

Abstract
Nowadays, the study of the peroxisome proliferators activated receptors (PPARs) as potential targets for cancer prevention and therapy has gained a strong interest. From a biological point of view, the overall responsibility of PPARs in cancer development and progression is still controversial since several studies report both antiproliferative and tumor-promoting actions for these signaling molecules in human cancer cells and animal models. In this paper, we discuss PPARs functions in the context of different types of gastrointestinal cancer.
1. Introduction
Since the discovery of the peroxisome proliferators activated receptors (PPARs) [1] in Xenopus frogs as receptors that induce the proliferation of peroxisomes in cells [2], three main forms transcribed from three different genes have been indentified: PPARα, PPARβ/δ, and PPARγ. Despite the little divergence of homology observed, each isoform possesses distinct biological activities and is expressed in different tissues [3]. PPARα is mainly expressed in the liver, the kidney, and the heart and is primarily involved in lipid metabolism. PPARγ is a master regulator of adipogenesis and fat storage: it regulates adipocyte differentiation and insulin sensitivity in adipose tissue. PPARβ/δ is found in a broad range of tissues but markedly expressed in brain, adipose tissue, and skin and its function awaits further exploration. PPARs are key mediators of energy homeostasis, lipid, and glucose metabolism although they have also been associated with other biological processes including development, differentiation, inflammation, atherosclerosis, wound healing, and tumor formation. All PPARs heterodimerize with the retinoid X receptor (RXR) to bind successively to specific DNA regions of target genes named PPREs (peroxisome proliferator hormone response elements). Like PPARs, RXR exists as three distinct isoforms: RXRα, β, and γ, all of which are activated by the endogenous agonist 9-cis retinoic acid [4]. Contrasting observations confer to PPARs a double-edge sword nature in cancerogenesis, considering that either tumor suppressing or stimulating effects have been evidenced for these nuclear receptors [5].
PPARs function is modified by the specific shape of their ligand-binding domain induced by ligand binding and by a number of coactivator and corepressor proteins, the presence of which can stimulate or inhibit receptor function, respectively [6]. Endogenous ligands for the PPARs include free fatty acids and eicosanoids. PPAR isoform-specific agonists, specifically fibrates for PPARα and thiazolidinediones for PPARγ, are currently prescribed as lipid and glucose-lowering drugs, respectively [7]. Although several reports highlight antiproliferative and prodifferentiative actions of PPARγ ligands in cancer cell lines and animal models of human neoplastic disease [8], more recent studies illustrating tumor-promoting effects of PPARγ, in particular in colon and breast cancer models, raise considerable concern about the practicability and safety of PPARγ ligands as anticancer drugs [9, 10]. In this paper we discuss PPARs functions in the context of different types of gastrointestinal cancer.
2. PPARs and Gastrointestinal Tract Cancer
Numerous studies in the last decade have focused on the effects of PPARs activity on gastrointestinal tract tumor biology, exploring mechanisms, target genes, clinical applications, and evaluating the potential therapeutic use in cancer treatment of PPARs agonists, which seemed promising as components of combination treatments in both in vitro and in vivo models of cancer [11–13]. In particular, a possible role for PPARγ as a tumor suppressor and as an inducer of differentiation of cancer stem cells has been explored, and its protein level in tumor specimens has been identified as a significant prognostic marker [14].
A recent meta-analysis has found an association between the PPARG polymorphism 34 C > G and colon cancer risk [15], and a PPARG germline mutation replacing serine 289 with cysteine in the mature protein (S289C) has been reported associated with dyslipidemia and colonic polyp formation progressing to full-blown adenocarcinoma [16]. Furthermore, studies performed in animal models challenged with procarcinogenic and anticarcinogenic agents have put in evidence that PPARγ signaling pathway is critically engaged in the antitumor activity of normal organisms [17]. Anyway, the role of PPARγ in the neoplastic diseases of the gastrointestinal tract remains controversial, as this nuclear receptor shows dissimilar growth-suppressive effects in different cancers. Moreover, PPARγ activation induces diverse growth inhibition in different cancer cell lines [18]. PPARγ inhibits tumor growth only in the presence of functional APC but not in cells with loss of APC function [19], and PPARγ agonists have been reported to have tumor-promoting effects in the ApcMin/+ mice [10], suggesting that loss of APC may alter the normal response of intestinal epithelial tumor cells to PPARγ agonists. The latter could be one important feature that can explain the discrepancies reported in the literature about the dual role of PPARγ in gastrointestinal cancer.
In the esophagus, the evaluation of PPARγ protein and mRNA expression levels in samples of normal esophageal squamous epithelium, Barrett's esophagus, and esophageal adenocarcinoma has shown a trend toward increased expression going from normal tissue to pathological samples and a trend towards increased PPARγ expression with decreasing levels of differentiation [20]. Similarly, PPARγ expression is increased in human gastric cancer tissue [21], and immunohistochemistry has evidenced its overexpression in gastric mucosal dysplasia and gastric carcinoma compared with chronic gastritis [22]. In addition, the presence of PPARγ protein has been evidenced in surgically resected specimens from well differentiated, moderately differentiated, and poorly differentiated gastric adenocarcinoma [23]. On the other hand, PPARγ agonists show dose-dependent inhibitory effects on the proliferation of gastric cancer cell lines, and this effect is augmented by the simultaneous addition of 9-cis retinoic acid; flow cytometry demonstrates G1 cell cycle arrest and a significant increase of annexin V-positive cells, suggesting that induction of apoptosis together with G1 cell cycle arrest may be one of the mechanisms of the antiproliferative effect of PPARγ activation in human gastric cancer cells [23].
Regarding the large bowel, high expression of PPARγ is detected in the normal mucosa of the colon and rectum, and a deficiency in intestinal PPARγ is associated with enhanced tumorigenicity in mouse small intestine and colon. A series of evidence suggests that PPARG is a tumor suppressor gene in colorectal cancer: (i) loss of function point mutations has been evidenced in one allele of PPARG in primary colorectal patients, and the mutations impair the function of PPARγ by affecting the ligand-binding domain, which results in an inability to bind ligands and control gene regulation; (ii) polymorphism in the PPARγ gene has been found in colorectal cancer patients; (iii) expression of PPARγ in colorectal cancer is associated with a good prognosis [24]. Anyway, decreased PPARγ expression compared with adjacent normal colonic mucosa is detected in a number of colorectal cancer patients [25], and PPARG inactivation seems to play a role in colorectal cancer progression, although the events involved are not yet clear. In a large series of primary colorectal cancers, about 60% of tumors showed PPARγ upregulation, whereas 35% of the tumours showed lower PPARγ levels compared to the nontumorous normal mucosa. A significant association was evidenced between low PPARγ expression and distant metastases and reduced patients' survival [26].
PPARG epigenetic silencing has been found to be coordinated by ubiquitin-like with PHD and RING finger domains 1 (UHRF1), a member of a subfamily of RING-finger-type E3 ubiquitin ligases, which mediates colorectal cancer progression. This protein is encoded by the UHRF1 gene and its expression peaks at late G1 phase and continues during G2 and M phases of the cell cycle, playing a major role in the G1/S transition by regulating topoisomerase II alpha and retinoblastoma gene expression and functioning in the p53-dependent DNA damage checkpoints. UHRF1 binds to specific DNA sequences and recruits a histone deacetylase to regulate gene expression, functioning as a cofactor that coordinates the epigenetic silencing of tumor suppressor genes. UHRF1 overexpression induces PPARG silencing through its recruitment on the PPARG promoter promoting DNA methylation and histone repressive modifications, and it is associated with a higher proliferative, clonogenic, and migration potential, and with phenotypic features resembling those occurring in the epithelial-mesenchymal transition [27]. PPARγ agonists such as thiazolidinediones, also known as glitazones (rosiglitazone, troglitazone, and pioglitazone), have been shown to induce apoptosis in human colon cancer cells, and the molecular mechanism involves glycogen synthase kinase-3β (GSK-3β), a crucial activator of nuclear factor-kappa B (NF-kappaB), which plays a critical role in the mediation of survival signals in cancer cells, with inhibition of NF-kappaB activity and GSK-3β expression in a dose-dependent manner. Glitazone treatment inhibits colon cancer cell growth, and cells are arrested in G(0)/G(1) phase followed by the induction of apoptosis with concomitant decrease in the expression of the G(0)/G(1) phase regulatory proteins Cdk2, Cdk4, cyclin B1, D1, and E, decrease in the antiapoptotic protein Bcl-2, and increase in the expression of the proapoptotic-associated proteins caspase-3, caspase-9, and Bax [28]. Similarly to the phenomenon evidenced in gastric cancer lines [23], the effect is augmented by the simultaneous addition of the RXRα ligand 9-cis retinoic acid [29].
On the other hand, inhibiting PPARγ prevents proliferation of human colon cancer HT-29 cells, as evidenced by challenge with cyclic phosphatidic acid (cPA), a structural analog of lysophosphatidic acid (LPA), and a specific, high-affinity PPARγ antagonist [30]. Moreover, synthetic and physiological agonists of PPARγ and PPARβ/δ induce expression of vascular endothelial growth factor (VEGF) in the colorectal tumor cell lines SW480 and HT29 [31]. Interestingly, PPARβ/δ is a promising drug target since its agonists promote terminal differentiation, but there are reports showing either pro- or anticarcinogenic effects of PPARβ/δ in cancer models [32]. Expression of PPARβ/δ mRNA and protein is lower in human and Apc (+/Min-FCCC) mouse colon tumors in respect of matched normal tissue, and stable overexpression of PPARβ/δ in human HT29 colon cancer cell lines enhances ligand activation of PPARβ/δ and inhibition of clonogenicity [33]. The role of PPARβ/δ in the pathogenesis of colorectal cancer has been evaluated in studies performed in vivo on rectal cancer patients and in vitro on colon cancer cell lines with different metastatic potentials. The intensity of PPARβ/δ expression has been found increased in human rectal cancer tissue compared to adjacent or distant normal mucosa [34], in rectal cancers with better differentiation than in those with poor differentiation, and in early-stage tumors than in advanced ones [35]. Besides, PPARβ knockdown in vitro has evidenced that PPARβ/δ may facilitate differentiation and inhibit the cell-fibronectin adhesion of colon cancer cell lines [35].
Anyway, some colorectal cancer cell lines are resistant to PPARγ agonists, because elevated PPARδ expression and/or activation of PPARδ antagonize the ability of PPARγ to induce colorectal carcinoma cell death, as a result of opposing effects of PPARδ and PPARγ in regulating programmed cell death mediated by survivin and caspase-3: activation of PPARγ results in decreased survivin expression and increased caspase-3 activity, whereas activation of PPARδ counteracts these effects [36]. In addition, the concomitant expression of PPAR β/δ and cyclooxygenase (COX)-2 in tumor tissues is associated with a higher incidence of liver metastasis and consequent poor prognosis in colorectal cancer patients [37].
PPARγ activation induces expression of Krüppel-like factor (KLF) 4, known also as gut-enriched Krüppel-like factor (GKLF), which acts as a transcriptional activator or repressor depending on the promoter context and/or cooperation with other transcription factors. KLF4 is a nodal player in the network of PPARγ-regulated genes, and treatment of colon cancer cells with PPARγ agonists influences KLF4 target genes, whose expression is decreased (cyclin D1) or increased (GPA33, encoding the glycoprotein A33 that is a colon cancer antigen, p21WAF1/Cip1, and keratin 19), respectively [38].
Epigenetic silencing of PPARG in colorectal cancer may be a significant prognostic marker of tumor progression, and methylation on a specific region of the promoter is strongly correlated with PPARγ lack of expression in primary colorectal cancers and with patients' poor prognosis [26]. The same methylation pattern is found in PPARγ negative colorectal cancer cell lines. Transcriptional silencing is due to the recruitment of methyl CpG binding protein 2 (MeCP2), histone deacetylase 1 (HDAC1), and histone-lysine N-methyltransferase (EZH2) that impart repressive chromatin signatures determining an increased cell proliferative and invasive potential [26].
As reported in this section, many clinical and experimental data support the critical role played by PPARs in gastro-intestinal tumorigenesis and neoplastic gut disease behavior, but the molecular mechanisms involved are still a matter of debate. Furthermore, the results of many studies are conflicting and lead to the conclusion that PPARs may have both tumor suppressor and procarcinogenic activity. These controversies may arise from methodological differences among the study protocols, anyway some evidence suggests that ligand-related PPARs activation induces growth arrest in cancer cells and tumor growth inhibition deriving from antiproliferative or proapoptotic effects. On the other hand, PPARs have been found to stimulate tumor cell proliferation and induce neo-angiogenesis, favoring cancer growth and spreading. PPARs agonists provoke several physiological modifications influencing lipid metabolism, glucose homeostasis, and inflammation signaling cascade, and considering that among the major risk factors for colorectal cancer are comprised obesity, metabolic derangement, and chronic inflammatory bowel disease, PPARs modulation could be a valuable tool in the prevention and treatment of colorectal cancer. A mandatory and preliminary condition is represented by the full understanding of the complex mechanisms involved in the regulation of PPARs transcriptional activity and unveiling of the intricacy of PPAR-dependent and PPAR-independent effects stimulated by the different ligands. The same PPAR is able to modulate different target genes and cooperate with other nuclear receptors and signalling molecules involved in cell proliferation and cell death, increasing the difficulty to dissect the role of the single players that take part in this physiologically basic but really intricate network.
3. PPARs and Liver Cancer
Hepatocellular carcinoma (HCC) is the most common type of liver cancer. HCC often arises from viral hepatitis infection (hepatitis B or C), cirrhosis, alcohol consumption being its most common cause. HCC has recently been linked to nonalcoholic fatty liver disease (NAFLD), the hepatic manifestation of obesity and metabolic syndrome. HCC presents with an aberrant lipid metabolism as revealed by quantitative profiling in patient plasma by using ultraperformance liquid chromatography coupled to mass spectrometry approaches [46]. Compared to other cancers, HCC is quite a rare tumor and, in countries where hepatitis is not endemic, most malignant cancers in the liver are not primary HCC but metastasis (spread) of cancer from elsewhere in the body, for example, colorectal cancer. A great bulk of evidence suggests a role for lipid-sensing nuclear receptors in the pathogenesis of NAFLD and HCC. Lipid sensing nuclear receptors, including PPARs, are the master transcriptional regulators of lipid and carbohydrate metabolism and inflammatory responses, thus standing as suitable therapeutic targets for both NAFLD and HCC [47, 48]. In the leptin-deficient ob/ob mouse model of metabolic syndrome, PPARγ is critical for the development of hepatic steatosis, through modulation of its target protein fat-specific protein 27 (Fsp27) [49]. Hepatic transcriptional effects of PPARα, PPARγ, and PPARδ are multiple and recent hypothesis-driven and unbiased genomewide high-throughput approaches in hepatocytes are continuously uncovering new target genes involved in lipid metabolism or confirming established ones, as ACSL3, ACOX1, SULT2A1, ACADL, CD36, IGFBP1, and G0S2 [50]. PPARs, as other nuclear receptors, can be activated in the liver by several hundreds of environmental chemicals and contaminants, and this has been demonstrated to contribute to the process of hepatocarcinogenesis as observed in in vitro and in vivo rodent models by large screening studies: however, the biological differences between rodents and humans and the distinct mode of actions make it difficult to extrapolate useful information for the clinics and to determine human carcinogenic risk upon exposure to environmental chemicals [51]. PPARα plays a dominant role in hepatocarcinogenesis induced by trichloroethylene (TCE), an industrial solvent and a widespread environmental contaminant [52]. Beinga central regulator of triglyceride homeostasis and mediating hepatocarcinogenesis in rodents, not surprisingly PPARα, contributes to steatosis and HCC induced by hepatitis C virus (HCV) in rodent models [53, 54]. In human hepatocarcinoma cells, PPARα is chiefly related to apoptosis as evidenced by determination of BAD, myc, and protein phosphatase 2A protein content and PPARγ is instead chiefly related to cell proliferation, evidenced by decreased cell number and increased number of cells in the G0/G1 phase of the cycle [55]. Mice lacking one allele of PPARG were more susceptible to liver cancer in a diethylnitrosamine (DEN)-induced HCC model: PPARγ suppressed tumor cell growth through reducing cell proliferation and inducing G(2)/M phase arrest, apoptosis, and upregulating growth differentiation factor-15 [56]. Consistently, troglitazone, a PPARγ ligand, inhibited growth and induced apoptosis of HepG2 cells in a dose-dependent manner [57]. Moreover, in the partial hepatectomy rat model of liver regeneration, it was shown that PPARγ signaling is a key negative regulator of hepatocyte proliferation and may be responsible for the inhibition of liver growth during regeneration [58]. PPARs actively crosstalk with other signaling mediators implicated in lipid metabolism and hepatocyte malignancy; for instance, AMP-activated protein kinase (AMPK), an energy sensing enzyme implicated in the transition from NAFLD to HCC [59], and whose activation has been reported to be lipid lowering and antitumoral in mice and in hepatoma cells [60, 61]. In HCC cells, AMPK activators AICAR and metformin inhibit directly transcriptional activities of PPARα and PPARγ to modulate energy generation through fatty acid oxidation process [62]. Mice with a combination of genetic inactivations for hepatic growth hormone and glucocorticoid receptor signaling effectors displayed upregulation of prolipogenic PPARγ and downstream transcription factor SREBP-1c, demonstrating a crosstalk between these molecular networks [63]. Mice with specific inactivation of the NF-kappaB essential modulator gene (NEMO (L-KO) mice) exposed to a high-fat diet display a worsened liver steatosis as a consequence of PPARα and increased PPARγ expression [64]. From a therapeutic perspective, PPARγ agonists, such as antidiabetic thiazolidinediones (TZD), have in vitro antiproliferative effect, have been associated with lower risk and a better prognosis in HCC, not only related to anti-NAFLD but also to antiviral hepatitis effects [65]. The effective anticancer properties and the underlying molecular mechanisms of these drugs in vivo remain unclear because the primary target of TZD is PPARγ, which is upregulated in HCC and seems to provide tumor-promoting responses. Reconciling this discrepancy, it may be that these established PPARs agonists exert a hypolipidemic and antitumoral action in liver cells through PPAR-independent pathways [66, 67].
As mentioned, when the liver is infected with hepatic viruses, this can ultimately result in liver cancer, and hepatitis viruses are one of the leading causes of chronic liver disease [68]. Hepatitis viruses are a global health problem if we consider approximately 200 million patients carrying a chronic HCV infection and about 350 million chronically infected with HBV [69].
PPARs were suggested as new therapeutic targets in the traditional treatment of HCV-induced liver injury when two studies found that PPARα drastically decreased in HCV-infected patients [70] together with its target gene carnitine palmitoyl acyl-CoA transferase 1A (CPT1A) [71]. The impaired PPARα expression was due to HCV core protein expression [71]. Successively, we and others have recently uncovered a role for PPARγ in HCV infection [42, 72, 73]. Granted that HCV is classified in six different major genotypes and that mechanisms involved in pathobiology of disease are genotype dependent [39, 73], from a biological point of view, reduced PPARγ levels found in in vitro models of HCV expressing the core protein genotype 3a are associated with increased fat accumulation and impaired insulin signaling [72, 73]. The latter impairs the sustained response rate to peg-interferon plus ribavirin in chronic hepatitis C patients [40]. PPARγ degrades IRS1 protein through suppressor of cytokine signaling protein 7 (SOCS-7) whose expression could be pharmacologically controlled by agonist and antagonist of PPARγ [41]. PPARγ agonists have already been suggested as an adjuvant therapy in chronic hepatitis C [74, 75]. In fact, there is the belief that correcting insulin resistance is a rational option in chronic hepatitis C patients [76]. However, new modalities of this correction have to be explored based on the mechanisms inducing insulin resistance, as insulin-sensitizing therapy should be tailored according to the infecting HCV genotype, as suggested [76].
Steatosis is a common histological feature of chronic infection between hepatitis C and B virus. Another common feature is the ability of both viruses in modulating PPARα and PPARγ activity/expression which are related to steatosis. As for HBV, in vitro studies using hepatoma cell lines and studies on transgenic mouse models for HBV have provided indication for a role of PPARs in HBV-related diseases and in controlling viral transcription and replication. Kim et al. [77] demonstrated that SREBP-1 and PPARγ were transcriptionally induced by HBV X protein (HBx) in order to provoke hepatic steatosis in HepG2-HBx stable cells and HBx-transgenic mice.
Moreover thiazolidinediones (TZD, class of PPARγ ligands) have been suggested as useful drugs for HCC chemoprevention and treatment as TZD administration in hepatitis B virus (HBV)-transgenic mice reduced tumor incidence in the liver, inhibiting hepatocyte proliferation and increasing apoptosis, probably through inhibition of nucleophosmin (NPM) protein and mRNA expression [68]. Furthermore it was also reported a role for PPARs in regulating HBV transcription and regulation in vivo [78] and in vitro [79]. Guidotti et al. [78] demonstrated that HBV transgenic mice treated with two synthetic PPARα ligands (Wy-14,643 and clofibric acid) resulted in an increased HBV transcription rates suggesting that in patients receiving these drugs who are also infected with HBV viral replication may be activated, and this could have potentially detrimental effects on the outcome of the viral infection. Conversely, Wakui et al. [79] demonstrated that the PPARα ligand bezafibrate had no effect on HBV replication within HepG2 cells whilst a PPARγ ligand, rosiglitazone, reduced the amount of HBV DNA, hepatitis B surface antigen (HBsAg), and hepatitis B e antigen (HBeAg) in the culture supernatant, suggesting that the combination therapy of rosiglitazone and nucleot(s)ide analogues or interferon could be a therapeutic rational option also for chronic HBV infection.
4. PPARs and Pancreatic Cancer
Pancreatic cancer (PC) is one of the most lethal malignant diseases with a really terrible prognosis and is ranked as the fourth leading cause of cancer-related deaths worldwide [80]. PC is referred to as a “silent killer” because early pancreatic cancer often does not cause symptoms and the later symptoms are usually nonspecific and varied. Despite many advances in modern medicine, the available therapeutic strategies based on surgery and conventional chemotherapy are still largely unsatisfactory in patients with pancreatic cancer. When patients present locally advanced or metastatic tumors (which render them ineligible for surgical resection), they are treated with the gold standard chemotherapy which is based on gemcitabine, an S-phase nucleoside cytidine analogue. The overall survival is unacceptably small, and novel therapeutic approaches to overcome the resistance of PC to conventional anticancer therapies are urgently needed. Scientists are also looking for an ideal combination partner in therapeutic settings that require the inhibition of tumor-protecting mechanisms/proteins to overcome treatment resistance.
PPARγ is commonly upregulated in pancreatic ductal adenocarcinoma and might be considered a prognostic marker in this disease [81].
To date several research groups have demonstrated the ability of thiazolidinedione (TZD, class of PPARγ ligands) to attenuate the growth of pancreatic cancer cells in vitro, which was associated to G1 cell cycle arrest and cell differentiation and to increased apoptotic cell death [43]. Moreover, Hashimoto et al. [82] suggest a double beneficial effect of TZD showing the dual advantage of inhibiting pancreatic cancer cell growth while reducing the invasiveness of the tumor cells. Moreover, TZD attenuated pancreatic cancer cell migration and invasion by modulation of actin organization and expression of matrix metalloproteinase-2 and plasminogen activator inhibitor-1, respectively [83, 84]. An increasing number of studies have implicated STAT activation, particularly STAT3, in transformation and tumor progression. Direct targeting of STAT3 in malignant tumors may represent another important therapeutic tool as STAT proteins are emerging as ideal targets for cancer therapy [44]. Vitale et al. [85] showed that, in pancreatic cancer cells, PPAR-γ agonist (troglitazone, TGZ) counteracts STAT3 protein potentiating the anticancer effects of IFN-β through the induction of cell cycle perturbations and the occurrence of autophagy cell death in pancreatic cancer cells. Co-incubation of pancreatic cancer cells with IFN-β and TGZ suppresses STAT3 activation and delays G0/G1-S phase progression that occurred together with an increase in p21 and p27 protein expression that was more evident after 24 hours of treatment with the pharmacological combination.
Even though we did not observe a PPARγ altered expression in 30 matched pairs of tumour and adjacent normal tissue samples collected from patients undergoing pancreatic resection [45], a recent study supports a role of PPARγ as an ideal partner of the standard therapy based on gemcitabine since the anticancer effect of gemcitabine can be enhanced by ligands for PPARγ such as pioglitazone (Pio) and rosiglitazone [86]. The authors demonstrate that Pio significantly inhibits the NF-κB transcriptional activity and potentiates the gemcitabine effect on the apoptosis rate in three different pancreatic cancer cell lines as demonstrated by cotreatment with Pio and Gem on caspase-3 and caspase-7 cleavage. The authors conclude that since Pio is widely used in the treatment of diabetes mellitus, it may become a possible partner of Gem-based chemotherapy. Considered the adverse effects associated with TZDs, such as weight gain, macular edema, bone loss, and heart failure in at-risk individuals [87, 88], scientists must press on investigating new analogs of PPARγ agonists in order to potentiate the beneficial effect while reducing the side effects (Figure 1 and Table 1).
Figure 1.
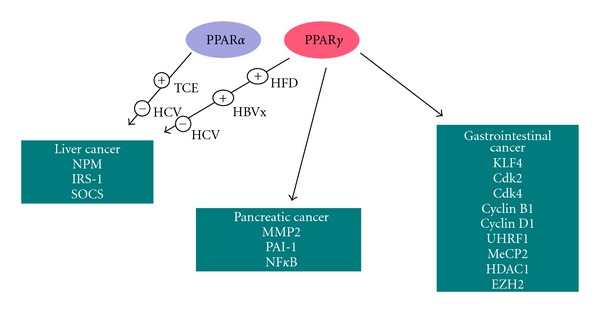
Schematic representation of the PPARs signaling operating in cancer. Krueppel-like factor 4 (KLF4); cyclin-dependent kinase (2, 4); cyclin B1, D1; ubiquitin-like, containing PHD and RING finger domains, 1; methyl CpG binding protein 2 (MeCP2); Histone deacetylase 1(HDAC1); histone-lysine N-methyltransferase (EZH2); matrix metallopeptidase 2 (MMP2); plasminogen activator inhibitor 1 (PAI-1) nuclear factor-kappaB (NFκB); nucleophosmin (NPM); insulin receptor substrate 1 (IRS-1); suppressor of cytokine signal (SOCS). For further explanations, please refer to the text.
Table 1.
Differential patterns of PPARs expression in gastrointestinal system disease.
| Organ | PPARs expression | Author and reference | |
|---|---|---|---|
| Esophagus | PPARγ ↑ | PPARγ overexpression influences the development of Barrett's esophagus and esophageal adenocarcinoma | Wang et al. [20] |
|
| |||
| Stomach | PPARγ ↑ | Crucial role of PPARγ in the pathogenesis of gastric carcinoma | Ma et al. [21] |
| PPARγ ↑ | PPARγ is upregulated in gastric adenocarcinoma | Yao et al. [22] | |
| PPARγ ↑ | PPARγ protein evidenced in gastric adenocarcinoma specimens and PPARγ agonists show dose-dependent inhibitory effects on the proliferation of gastric cancer cell lines | Sato et al. [23] | |
|
| |||
| PPARγ ↑ | PPARγ expression in colorectal cancer is associated with a good prognosis | Dai and Wang[24] | |
| Colon-rectum | PPARγ ↓ | PPARγ underexpression is detected in a number of colorectal cancer patients, and epigenetic silencing of PPARγ is a biomarker for colorectal cancer progression and adverse patients' outcome | Pancione et al. [26] |
| PPARγ ↓ | PPARγ epigenetic silencing is coordinated by UHRF1 mediating colorectal cancer progression, and a significant low PPARγ expression is associated with distant metastases and reduced patients' survival | Sabatino et al. [27] | |
|
| |||
| Liver | PPARα and PPARγ↓ | HCV decreases PPARs in order to induce triglycerides accumulation | Ripoli and Pazienza[39] |
| Romero-Gómez et al.[40] | |||
| Pazienza et al.[41] | |||
| PPARγ ↑ | HBx enhances C/EBPαthat in turn induces PPARγ expression and activation | Dharancy et al.[42] | |
| Tsujie et al.[43] | |||
|
| |||
| Pancreas | PPARγ ↑ | PPARγ is highly expressed in pancreatic cancer and is associated with shorter overall survival times | Yu and Jove[44] |
| PPARγ — | PPARγ is unaltered in PC but expression levels between PPARγ and DNMT1 and between DNMT1 and DNMT3B are highly correlated | Pazienza et al. [45] | |
5. Conclusion
A potential role for PPARs agonists in the adjuvant treatment of digestive system cancers is advisable, but further studies are warranted in order to better clarify the role of PPARs in gastrointestinal cancerogenesis. PPARs could have prognostic and/or therapeutic roles, but there is urgent need to shed light on the favorable potential or harmful risk of their modulators.
Conflict of Interests
The authors declare that they have no competing interests.
Acknowledgments
This work was supported by “Ministry of Italian Health” Grant through the Research Laboratory and Gastroenterology Unit (RC1203GA58) and Division of Internal Medicine and Chronobiology Unit (RC1203ME46), IRCCS Scientific Institute and Regional General Hospital “Casa Sollievo della Sofferenza”, Opera di Padre Pio da Pietrelcina, San Giovanni Rotondo, Italy, and by the “5x1000” voluntary contributions.
References
- 1.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 2.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68(5):879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 3.Escher P, Wahli W. Peroxisome proliferator-activated receptors: Insight into multiple cellular functions. Mutation Research. 2000;448(2):121–138. doi: 10.1016/s0027-5107(99)00231-6. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Borgmeyer U, Heyman RA, et al. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes and Development. 1992;6(3):329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- 5.Panigrahy D, Kaipainen A, Kieran MW, Huang S. PPARs: a double-edged sword in cancer yherapy? PPAR Research. 2008;2008:2 pages. doi: 10.1155/2008/350351.350351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochimica et Biophysica Acta. 2007;1771(8):936–951. doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Charoensuksai P. PPARs in rhythmic metabolic regulation and implications in health and disease. PPAR Research. 2010;2010:9 pages. doi: 10.1155/2010/243643.243643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eibl G. The role of PPAR-γ and its interaction with COX-2 in pancreatic cancer. PPAR Research. 2008;2008:6 pages. doi: 10.1155/2008/326915.326915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang K, Fan KH, Lamprecht SA, et al. Peroxisome proliferator-activated receptor γ agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1+/− double mutant mice. International Journal of Cancer. 2005;116(4):495–499. doi: 10.1002/ijc.21018. [DOI] [PubMed] [Google Scholar]
- 10.Pino MV, Kelley MF, Jayyosi Z. Promotion of colon tumors in C57BL/6J-APCmin/+ mice by thiazolidinedione PPARγ agonists and a structurally unrelated PPARγ agonist. Toxicologic Pathology. 2004;32(1):58–63. doi: 10.1080/01926230490261320. [DOI] [PubMed] [Google Scholar]
- 11.Voutsadakis IA. Peroxisome proliferator-activated receptor γ (PPARγ) and colorectal carcinogenesis. Journal of Cancer Research and Clinical Oncology. 2007;133(12):917–928. doi: 10.1007/s00432-007-0277-y. [DOI] [PubMed] [Google Scholar]
- 12.Bassaganya-Riera J, Carter AB, Misyak SA, Hontecillas R. Dietary modulation of inflammation-induced colorectal cancer through PPARγ . PPAR Research. 2009;2009:9 pages. doi: 10.1155/2009/498352.498352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nature Reviews Cancer. 2012;12(3):181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins GT, Nie D. PPAR gamma, bioactive lipids, and cancer progression. Frontiers in Bioscience. 2012;17:1816–1834. doi: 10.2741/4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu YL, Li GL, Huang HL, Zhong J, Dai LC. Peroxisome proliferator-activated receptor-γ 34C>G polymorphism and colorectal cancer risk: a meta-analysis. World Journal of Gastroenterology. 2010;16(17):2170–2175. doi: 10.3748/wjg.v16.i17.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capaccio D, Ciccodicola A, Sabatino L, et al. A novel germline mutation in peroxisome proliferator-activated receptor γ gene associated with large intestine polyp formation and dyslipidemia. Biochimica et Biophysica Acta. 2010;1802(6):572–581. doi: 10.1016/j.bbadis.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Dai Y, Qiao L, Kwok WC, et al. Peroxisome proliferator-activated receptor-γ contributes to the inhibitory effects of embelin on colon carcinogenesis. Cancer Research. 2009;69(11):4776–4783. doi: 10.1158/0008-5472.CAN-08-4754. [DOI] [PubMed] [Google Scholar]
- 18.Qiao L, Zou B, Wong BCY. Current understanding of the role of PPAR γ in gastrointestinal cancers. PPAR Research. 2009;2009:8 pages. doi: 10.1155/2009/816957.816957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Girnun GD, Smith WM, Drori S, et al. APC-dependent suppression of colon carcinogenesis by PPARγ . Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13771–13776. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang W, Wang R, Zhang Z, Li D, Yu Y. Enhanced PPAR-γ expression may correlate with the development of Barrett’s esophagus and esophageal adenocarcinoma. Oncology Research. 2011;19(3-4):141–147. doi: 10.3727/096504011x12935427587849. [DOI] [PubMed] [Google Scholar]
- 21.Ma XM, Yu H, Huai N. Peroxisome proliferator-activated receptor-γ is essential in the pathogenesis of gastric carcinoma. World Journal of Gastroenterology. 2009;15(31):3874–3883. doi: 10.3748/wjg.15.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao L, Liu F, Sun L, et al. Upregulation of PPARγ in tissue with gastric carcinoma. Hybridoma. 2010;29(4):341–343. doi: 10.1089/hyb.2010.0013. [DOI] [PubMed] [Google Scholar]
- 23.Sato H, Ishihara S, Kawashima K, et al. Expression of peroxisome proliferator-activated receptor (PPAR)γ in gastric cancer and inhibitory effects of PPARγ agonists. British Journal of Cancer. 2000;83(10):1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai Y, Wang WH. Peroxisome proliferator-activated receptor γ and colorectal cancer. The World Journal of Gastrointestinal Oncology. 2010;2(3):159–164. doi: 10.4251/wjgo.v2.i3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konstantinopoulos PA, Vandoros GP, Sotiropoulou-Bonikou G, Kominea A, Papavassiliou AG. NF-κB/PPARγ and/or AP-1/PPARγ ‘on/off’ switches and induction of CBP in colon adenocarcinomas: correlation with COX-2 expression. International Journal of Colorectal Disease. 2007;22(1):57–68. doi: 10.1007/s00384-006-0112-y. [DOI] [PubMed] [Google Scholar]
- 26.Pancione M, Sabatino L, Fucci A, et al. Epigenetic silencing of peroxisome proliferator- activated receptor γ is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0014229.e14229 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Sabatino L, Fucci A, Pancione M, et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. doi: 10.1038/onc.2012.3. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 28.Ban JO, Kwak DH, Oh JH, et al. Suppression of NF-κB and GSK-3β is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chemico-Biological Interactions. 2010;188(1):75–85. doi: 10.1016/j.cbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Papi A, Rocchi P, Ferreri AM, Orlandi M. RXRγ and PPARγ ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Letters. 2010;297(1):65–74. doi: 10.1016/j.canlet.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 30.Tsukahara T, Hanazawa S, Kobayashi T, Iwamoto Y, Murakami-Murofushi K. Cyclic phosphatidic acid decreases proliferation and survival of colon cancer cells by inhibiting peroxisome proliferator-activated receptor γ . Prostaglandins and Other Lipid Mediators. 2010;93(3-4):126–133. doi: 10.1016/j.prostaglandins.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Röhrl C, Kaindl U, Koneczny I, et al. Peroxisome-proliferator-activated receptors γ and β/δ mediate vascular endothelial growth factor production in colorectal tumor cells. Journal of Cancer Research and Clinical Oncology. 2011;137(1):29–39. doi: 10.1007/s00432-010-0856-1. [DOI] [PubMed] [Google Scholar]
- 32.Peters JM, Foreman JE, Gonzalez FJ. Dissecting the role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in colon, breast, and lung carcinogenesis. Cancer and Metastasis Reviews. 2011;30(3-4):619–640. doi: 10.1007/s10555-011-9320-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foreman JE, Chang WCL, Palkar PS, et al. Functional characterization of peroxisome proliferator-activated receptor-β/δ expression in colon cancer. Molecular Carcinogenesis. 2011;50(11):884–900. doi: 10.1002/mc.20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Zhang H, Zhou ZG, Yan H, Adell G, Sun XF. Biological function and prognostic significance of peroxisome proliferator-activated receptor δ in rectal cancer. Clinical Cancer Research. 2011;17(11):3760–3770. doi: 10.1158/1078-0432.CCR-10-2779. [DOI] [PubMed] [Google Scholar]
- 35.Yang L, Olsson B, Pfeifer D, et al. Knockdown of peroxisome proliferator-activated receptor-β induces less differentiation and enhances cell-fibronectin adhesion of colon cancer cells. Oncogene. 2010;29(4):516–526. doi: 10.1038/onc.2009.370. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, Ning W, Xie D, Guo L, DuBois RN. Peroxisome proliferator-activated receptor δ confers resistance to peroxisome proliferator-activated receptor γ-induced apoptosis in colorectal cancer cells. Oncogene. 2012;31(8):1013–1023. doi: 10.1038/onc.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshinaga M, Taki K, Somada S, et al. The expression of both peroxisome proliferator-activated receptor delta and cyclooxygenase-2 in tissues is associated with poor prognosis in colorectal cancer patients. Digestive Diseases and Sciences. 2011;56(4):1194–1200. doi: 10.1007/s10620-010-1389-9. [DOI] [PubMed] [Google Scholar]
- 38.Rageul J, Mottier S, Jarry A, et al. KLF4-dependent, PPARγ-induced expression of GPA33 in colon cancer cell lines. International Journal of Cancer. 2009;125(12):2802–2809. doi: 10.1002/ijc.24683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ripoli M, Pazienza V. Impact of HCV genetic differences on pathobiology of disease. Expert Review of Anti-Infective Therapy. 2011;9(9):747–759. doi: 10.1586/eri.11.94. [DOI] [PubMed] [Google Scholar]
- 40.Romero-Gómez M, Del Mar Viloria M, Andrade RJ, et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology. 2005;128(3):636–641. doi: 10.1053/j.gastro.2004.12.049. [DOI] [PubMed] [Google Scholar]
- 41.Pazienza V, Vinciguerra M, Andriulli A, Mangia A. Hepatitis C virus core protein genotype 3a increases SOCS-7 expression through PPAR-γ in Huh-7 cells. Journal of General Virology. 2010;91(7):1678–1686. doi: 10.1099/vir.0.020644-0. [DOI] [PubMed] [Google Scholar]
- 42.Dharancy S, Lemoine M, Mathurin P, Serfaty L, Dubuquoy L. Peroxisome proliferator-activated receptors in HCV-related infection. PPAR Research. 2009;2009:5 pages. doi: 10.1155/2009/357204.357204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsujie M, Nakamori S, Okami J, et al. Thiazolidinediones inhibit growth of gastrointestinal, biliary, and pancreatic adenocarcinoma cells through activation of the peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha pathway. Experimental Cell Research. 2003;289(1):143–151. doi: 10.1016/s0014-4827(03)00263-5. [DOI] [PubMed] [Google Scholar]
- 44.Yu H, Jove R. The stats of cancer—new molecular targets come of age. Nature Reviews Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 45.Pazienza V, Tavano F, Benegiamo G, et al. Correlations among PPARγ, DNMT1, and DNMT3B expression levels and pancreatic cancer. PPAR Research. 2012;2012:7 pages. doi: 10.1155/2012/461784.461784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patterson AD, Maurhofer O, Beyoglu D, et al. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Research. 2011;71(21):6590–6600. doi: 10.1158/0008-5472.CAN-11-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vacca M, Degirolamo C, Mariani-Costantini R, Palasciano G, Moschetta A. Lipid-sensing nuclear receptors in the pathophysiology and treatment of the metabolic syndrome. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2011;3(5):562–587. doi: 10.1002/wsbm.137. [DOI] [PubMed] [Google Scholar]
- 48.Li G, Guo GL. Role of class II nuclear receptors in liver carcinogenesis. Anti-Cancer Agents in Medicinal Chemistry. 2011;11(6):529–542. doi: 10.2174/187152011796011064. [DOI] [PubMed] [Google Scholar]
- 49.Matsusue K, Kusakabe T, Noguchi T, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARγ target gene Fsp27. Cell Metabolism. 2008;7(4):302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Meer DLM, Degenhardt T, Väisänen S, et al. Profiling of promoter occupancy by PPARα in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Research. 2010;38(9):2839–2850. doi: 10.1093/nar/gkq012.gkq012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guyton KZ, Chiu WA, Bateson TF, et al. A reexamination of the PPAR-α activation mode of action as a basis for assessing human cancer risks of environmental contaminants. Environmental Health Perspectives. 2009;117(11):1664–1672. doi: 10.1289/ehp.0900758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor α (PPARα in mouse liver tumor induction by trichloroethylene and metabolites. Critical Reviews in Toxicology. 2008;38(10):857–875. doi: 10.1080/10408440802209796. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARα activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. Journal of Clinical Investigation. 2008;118(2):683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor α in transgenic mice: Implications for HCV-associated hepatocarcinogenesis. International Journal of Cancer. 2008;122(1):124–131. doi: 10.1002/ijc.23056. [DOI] [PubMed] [Google Scholar]
- 55.Maggiora M, Oraldi M, Muzio G, Canuto RA. Involvement of PPARα and PPARγ in apoptosis and proliferation of human hepatocarcinoma HepG2 cells. Cell Biochemistry and Function. 2010;28(7):571–577. doi: 10.1002/cbf.1691. [DOI] [PubMed] [Google Scholar]
- 56.Yu J, Shen B, Chu ESH, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51(6):2008–2019. doi: 10.1002/hep.23550. [DOI] [PubMed] [Google Scholar]
- 57.Zhou YM, Wen YH, Kang XY, Qian HH, Yang JM, Yin ZF. Troglitazone, a peroxisome proliferator-activated receptor γ ligand, induces growth inhibition and apoptosis of HepG2 human liver cancer cells. World Journal of Gastroenterology. 2008;14(14):2168–2173. doi: 10.3748/wjg.14.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto Y, Ono T, Dhar DK, et al. Role of peroxisome proliferator-activated receptor-gamma (PPARγ) during liver regeneration in rats. Journal of Gastroenterology and Hepatology. 2008;23(6):930–937. doi: 10.1111/j.1440-1746.2008.05370.x. [DOI] [PubMed] [Google Scholar]
- 59.MartíNez-LóPez N, Varela-Rey M, FernáNdez-Ramos D, et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology. 2010;52(5):1621–1631. doi: 10.1002/hep.23860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vara D, Salazar M, Olea-Herrero N, Guzmán M, Velasco G, Díaz-Laviada I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death and Differentiation. 2011;18(7):1099–1111. doi: 10.1038/cdd.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhalla K, Hwang BJ, Dewi RE, et al. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prevention Research. 2012;5(4):544–552. doi: 10.1158/1940-6207.CAPR-11-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sozio MS, Lu C, Zeng Y, Liangpunsakul S, Crabb DW. Activated AMPK inhibits PPAR-α and PPAR-γ transcriptional activity in hepatoma cells. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2011;301(4):G739–G747. doi: 10.1152/ajpgi.00432.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mueller KM, Kornfeld JW, Friedbichler K, et al. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology. 2011;54(4):1398–1409. doi: 10.1002/hep.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wunderlich FT, Luedde T, Singer S, et al. Hepatic NF-κB essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1297–1302. doi: 10.1073/pnas.0707849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shim J, Kim BH, Kim YI, et al. The peroxisome proliferator-activated receptor γ ligands, pioglitazone and 15-deoxy-Δ12,14-prostaglandin J2, have antineoplastic effects against hepatitis B virus-associated hepatocellular carcinoma cells. International Journal of Oncology. 2010;36(1):223–231. [PubMed] [Google Scholar]
- 66.Yamasaki D, Kawabe N, Nakamura H, et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. European Journal of Cell Biology. 2011;90(8):657–664. doi: 10.1016/j.ejcb.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 67.Galli A, Ceni E, Mello T, et al. Thiazolidinediones inhibit hepatocarcinogenesis in hepatitis B virus-transgenic mice by peroxisome proliferator-activated receptor γ-independent regulation of nucleophosmin. Hepatology. 2010;52(2):493–505. doi: 10.1002/hep.23669. [DOI] [PubMed] [Google Scholar]
- 68.Saito I, Miyamura T, Ohbayashi A, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(17):6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dharancy S, Lemoine M, Mathurin P, Serfaty L, Dubuquoy L. Peroxisome proliferator-activated receptors in HCV-related infection. PPAR Research. 2009;2009:5 pages. doi: 10.1155/2009/357204.357204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng Y, Dharancy S, Malapel M, Desreumaux P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor α and carnitine palmitoyl acyl-CoA transferase 1A. World Journal of Gastroenterology. 2005;11(48):7591–7596. doi: 10.3748/wjg.v11.i48.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dharancy S, Malapel M, Perlemuter G, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128(2):334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 72.De Gottardi A, Pazienza V, Pugnale P, et al. Peroxisome proliferator-activated receptor-α and -γ mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Alimentary Pharmacology and Therapeutics. 2006;23(1):107–114. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 73.Pazienza V, Clément S, Pugnale P, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45(5):1164–1171. doi: 10.1002/hep.21634. [DOI] [PubMed] [Google Scholar]
- 74.Elgouhari HM, Cesario KB, Lopez R, Zein NN. Pioglitazone improves early virologic kinetic response to PEG IFN/RBV combination therapy in hepatitis C genotype 1 naive pts. Hepatology. 2008;48(article 383) [Google Scholar]
- 75.Overbeck K, Genné D, Golay A, Negro F. Pioglitazone in chronic hepatitis C not responding to pegylated interferon-α and ribavirin. Journal of Hepatology. 2008;49(2):295–298. doi: 10.1016/j.jhep.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 76.Negro F. Correction of insulin resistance in chronic hepatitis C patients not responding to the standard of care: more questions than answers. Journal of Hepatology. 2009;50(6):1271–1272. doi: 10.1016/j.jhep.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 77.Kim KH, Shin HJ, Kim K, et al. Hepatitis B Virus X Protein Induces Hepatic Steatosis Via Transcriptional Activation of SREBP1 and PPARγ . Gastroenterology. 2007;132(5):1955–1967. doi: 10.1053/j.gastro.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 78.Guidotti LG, Eggers CM, Raney AK, et al. In vivo regulation of hepatitis B virus replication by peroxisome proliferators. Journal of Virology. 1999;73(12):10377–10386. doi: 10.1128/jvi.73.12.10377-10386.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wakui Y, Inoue J, Ueno Y, et al. Inhibitory effect on hepatitis B virus in vitro by a peroxisome proliferator-activated receptor-β ligand, rosiglitazone. Biochemical and Biophysical Research Communications. 2010;396(2):508–514. doi: 10.1016/j.bbrc.2010.04.128. [DOI] [PubMed] [Google Scholar]
- 80.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer Journal for Clinicians. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 81.Kristiansen G, Jacob J, Buckendahl AC, et al. Peroxisome proliferator-activated receptor γ is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clinical Cancer Research. 2006;12(21):6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- 82.Hashimoto K, Farrow BJ, Evers BM. Activation and Role of MAP Kinases in 15d-PGJ2-Induced Apoptosis in the Human Pancreatic Cancer Cell Line MIA PaCa-2. Pancreas. 2004;28(2):153–159. doi: 10.1097/00006676-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 83.Galli A, Ceni E, Crabb DW, et al. Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARγ independent mechanisms. Gut. 2004;53(11):1688–1697. doi: 10.1136/gut.2003.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Motomura W, Nagamine M, Tanno S, et al. Inhibition of cell invasion and morphological change by troglitazone in human pancreatic cancer cells. Journal of Gastroenterology. 2004;39(5):461–468. doi: 10.1007/s00535-003-1324-3. [DOI] [PubMed] [Google Scholar]
- 85.Vitale G, Zappavigna S, Marra M, et al. The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnology Advances. 2012;30(1):169–184. doi: 10.1016/j.biotechadv.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Koga H, Selvendiran K, Sivakumar R, et al. PPARγ potentiates anticancer effects of gemcitabine on human pancreatic cancer cells. International Journal of Oncology. 2012;40(3):679–685. doi: 10.3892/ijo.2011.1237. [DOI] [PubMed] [Google Scholar]
- 87.Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opinion on Drug Safety. 2010;9(2):347–354. doi: 10.1517/14740331003623218. [DOI] [PubMed] [Google Scholar]
- 88.Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN. How safe is the use of thiazolidinediones in clinical practice? Expert Opinion on Drug Safety. 2009;8(1):15–32. doi: 10.1517/14740330802597821. [DOI] [PubMed] [Google Scholar]