

Abstract
Metalloregulators regulate transcription in response to metal ions. Many studies have provided insights into how transcription is activated upon metal binding by MerR-family metalloregulators. In contrast, how transcription is turned off after activation is unclear. Turning off transcription promptly is important, however, as the cells would not want to continue expressing metal resistance genes and thus waste energy after metal stress is relieved. Using single-molecule FRET measurements we studied the dynamic interactions of the copper efflux regulator (CueR), a Cu+-responsive MerR-family metalloregulator, with DNA. Besides quantifying its DNA binding and unbinding kinetics, we discovered that CueR spontaneously flips its binding orientation at the recognition site. CueR also has two different binding modes, corresponding to interactions with specific and nonspecific DNA sequences, which would facilitate recognition localization. Most strikingly, a CueR molecule coming from solution can directly substitute for a DNA-bound CueR or assist the dissociation of the incumbent CueR, both of which are unique examples for any DNA-binding protein. The kinetics of the direct protein substitution and assisted dissociation reactions indicate that these two unique processes can provide efficient pathways to replace a DNA-bound holo-CueR with apo-CueR, thus turning off transcription promptly and facilely.
Keywords: single-molecule imaging, protein–DNA interaction dynamics
Bacteria often dwell in environments with high concentrations of metals. Some of these metals are essential, but many are toxic. Even the essential metals, for example iron and copper, can become detrimental above a certain concentration inside cells. Many biological processes are thus present to regulate and maintain intracellular metal homeostasis (1–9). One of them is through metalloregulators, which respond to metal ions and regulate the transcription of genes that protect the bacteria from metal-induced stress (5–7, 10). The MerR-family metalloregulators respond to many metal ions with high selectivity and sensitivity, such as Hg2+ and Cu2+ (5, 11, 12).
All MerR-family metalloregulators are homodimeric proteins. They regulate transcription via a DNA distortion mechanism (5, 13–16). They recognize specific dyad-symmetric DNA sequences within a promoter, and both their apo and holo forms bind DNA tightly. In the absence of metal, the metalloregulator bends the DNA; in this configuration RNA polymerase (RNAp) cannot interact with both -10 and -35 sequences properly and transcription is repressed. Upon binding metal, the metalloregulator changes its conformation and further unwinds the DNA slightly to allow proper RNAp interactions with the -10 and -35 sequences; transcription is then activated.
Although the mechanisms of transcription activation by MerR-family metalloregulators are well-studied (5, 13–16), little is yet known about how transcription activation is reversed. Turning off transcription promptly is important, however, as the cells would not want to continue expressing metal resistance genes and thus waste energy after metal stress is relieved. Metal dissociation to convert a holo-metalloregulator to its apo form would be the simplest way to turn off transcription, but is unlikely as the metal is bound tightly (often by cysteine ligands) and metal−cysteine bond dissociation is slow (17). For example, CueR, the Cu+-responsive MerR-family metalloregulator in Escherichia coli, has a Cu+ binding affinity of approximately 10-21 M (18). Although thiol ligand exchange can possibly facilitate Cu+ removal from the binding site as observed for copper chaperones (19), no evidence exists that CueR can undergo similarly facile ligand exchange reactions. Then a holo-metalloregulator has to be replaced somehow by its apo-protein to turn off transcription. Here the simplest scenario would be for the holo-protein to unbind from DNA, followed by the binding of an apo-protein. What then are the timescales of the protein unbinding and the subsequent binding? Are there any alternative, and more efficient, pathways to turn off transcription?
Here we use single-molecule FRET (smFRET) measurements (20, 21) to address the above questions. We focus on CueR, which regulates the transcription of CopA, a membrane transporter that pumps Cu+ out of the cytoplasm, and CueO, a periplasmic multicopper oxidase that facilitates copper removal from the cell (6, 22). By examining CueR–DNA interactions at the single-molecule level, we found that apo-CueR could turn off transcription via a direct protein substitution pathway and an assisted protein dissociation pathway; both pathways are unprecedented and more efficient than the generic unbinding-plus-binding pathway. Moreover, CueR can spontaneously flip while staying bound on DNA and it has two different binding modes, both of which are advantageous for its regulatory function.
Results
Experimental Strategy.
We used smFRET to probe CueR interactions with a 25-bp dsDNA (SI Appendix, Fig. S1A). One end of this DNA had a FRET donor, Cy3, and the other end on a different strand had a biotin for surface immobilization, ensuring that only dsDNA was immobilized and fluorescently monitored in the experiments. The homodimeric CueR was labeled with a single FRET acceptor, Cy5, and supplied in a flowing solution. Upon CueR binding to DNA, FRET occurs, changing the Cy3 and Cy5 fluorescence intensities, which report the CueR–DNA interactions.
We used two types of DNA. One contains the dyad-symmetric sequence recognized by CueR and is referred to as the (specific) DNA; this 25-bp sequence also spans the entire footprint of CueR at the promoter region (23). The other does not contain the CueR-recognition sequence and is referred to as the nonspecific DNA. We made two CueR variants to place the Cy5 label (SI Appendix, Fig. S2A). In one variant, Cy5 is attached to C129 within the metal-binding domain of one monomer (i.e., CueRCy5-C129). The other has the Cy5 attached to E96C located at the α-helix dimerization domain (i.e., CueRCy5-E96C). In vitro transcription assay confirmed that both variants were as active as the wild-type CueR in activating transcription controlled by the copA promoter (SI Appendix, Fig. S1B and C).
Two Protein Binding Orientations on DNA.
Fig. 1A shows the EFRET trajectory for a surface-immobilized DNA molecule interacting with holo-CueRCy5-C129. Three EFRET states are clear at E0 ∼ 0.07, E1 ∼ 0.25, and E2 ∼ 0.92. Correspondingly, the histogram of the EFRET trajectories shows three peaks centered at E0, E1, and E2 (Fig. 1F); the relative peak areas reflect the relative stabilities of the chemical species associated with the respective EFRET states.
Fig. 1.

Dynamic CueR–DNA interactions. (A) Single-molecule EFRET trajectory of an immobilized Cy3-DNA interacting with holo-CueRCy5-C129 (2 nM). The cartoons on the right show CueRCy5-C129 in two binding orientations. The FRET donor (green sphere) and acceptor (red sphere) are drawn on the DNA and protein at their approximate locations. The gray line is original data; the red line is after nonlinear filtering. (B) Same as A, but with 2 nM holo-CueRCy5-E96C. (C) Same as A, but using the nonspecific DNA and 4 nM holo-CueRCy5-C129. Here, τ0 is denoted as τoff, and τ1 and τ2 are denoted together as τon. (D) Same as A, but with a mixture of holo-CueRCy5-C129 and holo-CueRCy5-E96C of 5 nM each. (E) Same as A, but with a mixture of apo-CueRCy5-C129 and holo-CueRCy5-E96C of 5 nM each. The blue arrows denote the transitions from the holo-protein bound states to the apo-protein bound states, and the black arrows denote the reverse transitions. (F) Histogram of EFRET trajectories of holo-CueRCy5-C129–DNA interactions at [holo-CueRCy5-C129] = 2 nM. Compiled from > 500 EFRET trajectories. Solid lines are fits of Voigt functions centered at about 0.07, 0.25, and 0.92, with percentage peak areas of 90.5 ± 0.1%, 4.9 ± 0.3%, and 4.6 ± 0.3%, respectively. Bin size, 0.01. (G) Same as F, but with 2 nM holo-CueRCy5-E69C. The three peaks center at about 0.07, 0.42, and 0.62, with percentage peak areas of 89.5 ± 0.1%, 5.5 ± 0.3%, and 5.0 ± 0.3%, respectively. (H) [CueRCy5-C129] dependence of the (E2 + E1)/E0 peak area ratios obtained from data such as in F. The solid lines are fits with Eqs. 8 (holo) and 9 (apo) (see also SI Appendix, section S15).
The E0 state has the lowest EFRET value, which indicates the farthest FRET donor–acceptor distance; it corresponds to a free DNA without a bound protein (i.e., the unbound state). Its assignment was verified by a control experiment where no protein was present. The dominance of the E0 peak in the EFRET histogram comes from the low concentrations of the protein (≤ 10 nM), where the DNA stays mostly at the unbound state.
The E1 and E2 states, whose larger values indicate FRET donor–acceptor proximities, can be assigned to the two orientations of holo-CueRCy5-C129 bound to DNA. CueR is a homodimer; the Cy5 label at one monomer makes the protein asymmetric. The distance between the Cy5 on the protein and the Cy3 on DNA is closer in one binding orientation than in the other, resulting in two different EFRET values (i.e., E2 and E1) (Fig. 1A, Right, cartoons). Consistently, the E1 and E2 peaks have about the same area in the EFRET histogram (Fig. 1F), as the two protein binding orientations are chemically equivalent except for the labels. Moreover, with increasing holo-CueRCy5-C129 concentration (i.e., [holo-CueRCy5-C129]), the E1 and E2 peak areas in the EFRET histogram increase relative to that of E0 (Fig. 1H), consistent with that increasing protein concentration should lead to higher populations of protein–DNA complexes. Similarly, using apo-CueRCy5-C129, three EFRET states were observed, corresponding to the unbound and the two protein-bound states with opposite binding orientations (SI Appendix, Fig. S4A and B).
To confirm the assignments of the E1 and E2 states, we further studied CueRCy5-E69C (SI Appendix, Section S3). The Cy3–Cy5 distance in its E2 binding orientation should be longer than that for CueRCy5-C129, whereas that in its E1 orientation should be shorter (Fig. 1B, Right, cartoons). Indeed, for CueRCy5-E96C–DNA interactions, E2 decreased to approximately 0.62 and E1 increased to approximately 0.42 (Fig. 1B and G).
When CueRCy5-C129 interacted with the nonspecific DNA, three EFRET states were still observed (Fig. 1C). Therefore, CueR can always bind in two orientations regardless of the DNA sequence. The E1 and E2 values here are slightly shifted compared with those in interactions with the specific DNA (Fig. 1A), likely because the protein can slide more freely along the DNA in the absence of the recognition sequence.
Protein Flipping on DNA.
In the EFRET trajectories of both holo- and apo-CueR–DNA interactions (Fig. 1A and SI Appendix, Fig. S4A), the E0↔E1 and E0↔E2 transitions correspond to the reversible protein–DNA binding and unbinding. Interestingly, direct E1↔E2 transitions also occur. These E1↔E2 transitions could be due to (i) protein flipping, i.e., the protein flips its binding orientation spontaneously without detaching from DNA completely, or (ii) protein unbinding followed by a rapid rebinding in a different orientation during which the unbound E0 state was not detected because of limited experimental time resolution (approximately 50 ms). The second scenario is unlikely, however, because these E1↔E2 transitions still occur at protein concentrations as low as 0.5 nM, where protein rebinding is slow and the unbound state should be readily observed. Therefore, the direct E1↔E2 transitions indicate that a DNA-bound CueR can spontaneously flip its orientation without complete detachment from DNA.
In contrast, no direct E1↔E2 transitions were observed for CueRCy5-C129 interacting with the nonspecific DNA (Fig. 1C). This contrast indicates that protein flipping occurs only when CueR binds to the specific DNA sequence, in which CueR distorts the DNA structure as shown by past structural studies on MerR-family regulators (5, 13–15).
Single-Step Protein Binding Kinetics.
In the EFRET trajectories (e.g., Fig. 1A), τ0 is the microscopic dwell time on the unbound E0 state; its statistical properties, such as its average and distribution, contain the information about the protein binding kinetics. Here, 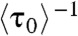 , where
, where 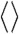 denotes averaging, represents the single-molecule rate of CueR binding to DNA. Expectedly,
denotes averaging, represents the single-molecule rate of CueR binding to DNA. Expectedly, 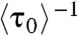 increases with increasing [holo- or apo-CueR] (Fig. 2A). Moreover, the distribution of τ0 follows a single-exponential decay in the presence of either holo- or apo-CueR (Fig. 2B). This distribution indicates that CueR binding to DNA follows simple single-step binding kinetics, i.e., containing only one rate-limiting step (see later for the derived probability density function of τ0, which quantitatively describes the distribution of τ0).
increases with increasing [holo- or apo-CueR] (Fig. 2A). Moreover, the distribution of τ0 follows a single-exponential decay in the presence of either holo- or apo-CueR (Fig. 2B). This distribution indicates that CueR binding to DNA follows simple single-step binding kinetics, i.e., containing only one rate-limiting step (see later for the derived probability density function of τ0, which quantitatively describes the distribution of τ0).
Fig. 2.

Single-step protein binding kinetics. (A) [CueR] dependence of 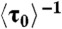 for CueRCy5-C129–DNA interactions. Solid lines are fits with Eq. 5 (see also SI Appendix, section S12). (B) Distributions of τ0 for holo- and apo-CueRCy5-C129 interactions with DNA, both at 0.5 nM protein concentration. Data compiled from about 500 (holo) and 600 (apo) EFRET trajectories. Bin size, 2 s. Both distributions were normalized to the first point for comparison. Solid lines are fits with a single exponential function Nγ exp(-γτ); γ(holo) = 0.038 ± 0.002 s-1, γ(apo) = 0.031 ± 0.004 s-1, and N is a scaling parameter.
for CueRCy5-C129–DNA interactions. Solid lines are fits with Eq. 5 (see also SI Appendix, section S12). (B) Distributions of τ0 for holo- and apo-CueRCy5-C129 interactions with DNA, both at 0.5 nM protein concentration. Data compiled from about 500 (holo) and 600 (apo) EFRET trajectories. Bin size, 2 s. Both distributions were normalized to the first point for comparison. Solid lines are fits with a single exponential function Nγ exp(-γτ); γ(holo) = 0.038 ± 0.002 s-1, γ(apo) = 0.031 ± 0.004 s-1, and N is a scaling parameter.
Two Different Binding Modes of Protein.
τ1 and τ2 are the microscopic dwell times on the E1 and E2 states, the two protein-bound states of opposite binding orientations (Fig. 1A and B). They contain the information about the kinetics of protein unbinding as well as what is happening kinetically during the protein-bound states. Interestingly, the distributions of τ1 and τ2 for holo-CueRCy5-C129–DNA interactions both follow a double-exponential decay (Fig. 3A and SI Appendix, Fig. S8A). This double-exponential distribution indicates that there are two major kinetic species within each protein-binding orientation, i.e., holo-CueR has two different binding modes on DNA. The distributions of τ1 and τ2 are identical within experimental error (SI Appendix, Fig. S8A), consistent with the E1 and E2 states being equivalent. Similar double-exponential distributions of τ1 and τ2 were also observed for apo-CueRCy5-C129–DNA interactions (Fig. 3A and SI Appendix, Fig. S4C).
Fig. 3.

Two different binding modes of CueR on DNA. (A) Distributions of τ2 for holo- and apo-CueRCy5-C129 interactions with specific DNA, both at 2 nM protein concentration. Data compiled from about 500 (holo) and 700 (apo) EFRET trajectories. Bin size, 0.15 s. The two distributions were normalized to the first data point for comparison. Solid lines are fits with a sum of two exponentials: N[Aγ1 exp(-γ1τ) + (1 - A)γ2 exp(-γ2τ)]. For holo: γ1 = 0.55 ± 0.01 s-1, γ2 = 6.4 ± 0.2 s-1, and A = 0.68 ± 0.01; for apo: γ1 = 0.35 ± 0.02 s-1, γ2 = 1.6 ± 0.06 s-1, and A = 0.57 ± 0.02. (B) Distributions of τ2→0 and τ2→1 at [holo-CueRCy5-C129] = 2 nM. Solid lines are global fits with a sum of two exponentials using the same parameters for the holo data in A. Bin size, 0.15 s. (C) Distribution of τon for holo-CueRCy5-C129 interactions with the nonspecific DNA at [holo-CueRCy5-C129] = 7 nM. To increase statistics, τ1 and τ2 from about 225 EFRET trajectories were combined here as τon. Bin size, 0.05 s. Solid line is a fit with Nγ exp(-γτ); γ = 12.8 ± 0.6 s-1.
In contrast, when the nonspecific DNA was used, the distribution of the dwell times on the protein-bound states follows a single-exponential decay (Fig. 3C), indicating that only one major kinetic species is present within each protein-binding orientation. Therefore, the two different binding modes of CueR on DNA are present only when CueR recognizes the specific DNA sequence.
Past studies have shown that MerR-family regulators distort DNA structure upon binding to the specific dyad-symmetric sequence (5, 13, 14). We can thus attribute one of the two kinetic species within each binding orientation to a complex in which CueR distorts the DNA structure. For the other kinetic species, we could attribute it to a CueR–DNA complex in which the CueR binds DNA in a way as if the DNA is nonspecific; this attribution is reasonable because CueR does bind nonspecific DNA and the binding mode here must be different from that of the specific DNA binding mode. No structural information is yet available about CueR, or any MerR-family regulators, in complex with a nonspecific DNA. Just for illustrative purposes, we drew the cartoon of this complex having the DNA structure undistorted (Fig. 1C, Right).
Moreover, τ2 can be divided into two subtypes: τ2→1 and τ2→0, depending on whether an E2 → E1 or E2 → E0 transition concludes a τ2 period in the EFRET trajectories. For holo-CueRCy5-C129–DNA interactions, τ2→1 and τ2→0 follow the same double-exponential distribution with identical exponents (Fig. 3B), indicating that upon leaving the E2 state, transitions to the E1 or E0 state must start from the same kinetic species of the two binding modes within the E2 state. Similar behaviors and conclusions also apply by analyzing τ1→2 versus τ1→0 (SI Appendix, Fig. S8D), as well as for apo-CueRCy5-C129–DNA interactions (SI Appendix, Fig. S4E and F).
Direct Protein Substitution and Assisted Protein Dissociation.
For the microscopic dwell time τ2 (or the equivalent τ1, SI Appendix, Fig. S5), its average 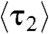 represents the lifetime of the protein-bound state E2. And,
represents the lifetime of the protein-bound state E2. And, 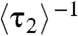 represents the rate of leaving the state E2. Surprisingly,
represents the rate of leaving the state E2. Surprisingly, 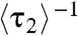 increases linearly with increasing [holo- or apo-CueR] (Fig. 4A). This dependence indicates that a protein molecule coming from the solution must disrupt the protein–DNA complex, thus shortening its lifetime.
increases linearly with increasing [holo- or apo-CueR] (Fig. 4A). This dependence indicates that a protein molecule coming from the solution must disrupt the protein–DNA complex, thus shortening its lifetime.
Fig. 4.

Direct protein substitution and assisted dissociation. (A) [CueR] dependence of 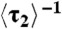 for CueRCy5-C129–DNA interactions. Solid lines are fits with Eqs. 6 (holo) and 7 (apo). (B) The [holo-CueR] dependence of
for CueRCy5-C129–DNA interactions. Solid lines are fits with Eqs. 6 (holo) and 7 (apo). (B) The [holo-CueR] dependence of 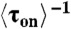 for holo-CueRCy5-C129 interactions with the nonspecific DNA; the τ1 and τ2 are combined here and denoted as τon. Solid line is a fit with a horizontal line at 5.9 ± 0.1 s-1. Each data point in A and B is an average of data from about 250 EFRET trajectories. (C) Dependence of N2→1/N2→0 on [holo-CueRCy5-C129]. Solid line is a fit with Eq. 3. (D) Dependence of N2→1/N2→0 on [apo-CueRCy5-C129]. Solid line is a fit with Eq. 4. (Inset) Same data but the y-value is the inverse, N2→0/N2→1.
for holo-CueRCy5-C129 interactions with the nonspecific DNA; the τ1 and τ2 are combined here and denoted as τon. Solid line is a fit with a horizontal line at 5.9 ± 0.1 s-1. Each data point in A and B is an average of data from about 250 EFRET trajectories. (C) Dependence of N2→1/N2→0 on [holo-CueRCy5-C129]. Solid line is a fit with Eq. 3. (D) Dependence of N2→1/N2→0 on [apo-CueRCy5-C129]. Solid line is a fit with Eq. 4. (Inset) Same data but the y-value is the inverse, N2→0/N2→1.
Leaving the E2 state can lead to either state E1, the other protein-bound state with the opposite protein orientation, or state E0, the unbound state. The former would involve the incoming protein to replace the incumbent protein on DNA and form a new complex with an opposite binding orientation (50% probability), namely a direct protein substitution. The latter would involve the incoming protein to carry away the incumbent protein, namely an assisted protein dissociation.
To differentiate these two possibilities, we examined the relative transition probabilities to the E1 and E0 states after the protein–DNA complex leaves the E2 state. This relative transition probabilities can be quantified by the ratio N2→1/N2→0, where N2→1 is the number of E2 → E1 transitions and N2→0 is that of E2 → E0 transitions observed in the EFRET trajectories. If the direct protein substitution dominates, N2→1/N2→0 would increase with increasing protein concentration because more E2 → E1 transitions are expected at higher protein concentrations. If the assisted protein dissociation dominates, N2→1/N2→0 would decrease with increasing protein concentration.
Strikingly, both behaviors were observed, but it depends on the metallation state of CueR. For holo-CueR–DNA interactions, N2→1/N2→0 increases with increasing protein concentration, whereas for apo-CueR, the opposite was observed (Fig. 4C and D). Therefore, both the direct protein substitution and the assisted protein dissociation operate when a CueR molecule interacts with a CueR–DNA complex. To our knowledge, these are unique examples of direct protein substitution and assisted protein dissociation known to any DNA-binding proteins.
To verify the direct protein substitution process by holo-CueR, we performed an experiment using a mixture of holo-CueRCy5-C129 and holo-CueRCy5-E96C. Direct transitions were indeed observed in the EFRET trajectories between the states of holo-CueRCy5-C129–DNA complexes (E1 ∼ 0.25, E2 ∼ 0.92) and those of holo-CueRCy5-E96C–DNA complexes (E1 ∼ 0.42, E2 ∼ 0.62), reporting the exchange of proteins on DNA (Fig. 1D and SI Appendix, Fig. S6).
Note that the direct protein substitution can lead to E1↔E2 transitions in the EFRET trajectories. These protein-substitution-caused E1↔E2 transitions do not preclude the presence of spontaneous protein flipping, however, because the E1↔E2 transitions still occur at very low protein concentrations, where protein substitution is negligible.
As a control, and in contrast, when the nonspecific DNA was used, the lifetime of the CueR–DNA complex is independent of the protein concentration, indicating no occurrence of either the direct protein substitution or the assisted protein dissociation (Fig. 4B). Therefore, these two processes only occur to a CueR–DNA complex in which CueR recognizes the specific sequence and distorts the DNA structure.
Interaction Mechanism and Quantitative Kinetics.
Combining the information determined above, we can formulate a minimal kinetic model of CueR–DNA interactions that contains the following processes (Fig. 5): (i) CueR binds to DNA reversibly (i.e., k1 and k-1) with single-step binding kinetics; its two binding orientations are distinguishable by asymmetrically labeling the protein; and the DNA that contains the recognition sequence can have a distorted structure in the complex (i.e., I1 and I2). (ii) CueR can flip its orientation spontaneously on DNA (i.e., k4); this flipping occurs only when CueR recognizes the specific sequence, where the DNA structure is distorted. (iii) In each binding orientation, CueR interacts with DNA in two different binding modes, which interconvert (i.e., k3 and k-3) and are observed only when CueR recognizes the specific DNA sequence. Besides the form in which DNA structure is distorted, we propose that the other form is similar to that when CueR binds to a nonspecific DNA (i.e., 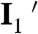 and
and 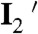 ). (iv) The CueR–DNA complex (I1 or I2) can undergo direct substitution, where a holo-CueR molecule from the solution replaces a bound holo-CueR protein directly (i.e., k2a), or undergo assisted dissociation, where an apo-CueR molecule from the solution helps carry away a bound apo-CueR molecule (i.e., k2b). For the direction substitution, the replacing protein has a 50% probability of ending up with the opposite orientation to the incumbent one. Either the direct substitution or the assisted dissociation occurs only when CueR recognizes the specific DNA sequence. And, all the transitions that depart from the E2 (or E1) state start from the same species I2 (or I1). Control experiments using vesicle trapping of a CueR–DNA pair further showed that the monomer-dimer equilibrium of CueR (if it exists) and the quartz surface effect were insignificant in considering the CueR–DNA interaction kinetics (SI Appendix, section S8). By removing the artificial differentiation of the two orientations by the FRET labels, this mechanistic model simplifies to SI Appendix, Fig. S12, in which only three distinct species exist: the free DNA, and the two CueR–DNA complexes of the two different binding modes.
). (iv) The CueR–DNA complex (I1 or I2) can undergo direct substitution, where a holo-CueR molecule from the solution replaces a bound holo-CueR protein directly (i.e., k2a), or undergo assisted dissociation, where an apo-CueR molecule from the solution helps carry away a bound apo-CueR molecule (i.e., k2b). For the direction substitution, the replacing protein has a 50% probability of ending up with the opposite orientation to the incumbent one. Either the direct substitution or the assisted dissociation occurs only when CueR recognizes the specific DNA sequence. And, all the transitions that depart from the E2 (or E1) state start from the same species I2 (or I1). Control experiments using vesicle trapping of a CueR–DNA pair further showed that the monomer-dimer equilibrium of CueR (if it exists) and the quartz surface effect were insignificant in considering the CueR–DNA interaction kinetics (SI Appendix, section S8). By removing the artificial differentiation of the two orientations by the FRET labels, this mechanistic model simplifies to SI Appendix, Fig. S12, in which only three distinct species exist: the free DNA, and the two CueR–DNA complexes of the two different binding modes.
Fig. 5.

Mechanistic model of CueR–DNA interactions. Here, k’s are the kinetic constants and described in the text. P denotes protein.
Using the kinetic mechanism in Fig. 5 we can derive the probability density functions, f, for the dwell times τ0, τ1, and τ2 (SI Appendix, sections S11 and S12); these f’s quantitatively describe the normalized distributions of the following dwell times:
| [1] |
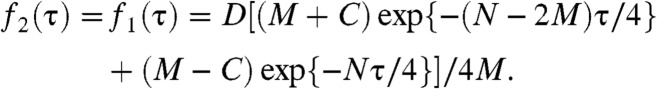 |
[2] |
Here [P] stands for protein concentration; M, N, C, and D are all functions of [P] and the k’s defined in Fig. 5. Here, f0(τ) is a single-exponential distribution function, as observed for the distribution of τ0 (Fig. 2B), and reflecting the single-step binding kinetics. The equivalent f2(τ) and f1(τ) are double-exponential distribution functions, consistent with experimental observations (Fig. 3A and SI Appendix, Fig. S4D) and reflecting the two kinetic species in both the E2 and E1 states associated with CueR’s two different binding modes on DNA. Note that for f2(τ) and f1(τ), either k2a or k2b is close to zero, depending on whether apo-CueR or holo-CueR is concerned.
Similarly, the probability density functions for τ2→0 and τ2→1 are predicted to also follow double-exponential distributions with identical exponents (SI Appendix, Eq. S7 and S8), as observed in Fig. 3B. From them we can derive N2→1/N2→0, the ratio between the numbers of E2 → E1 and E2 → E0 transitions in the EFRET trajectories:
| [3] |
| [4] |
Eq. 3 predicts that N2→1/N2→0 will increase linearly with increasing [P] when k2b = 0, consistent with Fig. 4C and supporting that the direct substitution process dominates for holo-CueR–DNA interactions (i.e., k2a≫k2b ≈ 0). On the other hand, Eq. 4 predicts that N2→1/N2→0 will decrease with increasing [P] when k2a = 0, consistent with Fig. 4D and supporting that the assisted protein dissociation dominates for apo-CueR–DNA interactions (i.e., k2b≫k2a ≈ 0).
From the probability density functions it follows:
| [5] |
| [6] |
| [7] |
where K3D = k3/k-3. Eq. 5 gives that 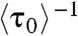 , which represents the protein binding rate, equals k1[P], as expected and observed (Fig. 2A). Regarding
, which represents the protein binding rate, equals k1[P], as expected and observed (Fig. 2A). Regarding 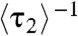 and
and 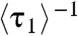 , Eq. 6 applies to holo-CueR–DNA interactions, and Eq. 7 to apo-CueR–DNA interactions; both are linear functions of [P], as observed (Fig. 4A and SI Appendix, Fig. S5). This kinetic model also gives the ratio of peak areas in the EFRET histograms (Fig. 1H; SI Appendix, section S15):
, Eq. 6 applies to holo-CueR–DNA interactions, and Eq. 7 to apo-CueR–DNA interactions; both are linear functions of [P], as observed (Fig. 4A and SI Appendix, Fig. S5). This kinetic model also gives the ratio of peak areas in the EFRET histograms (Fig. 1H; SI Appendix, section S15):
| [8] |
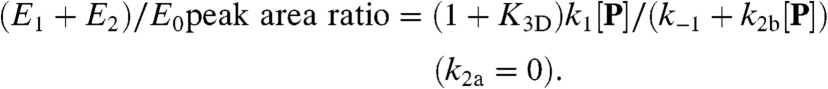 |
[9] |
Using the above equations to fit data, we determined the kinetic parameters for both holo-CueR and apo-CueR interactions with the specific DNA (Table 1 and SI Appendix, section S16). Strikingly, for holo-CueR, its rate constant for direct protein substitution (k2a ∼ 134 × 106 M-1 s-1) is about 15 times larger than its binding rate constant (k1 ∼ 9 × 106 M-1 s-1), whereas for apo-CueR, its rate constant for assisted dissociation (k2b ∼ 60 × 106 M-1 s-1) is 10 times larger than its binding rate constant (k1 ∼ 6 × 106 M-1 s-1). These larger rate constants indicate that the presence of a CueR on DNA facilitates another CueR molecule in finding the recognition sequence, leading to either direct protein substitution or assisted protein dissociation, both of which have functional advantages in transcription regulation (see Discussion).
Table 1.
Kinetic parameters for CueR interactions with specific DNA
| Processes | Kinetic parameters | Holo | Apo |
| Binding | k1( × 106 M-1 s-1) | 9 ± 4 | 6 ± 1 |
| Unbinding | k-1(s-1) | 1.1 ± 0.3 | 0.4 ± 0.1 |
| Dissociation constant | KD( = k-1/k1)(nM) | 122 ± 63 | 67 ± 20 |
| Interconversion | k3(s-1) | 2.0 ± 0.2 | 0.4 ± 0.1 |
| k-3(s-1) | 1.4 ± 0.1 | 0.54 ± 0.07 | |
| K3D = k3/k-3 | 1.4 ± 0.2 | 0.74 ± 0.2 | |
| Substitution | k2a( × 106 M-1 s-1) | 134 ± 20 | N/A |
| Assisted dissociation | k2b( × 106 M-1 s-1) | N/A | 54 ± 22 |
| Flipping | k4(s-1) | 0.13 ± 0.05 | 0.21 ± 0.03 |
N/A, not applicable.
Discussion
Using smFRET we have quantified the dynamic interactions of CueR with both specific and nonspecific DNA. We found that (i) both holo- and apo-CueR can flip their binding orientations spontaneously on DNA, a phenomenon that has also been observed for other DNA binding proteins (24); (ii) both holo- and apo-CueR have two different binding modes on DNA: one in which they recognize the specific sequence and distort the DNA structure and the other likely mimicking their interactions with nonspecific DNA; and (iii) when bound to DNA, holo-CueR can undergo direct substitution, whereas apo-CueR can undergo assisted dissociation, both by another protein molecule from the surrounding solution. Similar behaviors were also observed when a longer DNA sequence containing the entire promoter region was used (SI Appendix, section S19). To our knowledge, both the direct substitution and the assisted dissociation are unique examples known for any transcription factors as well as for DNA-binding proteins in general. Moreover, all these processes (i.e., flipping, two different binding modes, direct substitution, and assisted dissociation) occur only when CueR is interacting with a DNA that contains the recognition sequence.
All above features of CueR can provide advantages for its regulatory function. Being able to flip spontaneously on DNA indicates that the bound CueR is highly dynamic. This dynamic nature, especially for holo-CueR, may facilitate transcription initiation, which involves large structural rearrangements of associated proteins and DNA (16). The two different binding modes, in which CueR interacts with DNA, specifically or nonspecifically, are beneficial for CueR in its searching for the recognition sequence in the large bacterial chromosome [approximately 4.6 million base pairs for E. coli (25)]. Being able to interact with DNA nonspecifically would help CueR sliding along the chromosome; and upon locating the recognition sequence, the CueR–DNA complex can interconvert to the specific binding mode, thereby distorting the DNA structure to regulate transcription. This sliding along DNA via nonspecific interactions has long been realized to be advantageous for reducing the dimensionality of site search for DNA-binding proteins (26).
Both the direct protein substitution and the assisted dissociation can provide efficient pathways for CueR to turn off transcription after activation. As Cu+ dissociation from holo-CueR is unlikely due to its tight binding (18), holo-CueR, which is the activator, needs to be replaced by apo-CueR, which is the repressor, to turn off transcription. A generic way would be for holo-CueR to unbind from DNA first, followed by the binding of an apo-CueR, which would be the dominant form of the protein inside a cell after the activation of copper resistance genes. From the rate constant k-1 (1.1 s-1; Table 1), holo-CueR unbinding takes about 0.9 s (Fig. 6, step i). The timescale of the subsequent apo-CueR binding depends on the intracellular protein concentration. Depending on growth conditions, an E. coli cell has about 320–400 copies of CueR (27), corresponding to [P] ≈ 355–440 nM with a cell volume of approximately 1.5 fL (18). With the rate constant k1 ∼ 6 × 106 M-1 s-1 (Table 1), apo-CueR binding to DNA takes about 0.4–0.5 s (= 1/(k1[P]); Fig. 6, step ii). Therefore, this generic pathway takes a total of approximately 1.4 s to turn off transcription.
Fig. 6.

Pathways for turning off transcription by CueR. The timescales are denoted for relevant kinetic steps, including (i) unbinding, (ii) binding, (iii) direct substitution, and (iv) assisted dissociation.
Alternatively, the bound holo-CueR can be assisted to dissociate or be directly substituted by an apo-CueR (Fig. 6, steps iii and iv). It is reasonable to assume that apo-protein can either assist a holo-protein to unbind or substitute a holo-protein, as apo- and holo-proteins do not differ significantly in most of their kinetic parameters (Table 1). The direct substitution of holo-CueR by an apo protein was indeed observed when we used a mixture of apo-CueRCy5-C129 and holo-CueRCy5-E96C (Fig. 1E, and SI Appendix, section S21). Using the rate constant k2b (54 × 106 M-1 s-1), the assisted dissociation takes about 0.05 s (= 1/k2b[P]; Fig. 6, step iv), about 18 times faster than the spontaneous unbinding (about 0.9 s; Fig. 6, step i). Taking into account the subsequent apo-protein binding, the assisted dissociation would accelerate the unbinding-plus-binding pathway by approximately two times for turning off transcription. On the other hand, using the rate constant k2a (134 × 106 M-1 s-1; Table 1), the direct substitution takes about 0.02 s (= 1/k2a[P]) to reach the apo-protein bound, transcription-repressed state (Fig. 6, step iii), which is about 70 times faster than the unbinding-plus-binding pathway (Fig. 6, step i and ii). Therefore, the direct substitution of holo-CueR by apo-CueR would be the most efficient pathway for turning off transcription. It is worth noting that cellular conditions are not exactly the same as in our experiments and kinetic constants may thus differ; nevertheless, our results show that both the direct protein substitution and the assisted protein dissociation are possible pathways for turning off transcription.
Past studies have shown that for the prototype Hg2+-responsive metalloregulator MerR, a protein called MerD might mediate the unbinding of holo-MerR from DNA for turning off transcription (28). No evidence has yet been found, however, for a MerD homologue for CueR or other MerR-family regulators. As all known MerR-family metalloregulators share the DNA distortion mechanism for turning on transcription (5, 13, 14), it is likely that many of them share a common mechanism for turning off transcription. The direct substitution pathway and assisted dissociation pathway may thus be common mechanisms for MerR-family metalloregulators to turn off transcription efficiently after transcription activation.
Materials and Methods
Materials and methods are described in SI Appendix, section S1.
Supplementary Material
ACKNOWLEDGMENTS.
We thank C. Kinsland for access to the protein production facility and C. He and P.R. Chen for providing the plasmid and purification protocol of CueR. This research is supported by National Institutes of Health (NIH) Grant GM082939 and partly by National Science Foundation Grant CHE0645392, for developing the vesicle trapping combined with single-molecule FRET measurements. D.J.M. is an NIH Chemistry and Biology Interfaces trainee (5T32GM008500). A.G. and J.D.H acknowledge funding from NIH Grant GM059323.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1208508109/-/DCSupplemental.
References
- 1.Finney LA, O’Halloran TV. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 2.Andrews SC, Robinson AK, Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 3.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and metal sensing. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 4.Moore CM, Helmann JD. Metal ion homeostasis in Bacillus subtillis. Curr Opin Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR family of transcripitional regulators. FEMS Microbiol Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 6.Rensing C, Grass G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 7.Ma Z, Jacobsen FE, Giedroc DP. Coordination chemistry of bacterial metal transport and sensing. Chem Rev. 2009;109:4644–4681. doi: 10.1021/cr900077w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solioz M, Stoyanov JV. Copper homeostasis in Enterococcus hirae. FEMS Microbiol Rev. 2003;27:183–195. doi: 10.1016/S0168-6445(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 9.Nies DH. Efflux-mediated heavy meal resistance in prokaryotes. FEMS Microbiol Rev. 2003;27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 10.Dosanjh NS, Michel SL. Microbial nickel metalloregulation: NikRs for nickel ions. Curr Opin Chem Biol. 2006;10:123–130. doi: 10.1016/j.cbpa.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Ralston DM, O’Halloran TV. Ultrasensitivity and heavy-metal selectivity of the allosterically modulated merr transcription complex. Proc Natl Acad Sci USA. 1990;87:3846–3850. doi: 10.1073/pnas.87.10.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barkey T, Miler SM, Summers AO. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol Rev. 2003;27:355–384. doi: 10.1016/S0168-6445(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 13.O’Halloran TV, Frantz B, Shin MK, Ralston DM, Wright JG. The MerR heavy metal receptor mediates positive activation in a topologically novel transcription complex. Cell. 1989;56:119–129. doi: 10.1016/0092-8674(89)90990-2. [DOI] [PubMed] [Google Scholar]
- 14.Newberry KJ, Brennan RG. The structural mechanism for transcription activation by MerR family member multidrug transporter activation, N-terminus. J Biol Chem. 2004;279:20356–20362. doi: 10.1074/jbc.M400960200. [DOI] [PubMed] [Google Scholar]
- 15.Outten CE, Outten FW, O’Halloran TV. DNA distortion mechanism for transcriptional activation by ZntR, a Zn(II)-responsive MerR homologue in Escherichia coli. J Biol Chem. 1999;274:37517–37524. doi: 10.1074/jbc.274.53.37517. [DOI] [PubMed] [Google Scholar]
- 16.Frantz B, O’Halloran TV. DNA distortion accompanies transcriptional activation by the metal-responsive gene-regulatory protein MerR. Biochemistry. 1990;29:4747–4751. doi: 10.1021/bi00472a001. [DOI] [PubMed] [Google Scholar]
- 17.Holm RH, Kennepohl P, Solomon EI. Structural and functional aspects of metal sites in biology. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 18.Changela A, et al. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 19.Rosenzweig AC, O’Halloran TV. Structure and chemistry of the copper chaperone proteins. Curr Opin Chem Biol. 2000;4:140–147. doi: 10.1016/s1367-5931(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 20.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tinoco I, Jr, Gonzalez RL., Jr Biological mechanisms, one molecule at a time. Genes Dev. 2011;25:1205–1231. doi: 10.1101/gad.2050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoyanov JV, Hobman JL, Brown NL. CueR (YbbI) of Escherichia coli is a MerR family regulator controlling expression of the copper exporter CopA. Mol Microbiol. 2001;39:502–511. doi: 10.1046/j.1365-2958.2001.02264.x. [DOI] [PubMed] [Google Scholar]
- 23.Outten FW, Outten CE, Hale J, O’Halloran TV. Transcriptional activation of an Escherichia coli copper efflux regulation by the chromosomal MerR homologue, CueR. J Biol Chem. 2000;275:31024–31029. doi: 10.1074/jbc.M006508200. [DOI] [PubMed] [Google Scholar]
- 24.Abbondanzieri EA, et al. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.von Hippel PH, Berg OG. Facilitated target location in biological systems. J Biol Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 27.Yamamoto K, Ishihama A. Transcriptional response of Escherichia coli to external copper. Mol Microbiol. 2004;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 28.Hobman JL, Wilkie J, Brown NL. A design for life: Prokaryotic metal-bindng MerR family regulators. BioMetals. 2005;18:429–436. doi: 10.1007/s10534-005-3717-7. [DOI] [PubMed] [Google Scholar]
- 29.Andoy NM, et al. Single-molecule study of metalloregulator CueR-DNA interactions using engineered Holliday junctions. Biophys J. 2009;97:844–852. doi: 10.1016/j.bpj.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haran G. Noise reduction in single-molecule fluorescence trajectories of folding proteins. Chem Phys. 2004;307:137–145. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.