

Abstract
A grand challenge in synthetic biology is to use our current knowledge of RNA science to perform the automatic engineering of completely synthetic sequences encoding functional RNAs in living cells. We report here a fully automated design methodology and experimental validation of synthetic RNA interaction circuits working in a cellular environment. The computational algorithm, based on a physicochemical model, produces novel RNA sequences by exploring the space of possible sequences compatible with predefined structures. We tested our methodology in Escherichia coli by designing several positive riboregulators with diverse structures and interaction models, suggesting that only the energy of formation and the activation energy (free energy barrier to overcome for initiating the hybridization reaction) are sufficient criteria to engineer RNA interaction and regulation in bacteria. The designed sequences exhibit nonsignificant similarity to any known noncoding RNA sequence. Our riboregulatory devices work independently and in combination with transcription regulation to create complex logic circuits. Our results demonstrate that a computational methodology based on first-principles can be used to engineer interacting RNAs with allosteric behavior in living cells.
Keywords: post-transcriptional regulation, evolutionary computation, computational RNA design, RNA synthetic biology
The understanding of RNA interactions in living cells and their subsequent exploitation as regulators is providing new synthetic biology applications (1). RNA regulation is being studied from natural systems by the analysis of the interactions of small RNAs (sRNAs) with messenger RNAs (mRNAs) (2), proteins (3) or molecules (4). However, it is also possible to follow a forward engineering approach and attempt the de novo design of RNA regulators. Rational design techniques have been applied, in both prokaryotes and eukaryotes, for repression or activation of translation (5–10), mRNA degradation (11), riboswitches and ribozymes (12–14), transcription attenuation (15–18), and scaffolding (19). On the other hand, computational methods allowed designing nucleic-acid-based logic circuits in vitro (20–23), including the redesign of allosteric ribozymes (23), hence, challenging the current knowledge of nucleic acid structure and function. Previous RNA design approaches, however, have been mostly developed to work with in vitro systems or required incorporating fragments of natural sequences.
We now propose a fully automated sequence selection methodology to design general circuits based on RNA interactions to operate in living cells. Previous computational methodologies relied on the dominance of the Watson–Crick interactions (24), but they were not adapted to in vivo operations where RNA could be very unstable as it occurs in bacteria. Our approach consists of stabilizing RNA molecules by enforcing a given structure, as done in the inverse folding problem (25–27), together with targeted interactions and conformational changes. The analysis of natural systems unveils another challenge: The kinetics of RNA interactions is rate-limited by the initial interaction of solvent-accessible nucleotides from each binding partner, as illustrated in the kissing-loop mechanism (2). To make manageable the computational problem, here we also rely on Watson–Crick interactions (i.e., canonical purine-pyrimidine pairs plus the G–U pair), although it is possible to extend our approach to other types of interactions (28). Therefore, we are faced with the problem of designing a set of RNA species with predefined structures and with unspecified intermolecular interactions able to produce the intended allosteric regulation. Here, we show it is possible to solve such a combinatorial problem by developing a fully automated procedure that exploits physicochemical principles and structural constraints and that outputs the RNA sequences implementing the predefined interactions.
Results and Discussion
Computational Design of sRNA Circuits.
Our methodology (see details in SI Appendix) starts by choosing well-defined structures for all single species of the circuit. Because we focus on Watson–Crick interactions, we implicitly assume that the secondary structure already determines a stable three-dimensional architecture (29). We also hypothesize that the interaction between two species is nucleated by their unpaired nucleotides (Fig. 1A). In this interaction model, an initial intermolecular pairing driven by a small sequence (toehold or seed site) nucleates a downhill reaction where the size of the intermolecular pairs (represented as a reaction coordinate in Fig. 1A) rapidly increases until the hybridization ends. Initially, the algorithm assigns random nucleotides to the sequences of each RNA species, while respecting their designated secondary structure, which ensures a low 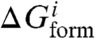 by solving an inverse folding problem (Fig. 1A). It then explores the space of allowed nucleotide sequences by using an objective function and a Monte Carlo simulated annealing (MCSA) search algorithm (30) (Fig. 1B). The convergence of our algorithm is coupled to the existence of large networks of neutral paths (of common structure) able to connect highly different sequences (31), and the algorithm scatters several initial random sequences along these networks to perform an efficient exploration of the sequence space. In addition, to enlarge such neutral paths and then improve the optimization, we allow non-neutral mutations perturbing, up to three base pairs, the structures specified for the single species.
by solving an inverse folding problem (Fig. 1A). It then explores the space of allowed nucleotide sequences by using an objective function and a Monte Carlo simulated annealing (MCSA) search algorithm (30) (Fig. 1B). The convergence of our algorithm is coupled to the existence of large networks of neutral paths (of common structure) able to connect highly different sequences (31), and the algorithm scatters several initial random sequences along these networks to perform an efficient exploration of the sequence space. In addition, to enlarge such neutral paths and then improve the optimization, we allow non-neutral mutations perturbing, up to three base pairs, the structures specified for the single species.
Fig. 1.

Schemes of methodology and designs. (A) Thermodynamic scheme of RNA interaction, showing the different free energies at play and the progression of the reaction. We define the reaction coordinate as the size of intermolecular pairs (d). (B) Optimization scheme followed to design the RNA devices. (C) Secondary structures specified for the single species to obtain different RNA devices. Nucleotides shown were maintained fixed; RBS sequence yellow colored. Different devices were designed by imposing different structures for the riboregulator.
The objective function is defined to minimize by MCSA two competing design goals: (i) free energy of complex formation and (ii) activation energy of complex formation. The first term accounts for the free energy difference between the interacting and free species for all possible interactions in the circuit (ΔGform). For the second term we consider a magnitude related to the kon of the RNA interaction, which we assume is determined by the length of the toehold sequence (α), where ΔGact (activation free energy) is proportional to C–α, C being a constant (21). To facilitate convergence, we incorporate into the objective function an additional term accounting for a given structural constraint (ΔGconstr). These three terms are then weighted resulting in a scalar optimization problem (Fig. 1B). Recent experimental screening of about 500 mutants (between cis and trans) of the IS10 antisense RNA system shows that ΔGform and ΔGact are the two principal predictors of RNA interaction (18), which provides significant support to our objective function. Here, we approximate the folding free energy of a given species (single or complex) from the minimum energy conformation (MEC), discarding the subdominant conformations of the corresponding thermodynamic ensemble. This is supported by the agreement between the MECs and the in vivo results recently reported for 3,000 yeast transcripts (32). Our sequence selection procedure optimizes simultaneously all RNA sequences of the circuit, and during the optimization we do not need to impose natural nucleotide compositions or specific loop sequences (1–3), thus providing unbiased synthetic sequences. In addition, because the evolutionary procedure is independent of the RNA model, new energetically modified models harnessing experimental data (33) can be employed to improve the prediction of the functionality of the riboregulatory devices.
Engineering Synthetic Riboregulation.
As a case study for our methodology we chose the challenging problem of engineering a synthetic riboregulator able to trans-activate the translation of a cis-repressed gene (SI Appendix, Fig. S4). This problem provides an assessment of the generality of our methodology because it requires the design of two RNA species that experience a conformational change after their mutual interaction. In this case, the quantification of the regulatory activity by differential protein expression already results in a characterization of the RNA interaction. Furthermore, our riboregulators enlarge the repertoire of exogenous activators of bacterial gene expression for creating gene circuits with complex dynamics (34). To design such riboregulatory devices, we implemented a natural mechanism (35, 36), where the sRNA activates translation by binding to the 5’-untranslated region (UTR) of a given mRNA. This binding is devised to produce a conformational change in the cis-repressed ribosome binding site (RBS) to become exposed to the solvent and, therefore, enable the docking of the 16S ribosomal unit during translation initiation (SI Appendix, Fig. S4). In our designs, we maintained as fixed the RBS sequence in the 5’-UTR. We selected an mRNA coding for a fluorescent protein (GFP) to facilitate the experimental characterization. The secondary structures for both sRNA and 5’-UTR of the mRNAGFP were specified as structural constraints (Fig. 1C), although these specifications are not strict and some perturbations in the structures are tolerated. For the sRNA, we picked several known structures to assay the versatility of our methodology: the predicted structures of the bacterial sRNAs SokC, FinP, and DsrA (T1, T2, and T3, respectively) and two artificial structures (T4 and T5) (6, 8). For the 5’-UTR of the mRNAGFP, we chose a conformation (C1) shown to produce the intended repression (6). Then, our algorithm explored all possible sequences compatible with such specifications (31, 37), searching a stable hybridization of the two RNAs where the RBS remained unpaired.
We experimentally validated in E. coli six solutions corresponding to different interaction models (Fig. 2) by exploring a space of 1040 sequences (SI Appendix, Table S7). The cis-repressing elements tightly reduced the protein expression to about 1–4% of the maximal expression (SI Appendix, Fig. S13). These strong repressions certainly support the formation of the predicted structures, which prevent the exposure of the RBS to the solvent. The designed riboregulators activated translation (monitored by fluorometry) in a range going from 2.8 (RAJ21) to 11.2 (RAJ11) of apparent activation fold (Fig. 3A). These values are similar to those reported for natural systems (35); therefore, our devices could be readily used to reprogram bacterial behavior. A flow cytometry analysis also revealed a statistically significant sRNA activation within the cell population in all designs (SI Appendix, Fig. S15), with average values presenting quantitative agreement with fluorometry results (SI Appendix, Fig. S12). The significant activation of protein expression in all cases supports the sRNA-mRNA interaction and the intended conformational change in the 5’-UTR of the mRNAGFP. Furthermore, manual design can be applied on top of our computational designs to enlarge the number of different devices (16). For instance, in system RAJ11 (SI Appendix, Fig. S6) a single point mutation would reduce the activity in 52% of the cases following our objective function (SI Appendix, Figs. S8 and S9). In particular, we identified that two compensatory mutations in the toehold would be sufficient to create an orthogonal device with similar specificity (SI Appendix, Table S8).
Fig. 2.

Schematic representation of the six different RNA devices we designed and engineered for riboregulation. Devices RAJ11 and RAJ12 were obtained by imposing the structure T4, device RAJ21 with T1, device RAJ22 with T2, device RAJ23 with T3, and device RAJ31 with T5. SI Appendix (Fig. S7) shows the helical structure of the different complexes together with the corresponding base-pairing probability matrixes. SI Appendix (Table S1) shows the sequences of species. SI Appendix (Table S4) shows the thermodynamic properties of the systems.
Fig. 3.

Experimental characterization of the RNA devices. Normalized fluorescence of the devices together with the apparent activation fold (Top), measured as the ratio of fluorescence in presence and absence of the riboregulator (see also SI Appendix, Fig. S15, for flow cytometry characterizations).
Context Analysis of Engineered Riboregulation.
Our design methodology does not consider cellular factors that could drive RNA interactions, such as the specific and nonspecific binding to endogenous RNAs or proteins. On the one hand, we could incorporate into the designed sequences specific recognition sites to known proteins, such as Hfq, provided its role in sRNA stabilization and catalysis of sRNA–mRNA pairing (38, 39). Instead, by expressing both RNAs within the same plasmid we may already promote local coexpression, avoiding intracellular RNA diffusion. Other cellular factors involve ribonucleases (RNases), which could be incorporated if their cleavage sequences are known. On the other hand, nonspecific interactions could jeopardize our predictions. For instance, bacterial RNase III is a potent and fast RNase that targets double stranded regions and we evaluated the effect of such an RNase on this system by replicating our characterizations in the corresponding knockout strain. Although we found an increase about the double in the apparent activation fold (from 11.2 to 26.7) when using a ΔRNase III strain (Fig. 4A), the real-time quantitative PCR (RT-qPCR) quantification of the ratio sRNA/mRNAGFP did not change in such a strain and it warrants further exploration (see SI Appendix).
Fig. 4.

Context analysis of the RNA devices. (A) Comparison of the activity of the device RAJ11 in the regular strain (JS006) and in a strain ΔRNase III (HT115). (B) Orthogonality analysis of the devices, showing the fraction of complex formation at the equilibrium (values provided in SI Appendix, Table S5; see also SI Appendix, Fig. S11). (C) Experimental validation of the orthogonality between the devices RAJ11 and RAJ12.
Because our methodology finds novel nucleotide sequences stabilizing a set of RNA structures and interactions, we would expect each sequence was sufficiently dissimilar to the others making cross-talk unlikely. Moreover, a Basic Local Alignment Search Tool showed that all sequences of our devices display no significant similarity to any known noncoding RNA sequence (SI Appendix, Table S6). To investigate the orthogonality of our riboregulatory devices, we checked the hybridization ability between possible combinations of cis-repressing and trans-activating RNAs. We estimated computationally the relative levels of the interaction complex at the equilibrium, showing that our devices, despite the homologies already present in the sequences and structures due to imposing a common RBS sequence, displayed low interactions between noncognate pairs (Fig. 4B; SI Appendix, Table S5; degree of orthogonality of the 98%). We also investigated the effect of higher RNA concentrations on the orthogonality, showing a notable dose-dependent cross-talk between devices RAJ12 and RAJ21 (SI Appendix, Fig. S11). We then performed an experimental validation of these orthogonality inferences with the devices RAJ11 and RAJ12. There we found that, despite having similar specificity, performance, and structure, the noncognate interaction (transRAJ11 and cisRAJ12) displayed no significant activity (Fig. 4C). We concluded that it was the large size of the sequence space (SI Appendix, Table S7) that permitted highly specific sRNAs to be obtained, which would allow them to operate within a same cellular compartment.
Engineering Modular AND Logic Gate.
Our devices can be then combined with transcription regulatory elements to engineer combinatorial logic gates (6, 7). Because transcription regulation is also very specific, we could create a large library of orthogonal logic gates. To exemplify this we designed an AND logic gate by placing the sRNA and the 5′-UTR-mRNAGFP under the control of tunable promoters (Fig. 5A). We used the PLtetO-1 and PLlacO-1 promoters together with a strain that constitutively expressed the transcription repressors TetR and LacI (40). Therefore, we could regulate the transcription of these promoters using the inducers anhydrotetracycline (aTc) and isopropyl β-D-1-thiogalactopyranoside (IPTG), respectively. To implement such a system, we considered the device RAJ11. The resulting AND logic gate had a very low leakage and displayed a sigmoidal response to both aTc and IPTG (Fig. 5 B and C and SI Appendix, Fig. S20). We obtained the transfer function of the device by using the apparent activation fold calculated from Fig. 3A. By setting high levels of IPTG, the concentration of aTc allows tuning the activity of the RNA device (SI Appendix, Fig. S19). Because riboregulators could be used with arbitrary promoters (6), we could create AND logic gates adapted to different applications.
Fig. 5.

The RNA device can work in combination with transcription regulation. (A) Scheme of a circuit coupling these two types of control (resulting in an AND logic gate), where IPTG and aTc are the two inputs and GFP the output. To implement this circuit we used the device RAJ11. (B) Experimental characterization of the circuit showing the dynamics of the normalized fluorescence after introduction of inducers at time 0 (see also SI Appendix, Fig. S19, for the full set of dynamics at different levels of IPTG and aTc). (C) Arbitrary units of GFP (colored circles) are estimated by taking the maximum value of the dynamics shown in SI Appendix (Fig. S19), ensuring OD600 < 0.4, and dividing this value by the one obtained in case of no inducers in the medium. Solid lines are obtained with the mathematical model presented in SI Appendix (Eq. S7), with parameters fitted against these experimental data.
Conclusions
In summary, we have presented a general methodology based on theoretical principles and combinatorial optimization to design interacting RNAs. The ability of our simple model to guide the design of fully synthetic riboregulation suggests that intracellular RNA interactions are predominantly governed by the energy of formation and the activation energy. Our de novo design approach relies on the enforcement of given structures to each RNA species, which provides the required stability in the cytoplasm and guides allosteric regulation. However, our objective function promotes the desired interactions, and the allosteric behavior is enforced through a constraints term. We have validated the methodology by engineering in bacteria different interaction models implementing translation activators. These tests already have addressed important challenges in the design of complex RNA interaction circuits, such as the design of precise conformational changes and the interface with the cellular machinery. Although further tests of the methodology would strengthen the performance of the algorithm, its basis on physicochemical principles would allow us to apply it in many different frameworks and obtain more sophisticated systems. For instance, we designed a pair of riboregulators able to synergistically activate protein expression (SI Appendix, Fig. S21), illustrating the versatility of our approach and the ability to explore even larger spaces.
As our automated methodology uses few specifications as inputs, it could also be used to test new mechanisms and hypotheses despite the lack of a complete molecular understanding of the living cell. It would then be possible to average out the effects of unknown natural systems by designing a large number of systems implementing a given set of specifications. On the other hand, the engineered RNA devices can be exploited in biotechnology, in particular for metabolic control purposes, where the enzymatic expression is cis-repressed (10) and the riboregulator is even combined with aptamers that sense a given metabolite (17, 41). As a result, the synthetic circuit can trigger the expression of enzymes for regulating the activity of the pathway. Also in bacteria, we could exploit other mechanisms such as ribozymes (14) or antiterminators (15), and high-throughput analyses could be addressed using a library of automatically designed sRNAs. Our methodology does not require any ribosome involvement, where the RBS sequence could be replaced by the corresponding binding sequence motif of an RNA-binding protein (or molecule) if known (19, 42). In addition, we could improve our methodology (see SI Appendix) by incorporating cellular factors (e.g., Hfq, RNase III, RNase E, or 16S rRNA) into the physicochemical model, by considering non-Watson–Crick interactions (28), or by adding predictors of translation initiation by 5’-UTR sequences (43). Furthermore, we could design riboregulators for eukaryotic hosts by replacing the RBS by the Kozak sequence, or alternatively, if known, by the internal ribosome entry site (44). In yeast, the sRNA would interact with the 5’-UTR of certain mRNA to alter the cap-dependent scanning of the 40S subunit (45). In higher eukaryotes, the sRNA could target a given RNA, leading to a conformational change that triggers the intended function (9). Yet, the proposed automatic algorithm to design sRNA circuits in living cells will open new venues for RNA synthetic biology (1) and for quantitative testing of our assumptions about RNA function.
Materials and Methods
Plasmid Construction.
The SI Appendix (Figs. S1 and S2) outlines the two plasmid templates, pSTC0 and pSTC1, used in this work. The SI Appendix (Table S11) shows all plasmids constructed. The target mRNA was placed in 5’ sense under the control of the PLlacO-1 promoter (regulated by LacI and IPTG) and the sRNA in 3’ sense under the PLtetO-1 promoter (regulated by TetR and aTc) (40). The RNA devices (from the terminator of the sRNA to the 5’-UTR of the mRNA) were made by DNA synthesis (DNA 2.0) and then inserted by ligation into the plasmid templates. From the resulting plasmids, the sRNA was removed to generate systems bearing only the mRNA operon. pSTC0 had the pMB1 origin, kanamycin resistance, and GFPmut3b as a reporter gene. pSTC1 had the pSC101 origin, kanamycin resistance, and superfolder GFP as a reporter gene (see details in SI Appendix).
Strains, Reagents, and Cell Culture.
E. coli TOP10 (Invitrogen) was used for routine transformation, as described in the protocol (46). Characterization experiments were performed in E. coli K-12 JS006 cells (MG1655 ΔaraC ΔlacI KanS) (47), in E. coli K-12 HT115 cells (W3110 rnc-14::Tn10) as RNase III depleted environment (48), and in E. coli K-12 MG1655-Z1 cells (MG1655 lacI+ tetR+ araC+ SpR) for constitutive control over the PLlacO-1 and PLtetO-1 promoters (49). Cells were grown aerobically in Luria–Bertani broth or in a modified M9 minimum media comprising M9 minimum salts (Sigma M6030) supplemented with glycerol at 0.8% (vol/vol) as the only carbon source, CaCl2 at 100 μM, MgSO4 at 2 mM, and FeSO4 at 100 μM for higher growth yield (50). Casamino acids were avoided because of their natural green fluorescence that produces too high a fluorescence background for fluorometer measurements. Cultures were grown over-night at 37 °C and at 225 rpm from single-colony isolates before being diluted. The following concentrations of antibiotics were used when appropriate: kanamycin (50 μg/mL), tetracycline (15 μg/mL), spectinomycin (100 μg/mL). When using E. coli K-12 MG1655-Z1 cells, 1 mM of IPTG was used for full activation of the PLlacO-1 promoter when needed, and 100 ng/mL of aTc was used for full activation of the PLtetO-1 promoter when needed.
Fluorescence Quantification Using Fluorometry.
Before characterization experiments, cells were grown in M9 over two nights in order to reach stationary phase. Cultures were then diluted 200 times in 200 μL of M9 within each well of the plate (Custom Corning Costar 96-well microplate, black transparent bottom with lid). The plate was incubated in an Infinite F500 multiwell fluorometer (TECAN) at 37 °C with shaking (orbital mode, frequency of 33 rpm, 2 mm of amplitude) and assayed with an automatically repeating protocol of absorbance measurements (600 nm absorbance filter) and fluorescence measurements (480/20 nm excitation filter–530/25 nm emission filter for GFP and 580/20 nm excitation filter–610/10 nm emission filter for RFP). Time between repeated measurements was 15 min. All samples were present in 3–6 replicated on the plate. Each measurement was repeated two to three times on independent days to verify reproducibility in the results. All data analyses were done using values harvested when cells were in exponential growth phase (OD600 between 0.1 and 0.4). Growth rates were obtained as the slope of a linear regression between the values of log(OD600) and time. Similar growth rates were observed in all experiments, except for the HT115 strain growing naturally slower. The steady-state protein expression value was obtained as the slope of a linear regression between the values of fluorescence and OD600. For the dynamical analysis of protein expression, the absolute fluorescence was divided by OD600 to have a magnitude per cell.
Fluorescence Quantification Using Flow Cytometry.
All expression data were collected using a Becton Dickinson FACSCanto II flow cytometer with a 488 nm argon laser and a 530/30 nm emission filter (GFP) and a 695/40 nm emission filter (RFP). Overnight cultures in M9 were diluted 200 times in 200 μL of fresh medium and incubated to reach an OD600 of about 0.1. Cells from M9 cultures were fixed using 4% paraformaldehyde (PFA)—culture cells were pelleted and washed in filtered PBS (Biorad), washed 15 min with PFA, washed in filtered PBS, and finally kept at 4 °C until measurement. Fluorescence measurement of gene expression from each sample was obtained from > 20,000 cells. We analyzed the data using FCS express 4 (Denovo software), and we gated the events using narrow forward and side scatter range. We then represented the fluorescence distributions in log scale.
Supplementary Material
ACKNOWLEDGMENTS.
We thank J. Hasty for providing the E. coli strain JS006, N. Guillén for the strain HT115, and M.B. Elowitz for the strain MG1655-Z1. We thank F. de la Cruz, R. Ruiz, and I. del Campo for practical support with flow cytometry measurements and S.F. Elena and M.P. Zwart for assistance in RT-qPCR assays. We also thank H. Isambert, J.J. Collins, F.J. Isaacs, B.F. Luisi, and E. Westhof for useful comments. Work was supported by the FP7-ICT-043338 (Bacterial Computing with Engineered Populations), the Actions Thématiques Incitatives de Genopole, and the Fondation pour la Recherche Medicale grants (to A.J.). G.R. is supported by an European Molecular Biology Organization long-term fellowship cofunded by Marie Curie actions (ALTF-1177-2011), and T.E.L. by a PhD fellowship from the AXA Research Fund.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203831109/-/DCSupplemental.
References
- 1.Isaacs FJ, Dwyer DJ, Collins JJ. RNA synthetic biology. Nat Biotechnol. 2006;24:545–554. doi: 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- 2.Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr Opin Microbiol. 2007;10:102–109. doi: 10.1016/j.mib.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Rajkowitsch L, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4:118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 4.Gallivan JP. Toward reprogramming bacteria with small molecules and RNA. Curr Opin Chem Biol. 2007;11:612–619. doi: 10.1016/j.cbpa.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfleger BF, et al. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24:1027–1032. doi: 10.1038/nbt1226. [DOI] [PubMed] [Google Scholar]
- 6.Isaacs FJ, et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol. 2004;22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 7.Callura JM, Dwyer DJ, Isaacs FJ, Cantor CR, Collins JJ. Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. Proc Natl Acad Sci USA. 2010;107:15898–15903. doi: 10.1073/pnas.1009747107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima N, Tamura T. Conditional gene silencing of multiple genes with antisense RNAs and generation of a mutator strain of Escherichia coli. Nucleic Acids Res. 2009;37:e103. doi: 10.1093/nar/gkp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkataraman S, Dirks RM, Ueda CT, Pierce NA. Selective cell death mediated by small conditional RNAs. Proc Natl Acad Sci USA. 2010;107:16777–16782. doi: 10.1073/pnas.1006377107. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Callura JM, Cantor CR, Collins JJ. Genetic switchboard for synthetic biology applications. Proc Natl Acad Sci USA. 2012;109:5850–5855. doi: 10.1073/pnas.1203808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rinaudo K, et al. A universal RNAi-based logic evaluator that operates in mammalian cells. Nat Biotechnol. 2007;25:795–801. doi: 10.1038/nbt1307. [DOI] [PubMed] [Google Scholar]
- 12.Beisel CL, Bayer TS, Hoff KG, Smolke CD. Model-guided design of ligand-regulated RNAi for programmable control of gene expression. Mol Syst Biol. 2008;4:224. doi: 10.1038/msb.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carothers JM, Goler JA, Juminaga D, Keasling JD. Model-driven engineering of RNA devices to quantitatively program gene expression. Science. 2011;334:1716–1719. doi: 10.1126/science.1212209. [DOI] [PubMed] [Google Scholar]
- 15.Dawid A, Cayrol B, Isambert H. RNA synthetic biology inspired from bacteria: Construction of transcription attenuators under antisense regulation. Phys Biol. 2009;6:025007. doi: 10.1088/1478-3975/6/2/025007. [DOI] [PubMed] [Google Scholar]
- 16.Lucks JB, Qi L, Mutalik VK, Wang D, Arkin AP. Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc Natl Acad Sci USA. 2011;108:8617–8622. doi: 10.1073/pnas.1015741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi L, Lucks JB, Liu CC, Mutalik VK, Arkin AP. Engineering naturally occurring trans-acting non-coding RNAs to sense molecular signals. Nucleic Acids Res. 2012;40:5775–5786. doi: 10.1093/nar/gks168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mutalik VK, Qi L, Guimaraes JC, Lucks JB, Arkin AP. Rationally designed families of orthogonal RNA regulators of translation. Nat Chem Biol. 2012;8:447–454. doi: 10.1038/nchembio.919. [DOI] [PubMed] [Google Scholar]
- 19.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Organization of intracellular reactions with rationally designed RNA assemblies. Science. 2011;333:470–474. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 20.Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-free nucleic acid logic circuits. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- 21.Yin P, Choi HM, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- 22.Ran T, Kaplan S, Shapiro E. Molecular implementation of simple logic programs. Nat Nanotechnol. 2009;4:642–648. doi: 10.1038/nnano.2009.203. [DOI] [PubMed] [Google Scholar]
- 23.Penchovsky R, Breaker RR. Computational design and experimental validation of oligonucleotide-sensing allosteric ribozymes. Nat Biotechnol. 2005;23:1424–1433. doi: 10.1038/nbt1155. [DOI] [PubMed] [Google Scholar]
- 24.Ellington AD. What’s so great about RNA? ACS Chem Biol. 2007;2:445–448. doi: 10.1021/cb700139t. [DOI] [PubMed] [Google Scholar]
- 25.Dahiyat BI, Mayo SL. De novo protein design: Fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 26.Jaramillo A, Wernisch L, Héry S, Wodak SJ. Folding free energy function selects native-like protein sequences in the core but not on the surface. Proc Natl Acad Sci USA. 2002;99:13554–13559. doi: 10.1073/pnas.212068599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koehl P, Levitt M. Protein topology and stability define the space of allowed sequences. Proc Natl Acad Sci USA. 2002;99:1280–1285. doi: 10.1073/pnas.032405199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leontis NB, Stombaugh J, Westhof E. The non-Watson–Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flamm C, Fontana W, Hofacker IL, Schuster P. RNA folding at elementary step resolution. RNA. 2000;6:325–338. doi: 10.1017/s1355838200992161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirkpatrick S, Gelatt CD, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 31.Schuster P. How to search for RNA structures. Theoretical concepts in evolutionary biotechnology. J Biotechnol. 1995;41:239–257. doi: 10.1016/0168-1656(94)00085-q. [DOI] [PubMed] [Google Scholar]
- 32.Kertesz M, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Washietl S, Hofacker IL, Stadler PF, Kellis M. RNA folding with soft constraints: Reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res. 2012;40:4261–4272. doi: 10.1093/nar/gks009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodrigo G, Carrera J, Jaramillo A. Computational design of synthetic regulatory networks from a genetic library to characterize the designability of dynamical behaviors. Nucleic Acids Res. 2011;39:e138. doi: 10.1093/nar/gkr616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lease RA, Belfort M. A trans-acting RNA as a control switch in Escherichia coli: DsrA modulates function by forming alternative structures. Proc Natl Acad Sci USA. 2000;97:9919–9924. doi: 10.1073/pnas.170281497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci USA. 2010;107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sim AYL, Levitt M. Clustering to identify RNA conformations constrained by secondary structure. Proc Natl Acad Sci USA. 2011;108:3590–3595. doi: 10.1073/pnas.1018653108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papenfort K, Bouvier M, Mika F, Sharma CM, Vogel J. Evidence for an autonomous 5’ target recognition domain in an Hfq-associated small RNA. Proc Natl Acad Sci USA. 2010;107:20435–20440. doi: 10.1073/pnas.1009784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roth A, Breaker RR. The structural and functional diversity of metabolite-binding riboswitches. Annu Rev Biochem. 2009;78:305–334. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 45.Berthelot K, et al. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol Microbiol. 2004;51:987–1001. doi: 10.1046/j.1365-2958.2003.03898.x. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47.Stricker J, et al. A fast, robust and tunable synthetic gene oscillator. Nature. 2008;456:516–519. doi: 10.1038/nature07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takiff HE, Chen SM, Court DL. Genetic analysis of the rnc operon of Escherichia coli. J Bacteriol. 1989;171:2581–2590. doi: 10.1128/jb.171.5.2581-2590.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunlop MJ, Cox RS, 3rd, Levine JH, Murray RM, Elowitz MB. Regulatory activity revealed by dynamic correlations in gene expression noise. Nat Genet. 2008;40:1493–1498. doi: 10.1038/ng.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paliy O, Gunasekera TS. Growth of E. coli BL21 in minimal media with different gluconeogenic carbon sources and salt contents. Appl Microbiol Biotechnol. 2007;73:1169–1172. doi: 10.1007/s00253-006-0554-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.