA 96:3540–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakashima T, Takayanagi H. 2009. Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol 29:555–567 [DOI] [PubMed] [Google Scholar]
- 40. Mahalingam CD, Datta T, Patil RV, Kreider J, Bonfil RD, Kirkwood KL, Goldstein SA, Abou-Samra AB, Datta NS. 2011. Mitogen-activated protein kinase phosphatase 1 regulates bone mass, osteoblast gene expression, and responsiveness to parathyroid hormone. J Endocrinol 211:145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leenders JJ, Pinto YM, Creemers EE. 2011. Tapping the brake on cardiac growth-endogenous repressors of hypertrophic signaling. J Mol Cell Cardiol 51:156–167 [DOI] [PubMed] [Google Scholar]
- 42. Manetsch M, Che W, Seidel P, Chen Y, Ammit AJ. 2012. MKP-1: A negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal 24:907–913 [DOI] [PubMed] [Google Scholar]
- 43. Franklin CC, Srikanth S, Kraft AS. 1998. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci USA 95:3014–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu H, Li Q, Herbert B, Zinna R, Martin K, Junior CR, Kirkwood KL. 2011. Anti-inflammatory effect of MAPK phosphatase-1 local gene transfer in inflammatory bone loss. Gene Ther 18:344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. 2006. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA 103:2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. 2006. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176:1899–1907 [DOI] [PubMed] [Google Scholar]
- 47. Galli C, Fu Q, Wang W, Olsen BR, Manolagas SC, Jilka RL, O'Brien CA. 2009. Commitment to the osteoblast lineage is not required for RANKL gene expression. J Biol Chem 284:12654–12662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. 1998. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95:3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sutton AL, MacDonald PN. 2003. Vitamin D: more than a “bone-a-fide” hormone. Mol Endocrinol 17:777–791 [DOI] [PubMed] [Google Scholar]
- 50. Nagpal S, Na S, Rathnachalam R. 2005. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev 26:662–687 [DOI] [PubMed] [Google Scholar]
- 51. Yasmin R, Williams RM, Xu M, Noy N. 2005. Nuclear import of the retinoid X receptor, the vitamin D receptor, and their mutual heterodimer. J Biol Chem 280:40152–40160 [DOI] [PubMed] [Google Scholar]