

Abstract
Leptin is an obesity-associated cytokine-like hormone encoded by the ob gene. Recent studies reveal that leptin promotes proliferation and differentiation of chondrocytes, suggesting a peripheral role of leptin in regulating growth plate function. Peroxisome proliferator-activated receptor-γ (PPARγ) is a transcriptional regulator of adipogenesis. Locally, PPARγ negatively regulates chondrogenic differentiation and terminal differentiation in the growth plate. The aim of this study was to test the hypothesis that leptin may suppress the inhibitory effects of PPARγ on growth plate chondrocytes. Chondrocytes were collected from distal femoral growth plates of newborn rats and were cultured in monolayer or cell pellets in the presence or absence of leptin and the PPARγ agonist ciglitazone. The results show that leptin attenuates the suppressive effects of PPARγ on chondrogenic differentiation and T3-mediated chondrocyte hypertrophy. Leptin treatment also leads to a mild downregulation of PPAR mRNA expression and a significant MAPK/ERK-dependent PPARγ phosphorylation at serine 112/82. Blocking MAPK/ERK function with PD98059 confirmed that leptin antagonizes PPARγ function in growth plate chondrocytes through the MAPK/ERK signaling pathway. Furthermore, leptin signaling in growth plate cells is also negatively modulated by activation of PPARγ, implying that these two signaling pathways are mutually regulated in growth plate chondrocytes.
1. Introduction
The process of longitudinal bone growth is under endocrine regulation. Some of the endocrine signals act locally in regulating growth plate chondrocyte proliferation and differentiation [1]. Leptin is a cytokine-like hormone that is the product of the ob gene and is expressed predominantly in adipocytes [2]. Leptin controls body fat tissue and body weight by reducing food intake and increasing thermogenesis [3] and functions via the leptin receptor (OB-R), the long form of which (OB-Rb) is the most abundantly expressed and only biologically active isoform [4]. Binding of leptin to its receptor triggers activation of janus kinases (JAKs) [5], leading to phosphorylation and activation of signal transducer and activator of transcription 3 (STAT3) [6]. Suppressor of cytokine signaling 3 (SOCS3) protein acts as a feedback inhibitor of the JAK/STAT3 pathway, inhibiting STAT3 phosphorylation [7].
Growth plate chondrocytes synthesize and secrete leptin and express the leptin receptor OB-Rb [8]. Leptin is involved in bone remodeling and has a direct peripheral effect on growth plate chondrocytes [8]. Organ cultures of mouse mandibular condyles reveal that leptin induces the proliferation and maturation of growth plate chondrocytes and stimulates endochondral bone growth directly at the level of the bone growth centers [9]. Studies of leptin-deficient ob/ob mice demonstrate that lack of leptin protein not only causes obesity in mice [2], but also results in disturbed columnar structure, decreased type X collagen expression, increased apoptosis, and premature mineralization in the growth plates [10]. Administration of leptin to ob/ob mice increases bone growth as well as indices of bone formation [2, 10]. Previously, we also reported that leptin synergizes with thyroid hormone in modulating terminal differentiation of growth plate chondrocytes [11], suggesting that peripheral leptin signaling plays an essential role in endochondral ossification at the growth plate.
PPARγ is a key transcriptional regulator of adipocyte differentiation. It regulates metabolism and storage of fat and is thought to be involved in the development of high fat diet-induced obesity [12]. Our previous studies revealed that PPARγ is expressed in growth plate chondrocytes, and activation of PPARγ promotes adipogenic transdifferentiation of growth plate chondrocytes, while attenuating both chondrogenic differentiation and terminal differentiation [13, 14].
Since leptin and PPARγ are both localized in growth plate cartilage and locally modulate chondrocyte function, the object of this study was to investigate the interaction between these two signaling pathways in growth plate chondrocytes. We hypothesized that leptin might prevent the inhibitory effects of PPARγ on chondrogenic differentiation and terminal differentiation of growth plate cells.
2. Materials and Methods
2.1. Cell Culture
Chondrocytes were isolated from the distal femoral growth plates of 3-day old neonatal Sprague-Dawley rats by sequential digestion in trypsin/EDTA (Invitrogen, Carlsbad, CA) for 1 h at 37°C, followed by 0.3% collagenase type I (Worthington, Lakewood, NJ) for 4 h at 37°C [15]. Cells were resuspended in DMEM/F12 medium (Invitrogen) supplemented with a defined media supplement (ITS+1, Sigma, St. Louis, MO) and plated in monolayer at a density of 5 × 105 cells/cm2, or in a pellet culture of 1 × 105 cells/mL. Tri-iodothyronine (T3, Sigma), leptin (Sigma), and ciglitazone (BioMol, Plymouth Meeting, PA) were added to the medium at concentrations of 100 ng/mL, 1 μg/mL, and 10 μM, respectively, except where specifically indicated. The MAPK/ERK inhibitor PD98059 (20 μM, Cell Signaling Technology, Danvers, MA) and the JNK inhibitor SP600125 (10 μM, Cell Signaling) were added to the medium 30 min before the leptin treatment.
Recombinant adenovirus carrying PPARγ1 (Ad-PPARγ) was kindly provided by Dr. Jameson (Northwestern University Medical School, Chicago, IL) [16] and was used at an MOI (multiplicity of infection) of 100. A structurally similar adenovirus containing the CMV promoter was used as a negative control.
2.2. Transient Transfection
PPAR transcriptional activity was evaluated by cotransfecting the cells with the peroxisome proliferator activator response element (PPRE) reporter plasmid (phRG-TK-PPRE2) [14] and PPARγ expression plasmid (pCMX-PPARγ) (provided by R. Evans, Salk Institute, La Jolla, CA) using lipofection (Fugene 6, Roche, Indianapolis, IN) [14]. The transfection mixture was replaced the following day with original medium containing leptin and/or ciglitazone. After 48 hours, cells were harvested and assayed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). The firefly luciferase expression vector pCMV-Luc (Promega) was used as an internal control.
2.3. Quantitative Real-Time RT-PCR
Total RNA was isolated from cultured growth plate chondrocytes using the RNeasy Kit (Qiagen, Valencia, CA). Reverse transcription was performed using random primers and Superscript III (Invitrogen). Real-time PCR reactions were conducted in an ABI Prism 7700 Sequence Detection System using SYBR Green PCR core reagents (Applied Biosystems, Foster City, CA). The forward and reverse primers for the amplifications are listed below:
-
18s: 5′-AGTCCCTGCCCTTTGTACACA-3′ and 5′-GATCCGAGGGCCTCACTAAAC-3′;
-
Col2a1: 5′-GGTGGAGCAGCAAGAGCAA-3′ and 5′-CGTCGCCGTAGCTGAAGTG-3′;
-
Aggrecan: 5′-CTAGCTGCTTAGCAGGGATAACG-3′ and 5′-CCGCAGAGTCACAAAGACCAA-3′;
-
Col10a1: 5′-GATCATGGAGCTCACGGAAAA-3′ and 5′-CCGTTCGATTCCGCATTG-3′;
-
PPARγ: 5′-TGACCAGGGAGTTCCTCAAAA-3′ and 5′-AGCAAACTCAAACTTAGGCTCCAT-3′;
-
Leptin: 5′-CACACACGCAGTCGGTATCC-3′ and 5′-TGAAGCCCGGGAATGAAGT-3′;
-
Leptin receptor (Ob-Rb): 5′-CTTAAGAACCCCTTCAAGAATTATGACT-3′ and 5′-GGGCAGAGGCAAATCATCTATAAC-3′;
-
SOCS3: 5′-CCTCAAGACCTTCAGCTCCAA-3′ and 5′-TCCGCTCTCCTGCAGCTT-3′.
2.4. Alkaline Phosphatase (ALP) Activity Assay
Chondrocyte pellets were homogenized and alkaline phosphatase activity determined as previously described using p-nitrophenyl phosphate (Sigma) as a substrate [15]. One unit of alkaline phosphatase was defined as the enzyme activity that liberated 1 μmol p-nitrophenol per 30 min at 37°C per mg of protein.
2.5. Histochemical Stainings
Histological stainings were performed on the chondrocytes cultured in monolayer. Cells were fixed in 3.7% formaldehyde at room temperature for 10 min and rinsed with PBS. For Alcian blue staining, cells were stained with a 4 : 1 ratio of 0.1 M HCl/0.5% Alcian blue stock [0.5% Alcian blue 8GX (Sigma) in 95% ethanol] overnight at 37°C in a humidified atmosphere. For alkaline phosphatase staining, cells were stained in the dark for 30 min in a 0.1 M Tris-HCl solution (pH 8.5) containing 0.2 mg/mL of Napthol AS-MX phosphate (Sigma) and 0.6 mg/mL of Fast Blue BB salt (Sigma).
2.6. Immunoblotting
Whole cell extracts were prepared from growth plate chondrocytes using RIPA buffer. An equal amount of protein was subjected to SDS-PAGE and transferred onto nitrocellulose membranes. The blots were incubated with anti-phospho-PPARγ (Ser112 of PPARγ2 and Ser82 of PPARγ1) (Assay Biotechnology Inc, Sunnyvale, CA), anti-PPARγ (H100, Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (Cell Signaling), anti-p44/42 MAPK (ERK1/2) (Cell Signaling), anti-phospho-Stat3 (Tyr705) (Cell Signaling), anti-Stat3 (Cell Signaling), and anti-β-actin (Sigma), followed by a HRP-conjugated secondary antibody. Immunoreactive proteins were visualized by Western Blotting Chemiluminescence Luminol Reagent (Santa Cruz).
2.7. Statistical Analysis
The results are represented as mean ± standard deviation. Data were analyzed by one-way ANOVA with post-hoc Tukey's HSD test or paired Student's t-test using JMP 8 software (SAS Institute Inc., Cary, NC). Statistical significance was set at P < 0.05.
3. Results
3.1. Leptin Attenuates PPARγ-Induced Inhibition in Chondrogenic Differentiation and T3-Mediated Hypertrophy
Incubating the growth plate chondrocytes with the PPARγ agonist ciglitazone for 5 days decreased both Col2a1 and aggrecan mRNA expression (Figures 1(a) and 1(b)), and reduced Alcian blue staining, an index for proteoglycan matrix accumulation (Figure 1(c)). Coaddition of leptin reduced the ciglitazone-induced inhibition of these chondrogenic differentiation markers. As previously observed [14], ciglitazone also inhibited T3-mediated chondrocyte hypertrophy, as shown by decreased Col10a1 mRNA expression (Figure 1(d)) and ALP activity (Figure 1(e)), as well as reduced ALP staining (Figure 1(f)). These decreases were also alleviated by coincubation with leptin (Figures 1(d)–1(f)).
Figure 1.
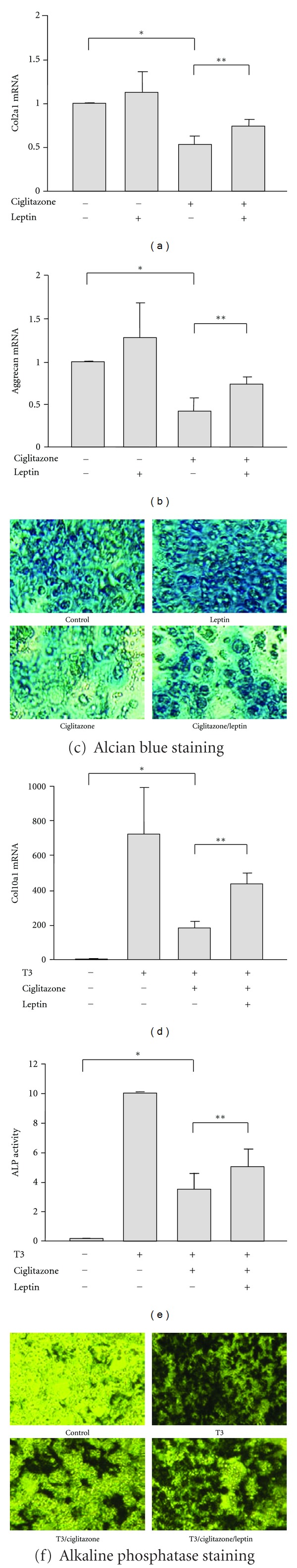
Leptin suppresses the effects of PPARγ on chondrogenic differentiation and chondrocyte hypertrophy in growth plate chondrocytes. ((a), (b)) Quantitative real-time RT-PCR analysis of Col2a1 (a) and aggrecan (b) mRNA expression in chondrocytes treated with ciglitazone and/or leptin for 5 days. *P < 0.05 versus the expression in control cells. **P < 0.05 versus the expression in the cells treated with ciglitazone alone. (c) Alcian blue staining of growth plate chondrocytes in monolayer cultures after 5 days of treatment with ciglitazone and/or leptin. ((d), (e)) Expression of Col10a1 mRNA expression (d) and alkaline phosphatase activity (e) of growth plate chondrocytes treated with ciglitazone and/or leptin for 5 days. *P < 0.05 versus the cells treated with T3 alone. **P < 0.05 versus the chondrocytes treated with both T3 and ciglitazone. (f) Alkaline phosphatase staining of chondrocytes cultured in monolayer and treated with ciglitazone and/or leptin for 5 days.
3.2. Leptin Inhibits PPARγ Signaling by Activating MAPK/ERK Pathway
In the dose dependent experiments, leptin was used at a range from 0.1 μg/mL to 1 μg/mL. Treatment with 1 μg/mL of leptin modestly inhibited PPARγ/ciglitazone-increased PPRE transcriptional activity (Figure 2(a)) and downregulated PPARγ mRNA expression (Figure 2(b)).
Figure 2.
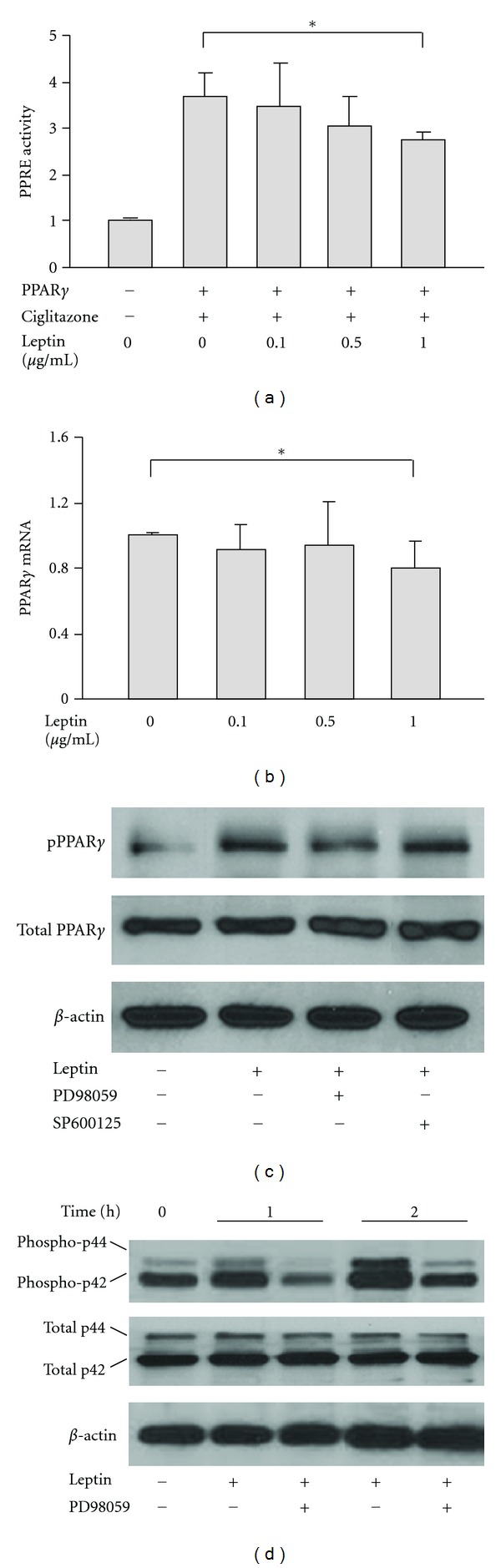
Leptin inhibits PPARγ signaling in growth plate chondrocytes by enhancing PPARγ phosphorylation via MAPK/ERK pathway. (a) PPARγ-mediated PPRE transcriptional activity in chondrocytes treated with leptin at a concentration ranged from 0.1 to 1 μg/mL. *P < 0.05 versus the samples untreated with leptin. (b) PPARγ mRNA expression of growth plate chondrocytes in response to leptin treatment. (c) Immunoblotting analysis of the levels of total and phosphorylated (Ser112/82) PPARγ protein in chondrocytes treated with leptin for 2 h. For ERK or JNK inhibition experiments, the cells were preincubated with PD98059 or SP600125 for 30 min before leptin treatment. (d) Expression of total and phosphorylated ERK proteins (ERK1/2) in growth plate chondrocytes treated with leptin for 1 h and 2 h.
Immunoblotting analysis showed that leptin treatment led to no significant change in total PPARγ protein level, but did lead to an increase in phosphorylated PPARγ, which was detected by an antibody directly against the phosphorylation site at Ser112/82 of PPARγ (Figure 2(c)). These leptin-induced increases in phosphorylated PPARγ were blocked by coincubation with the MAPK/ERK inhibitor PD98059, but not by the JNK inhibitor SP600125 (Figure 2(c)).
Activation of MAPK/ERK signaling by leptin was confirmed by examining the phosphorylation of ERK proteins p42 and p44 (ERK1/2) using immunoblotting. Cell lysates were collected from chondrocytes treated with leptin for 1 h and 2 h. Compared with the leptin untreated controls, leptin increased the levels of both phosphorylated p42 and p44 ERK, and these increases were blocked by PD98059 (Figure 2(d)).
3.3. Leptin Regulates PPARγ Effects on Growth Plate Chondrocytes through the MAPK/ERK Signaling Pathway
To evaluate the role of MAPK/ERK signaling in the leptin-induced suppression of PPARγ inhibitory effects on growth plate cell chondrogenic differentiation and chondrocyte hypertrophy, MAPK/ERK inhibitor PD98059 was presupplemented in the medium 30 min before the treatment with leptin and/or ciglitazone for 5 days. As shown in Figures 3(a) and 3(b), PD98059 abolished the leptin-induced increases of Col2a1 and Col10a1 mRNA expression in ciglitazone-treated growth plate chondrocytes (Figures 3(a) and 3(b)).
Figure 3.
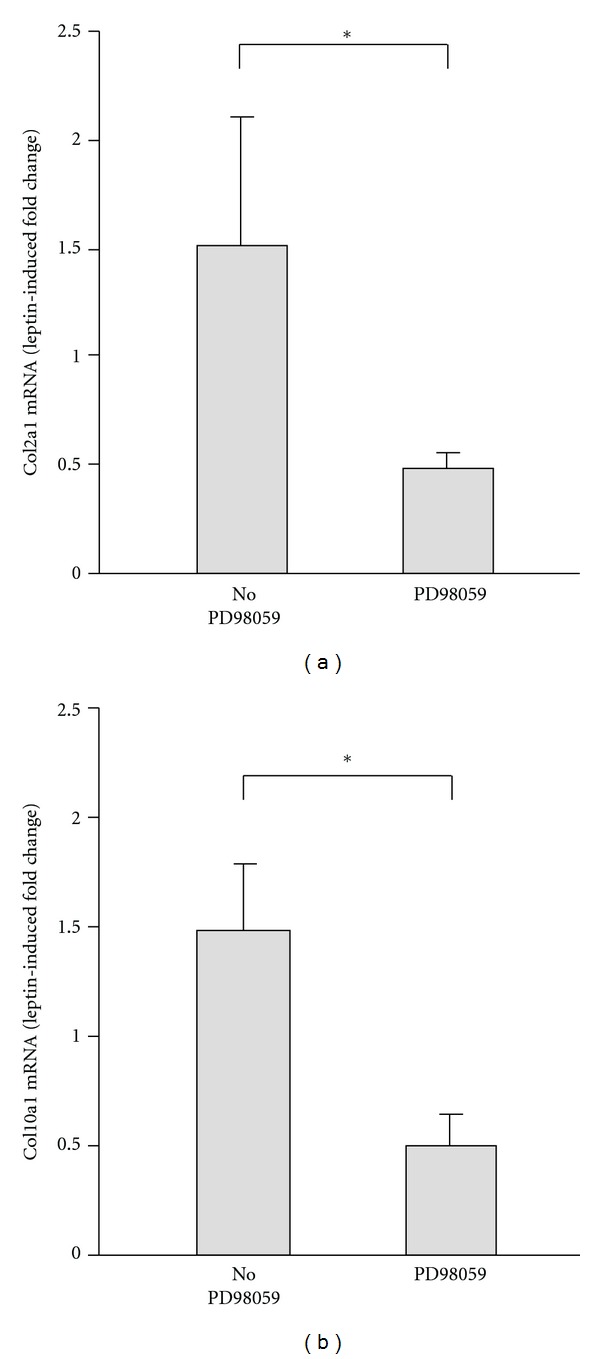
Leptin suppresses the PPARγ inhibitory effects on growth plate chondrocytes by activating MAPK/ERK signaling pathway. ((a), (b)) Gene expression of chondrogenic differentiation marker Col2a1 (a) and terminal differentiation marker Col10a1 (b) in chondrocytes treated with ciglitazone and leptin for 5 days in the presence and absence of PD98059. T3 was added to induce the chondrocyte terminal differentiation. Data are presented as the leptin-induced fold changes in ciglitazone-treated chondrocytes, normalized to the expression in cells treated with ciglitazone alone. *P < 0.05 versus expression changes in the chondrocytes untreated with PD98059.
3.4. Leptin Signaling Is Regulated by PPARγ in Growth Plate Chondrocytes
The influence of PPARγ on leptin signaling was analyzed in growth plate chondrocyte pellet cultures treated with ciglitazone and/or infected with Ad-PPARγ. Treatment of growth plate cells with ciglitazone and/or Ad-PPARγ for 5 days led to decreases in leptin mRNA expression (Figure 4(a)). Leptin receptor (Ob-Rb) expression was also downregulated when treating the cells with both ciglitazone and Ad-PPARγ (Figure 4(b)). Immunoblotting analysis of the chondrocytes treated with leptin and/or ciglitazone for 5 days demonstrated that leptin increased phosphorylation of STAT3 (Figure 4(c)). Incubation of the cells with ciglitazone decreased the phosphorylation of STAT3 and inhibited leptin-induced STAT3 activation (Figure 4(c)). Expression of SOCS3, a leptin signaling inhibitor, was increased after ciglitazone and/or Ad-PPARγ treatment (Figure 4(d)).
Figure 4.
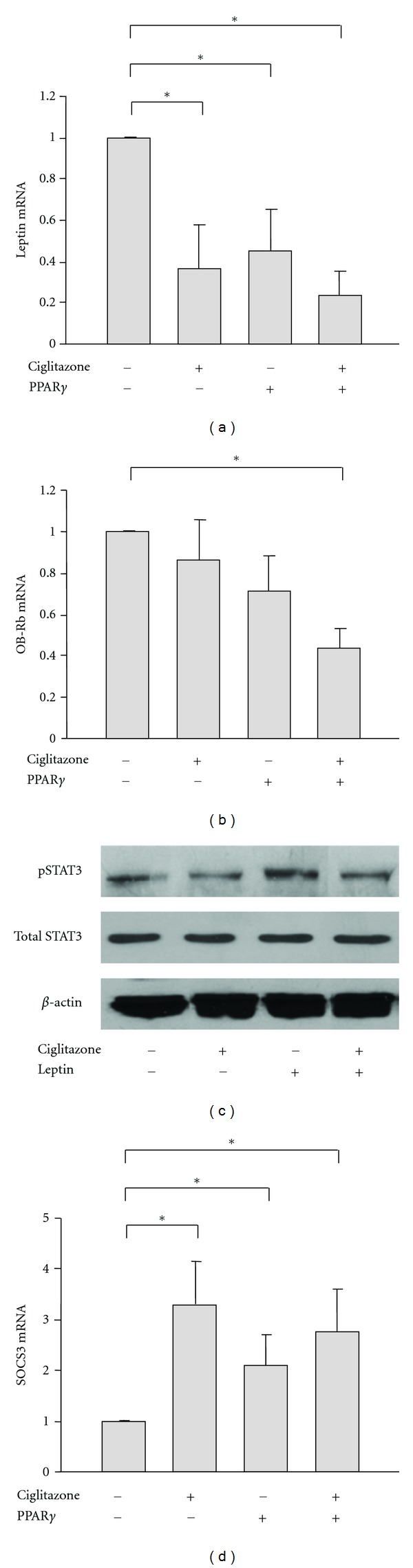
. Leptin signaling is regulated by PPARγ activation in growth plate chondrocytes. (a) and (b) Real-time RT-PCR analysis of gene expression of leptin ligand (a) and leptin receptor Ob-Rb (b) in growth plate chondrocytes treated with ciglitazone and/or Ad-PPARγ for 5 days. *P < 0.05 versus the expression in control cells. (c) Immunoblotting analysis of the levels of total and phosphorylated STAT3 in the lysates of chondrocytes treated with ciglitazone and/or leptin for 5 days. (d) SOCS3 mRNA expression in growth plate chondrocytes treated with ciglitazone and/or Ad-PPARγ for 5 days. *P < 0.05 versus the expression in control cells.
4. Discussion
Childhood obesity has become one of the most serious public health problems in recent decades [17]. Several pediatric orthopedic conditions are known to be related to obesity and involve the growth plates, including slipped capital femoral epiphysis (SCFE), adolescent Blount's disease, and increased risk of growth plate fracture [18, 19]. Disorganization of the normal columnar architecture and impaired differentiation into hypertrophic cells have been observed in the growth plates of both SCFE and Blount's disease patients [20], suggesting that dysfunction of the growth plate in obese children may contribute to the skeletal developmental abnormalities in addition to the mechanical stress resulting from increased body weight.
The relationship between leptin and pediatric obesity has been widely reported. Similar to mouse models, mutations in human leptin and/or leptin receptor genes are associated with early-onset childhood obesity [21]. Leptin levels are increased in obese children in direct proportion to the increase in body mass index [22]. The elevated circulating levels of leptin are thought to be important for the obese children to have normal rates of longitudinal growth, despite of their low levels of growth hormone [23].
In this study, we investigated the interaction between leptin and PPARγ, another important regulator of adiposity and energy balance. We show that addition of leptin partially releases the suppressive effect of PPARγ on chondrogenic differentiation and terminal differentiation in growth plate chondrocytes. The finding that addition of leptin only modestly decreases PPARγ expression at the mRNA level suggests that leptin might suppress PPARγ activity also by posttranslational modifications.
Genomic activity of PPARγ is regulated by various cellular processes. PPARγ is a phosphoprotein which is phosphorylated by MAPK signaling [24, 25]. Epidermal growth factor and platelet-derived growth factor have been reported to decrease the transcriptional activity of PPARγ by increasing its phosphorylation through MAPK signaling [25]. Both MAPK/ERK and MAPK/JNK signaling can phosphorylate PPARγ at a consensus MAPK phosphorylation site, serine 82 of mouse PPARγ1 and serine 112 of mouse PPARγ2 [26]. MAPK-mediated phosphorylation inhibits PPARγ transactivation function by attenuating PPARγ ligand-binding affinity [27], PPARγ nuclear export [28], and PPARγ inactivation by proteasomal degradation [29].
Our present study in rat growth plate chondrocytes reveals that leptin induces PPARγ phosphorylation at serine 112/82. The fact that this PPARγ phosphorylation is blocked by the ERK inhibitor PD98059 but not the JNK inhibitor SP600125 indicates that MAPK/ERK but not MAPK/JNK signaling is involved in the regulation of PPARγ by leptin in growth plate chondrocytes. These data are in agreement with findings in the ATDC5 chondrogenic cell-line, in which leptin has been reported to increase phosphorylation of ERK1/2 in a time- and dose-dependent manner, but not phosphorylation of JNK [30]. Furthermore, inhibition of MAPK/ERK by PD98059 abolishes the preventive effects of leptin on PPARγ-reduced chondrogenic differentiation and terminal differentiation, implying that the effects of leptin on PPARγ function in growth plate chondrocytes may result from MAPK/ERK-mediated PPARγ phosphorylation.
The MAPK/ERK pathway plays a role in chondrocyte differentiation and proliferation by mediating the upregulation of Sox9 and cyclin D1 expression [31, 32] and also has been reported to be a negative regulator of endochondral bone growth by inhibiting hypertrophic differentiation of chondrocytes [33, 34]. We previously reported that leptin promotes growth plate chondrocyte proliferation and terminal differentiation in part through IGF-1/IGF1R and Wnt/β-catenin signaling pathways [11]. The results of this study suggest that activation of MAPK/ERK by leptin may also promote chondrocyte proliferation and contribute to chondrocyte terminal differentiation by enhancing the number of proliferative cells. The finding that leptin-induced changes in Col2a1 and Col10a1 expression in the ciglitazone-treated chondrocytes are decreased by PD98059 indicate that leptin-activated MAPK/ERK signaling is involved in the inhibition of PPARγ activity and the negative effects of PPARγ on growth plate chondrocytes.
Activation of PPARγ has been reported to inhibit leptin gene expression in adipocytes [35, 36]. Heterozygous PPARγ-deficient mice exhibit high bone mass with higher leptin levels than wildtype littermates [37]. The inhibition of leptin by PPARγ may result from functional antagonism of liganded PPARγ on the CCAAT/enhancer binding protein α (C/EBPα) transactivation of the leptin promoter [36].
In our study, activation of PPARγ also negatively regulates leptin signaling. PPARγ and its agonist ciglitazone downregulate leptin, and its receptor mRNA expression, inhibit leptin-induced STAT3 phosphorylation and activation and increase STAT3 inhibitor SOCS3 expression. These findings indicate that PPARγ and leptin signaling pathways are mutually regulated in growth plate chondrocytes. The imbalance between the levels of PPARγ and leptin may facilitate the dysfunction of the growth plate observed in obese children.
Conflict of Interests
The authors declare no conflict of interests in this study.
Acknowledgment
This work was supported by a Grant from the National Institutes of Health to RTB (1 R01 AR47955).
Abbreviations
- PPARγ:
Peroxisome proliferator-activated receptor-γ
- MAPK:
Mitogen-activated protein kinase
- ERK:
Extracellular signal-regulated kinase
- JNK:
c-Jun N-terminal kinase
- JAK/STAT3:
Janus kinase/signal transducer and activator of transcription 3
- SOCS3:
Suppressor of cytokine signaling 3.
References
- 1.Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Hormone Research. 2005;64(4):157–165. doi: 10.1159/000088791. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 3.Cinti S, Frederich RC, Zingaretti MC, De Matteis R, Flier JS, Lowell BB. Immunohistochemical localization of leptin and uncoupling protein in white and brown adipose tissue. Endocrinology. 1997;138(2):797–804. doi: 10.1210/endo.138.2.4908. [DOI] [PubMed] [Google Scholar]
- 4.Houseknecht KL, Baile CA, Matteri RL, Spurlock ME. The biology of leptin: a review. Journal of Animal Science. 1998;76(5):1405–1420. doi: 10.2527/1998.7651405x. [DOI] [PubMed] [Google Scholar]
- 5.Heshka JT, Jones PJH. A role for dietary fat in leptin receptor, OB-Rb, function. Life Sciences. 2001;69(9):987–1003. doi: 10.1016/s0024-3205(01)01201-2. [DOI] [PubMed] [Google Scholar]
- 6.Vaisse C, Halaas JL, Horvath CM, Dernell JE, Jr., Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nature Genetics. 1996;14(1):95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 7.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Seminars in Cell and Developmental Biology. 2008;19(4):414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gat-Yablonski G, Phillip M. Leptin and regulation of linear growth. Current Opinion in Clinical Nutrition and Metabolic Care. 2008;11(3):303–308. doi: 10.1097/MCO.0b013e3282f795cf. [DOI] [PubMed] [Google Scholar]
- 9.Maor G, Rochwerger M, Segev Y, Phillip M. Leptin acts as a growth factor on the chondrocytes of skeletal growth centers. Journal of Bone and Mineral Research. 2002;17(6):1034–1043. doi: 10.1359/jbmr.2002.17.6.1034. [DOI] [PubMed] [Google Scholar]
- 10.Kishida Y, Hirao M, Tamai N, et al. Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification. Bone. 2005;37(5):607–621. doi: 10.1016/j.bone.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Shao YY, Ballock RT. Leptin synergizes with thyroid hormone signaling in promoting growth plate chondrocyte proliferation and terminal differentiation in vitro. Bone. 2011;48(5):1022–1027. doi: 10.1016/j.bone.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes and Development. 2000;14(11):1293–1307. [PubMed] [Google Scholar]
- 13.Wang L, Shao YY, Ballock RT. Peroxisome proliferator-activated receptor-γ promotes adipogenic changes in growth plate chondrocytes in vitro. PPAR Research. 2006;2006:8 pages. doi: 10.1155/PPAR/2006/67297.67297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Shao YY, Ballock RT. Peroxisome proliferator activated receptor-γ (PPARγ) represses thyroid hormone signaling in growth plate chondrocytes. Bone. 2005;37(3):305–312. doi: 10.1016/j.bone.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 15.Ballock RT, Reddi AH. Thyroxine is the serum factor that regulates morphogenesis of columnar cartilage from isolated chondrocytes in chemically defined medium. Journal of Cell Biology. 1994;126(5):1311–1318. doi: 10.1083/jcb.126.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park Y, Freedman BD, Lee EJ, Park S, Jameson JL. A dominant negative PPARγ mutant shows altered cofactor recruitment and inhibits adipogenesis in 3T3-L1 cells. Diabetologia. 2003;46(3):365–377. doi: 10.1007/s00125-003-1037-4. [DOI] [PubMed] [Google Scholar]
- 17.de Onis M, Blössner M, Borghi E. Global prevalence and trends of overweight and obesity among preschool children. American Journal of Clinical Nutrition. 2010;92(5):1257–1264. doi: 10.3945/ajcn.2010.29786. [DOI] [PubMed] [Google Scholar]
- 18.Gettys FK, Jackson JB, Frick SL. Obesity in pediatric orthopaedics. Orthopedic Clinics of North America. 2011;42(1):95–105. doi: 10.1016/j.ocl.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Lee RJ, Cullen C, Baca-Asher JS, Lazar-Antman MA, Leet AI. The effect of obesity on pediatric fractures. AAOS Annual Meeting; 2012; Presentation Number: 239. [Google Scholar]
- 20.Carter JR, Leeson MC, Thompson GH, Kalamchi A, Kelly CM, Makley JT. Late-onset tibia vara: a histopathologic analysis. A comparative evaluation with infantile tibia vara and slipped capital femoral epiphysis. Journal of Pediatric Orthopaedics. 1988;8(2):187–195. [PubMed] [Google Scholar]
- 21.Farooqi IS, O’Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nature Clinical Practice Endocrinology and Metabolism. 2008;4(10):569–577. doi: 10.1038/ncpendmet0966. [DOI] [PubMed] [Google Scholar]
- 22.Hassink SG, Sheslow DV, de Lancey E, Opentanova I, Considine RV, Caro JF. Serum leptin in children with obesity: relationship to gender and development. Pediatrics. 1996;98(2, part 1):201–203. [PubMed] [Google Scholar]
- 23.Phillip M, Moran O, Lazar L. Growth without growth hormone. Journal of Pediatric Endocrinology and Metabolism. 2002;15(5):1267–1272. [PubMed] [Google Scholar]
- 24.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ . Science. 1996;274(5295):2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 25.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochimica et Biophysica Acta. 2007;1771(8):952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. Journal of Biological Chemistry. 1997;272(8):5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 27.Shao D, Rangwala SM, Bailey ST, Krakow SL, Reginato MJ, Lazar MA. Interdomain communication regulating ligand binding by PPAR-γ . Nature. 1998;396(6709):377–380. doi: 10.1038/24634. [DOI] [PubMed] [Google Scholar]
- 28.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ . Molecular and Cellular Biology. 2007;27(3):803–817. doi: 10.1128/MCB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Floyd ZE, Stephens JM. Interferon-γ-mediated activation and ubiquitin-proteasome-dependent degradation of PPARγ in adipocytes. Journal of Biological Chemistry. 2002;277(6):4062–4068. doi: 10.1074/jbc.M108473200. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Eliezer M, Phillip M, Gat-Yablonski G. Leptin regulates chondrogenic differentiation in ATDC5 cell-line through JAK/STAT and MAPK pathways. Endocrine. 2007;32(2):235–244. doi: 10.1007/s12020-007-9025-y. [DOI] [PubMed] [Google Scholar]
- 31.Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(3):1113–1118. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimata M, Michigami T, Tachikawa K, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone. 2010;47(5):938–947. doi: 10.1016/j.bone.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, De Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes and Development. 2004;18(3):290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sebastian A, Matsushita T, Kawanami A, MacKem S, Landreth GE, Murakami S. Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. Journal of Orthopaedic Research. 2011;29(3):375–379. doi: 10.1002/jor.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kallen CB, Lazar MA. Antidiabetic thiazolidinediones inhibit leptin (ob) gene expression in 3T3-L1 adipocytes. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):5793–5796. doi: 10.1073/pnas.93.12.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hollenberg AN, Susulic VS, Madura JP, et al. Functional antagonism between CCAAT/enhancer binding protein-α and peroxisome proliferator-activated receptor-γ on the leptin promoter. Journal of Biological Chemistry. 1997;272(8):5283–5290. doi: 10.1074/jbc.272.8.5283. [DOI] [PubMed] [Google Scholar]
- 37.Akune T, Ohba S, Kamekura S, et al. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. Journal of Clinical Investigation. 2004;113(6):846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]