

Abstract
In pancreatic β cells, mitochondrial metabolism translates glucose sensing into signals regulating insulin secretion. Chronic exposure of β cells to excessive nutrients, namely, glucolipotoxicity, impairs β-cell function. This is associated with elevated ROS production from overstimulated mitochondria. Mitochondria are not only the major source of cellular ROS, they are also the primary target of ROS attacks. The mitochondrial uncoupling protein UCP2, even though its uncoupling properties are debated, has been associated with protective functions against ROS toxicity. Hormesis, an adaptive response to cellular stresses, might contribute to the protection against β-cell death, possibly limiting the development of type 2 diabetes. Mitochondrial hormesis, or mitohormesis, is a defense mechanism observed in ROS-induced stress-responses by mitochondria. In β cells, mitochondrial damages induced by sublethal exogenous H2O2 can induce secondary repair and defense mechanisms. In this context, UCP2 is a marker of mitohormesis, being upregulated following stress conditions. When overexpressed in nonstressed naïve cells, UCP2 confers resistance to oxidative stress. Whether treatment with mitohormetic inducers is sufficient to restore or ameliorate secretory function of β cells remains to be determined.
1. Introduction
Type 2 diabetes (T2D) is characterized by insufficient insulin release from pancreatic β cells that should compensate for peripheral insulin resistance [1]. Pancreatic β cell dysfunction and, eventually, death are considered to occur in response to metabolic stresses, which trigger mitochondrial oxidative damages, consequently interfering with glucose metabolism responsible for induction of insulin exocytosis. Accordingly, β cell should be equipped with efficient defense and adaptive mechanisms against chronic over stimulation of mitochondria, counteracting the adverse effects of oxidative stress. Emerging evidence indicate a hormetic nature of mitochondrial defensive response, that is, a cellular defense adaptation promoted by ROS-triggered signaling. Here, we discuss the putative mitohormetic role of mitochondrial uncoupling protein 2 (UCP2) in β cells and protection from mitochondrial oxidative damages.
2. Mitochondrial Metabolism, Glucose Sensing, and the Secretory Response
Pancreatic β cells function as glucose sensors to adjust insulin secretion to blood glucose levels, thereby maintaining glucose homeostasis. Translating nutrient signals into regulated insulin exocytosis relies on optimally tuned mitochondrial function [4]. Although glucose is the chief secretagogue for the β cell, metabolic profile of mitochondria is modulated by the relative contribution of glucose and lipid products for oxidative catabolism [5]. In the mitochondrion, substrates derived from glucose and fatty acids are oxidized and converted to ATP by the mitochondrial electron transport chain located in the inner mitochondrial membrane. Synthesized ATP is subsequently translocated to the cytosol, triggering insulin exocytosis thanks to calcium elevation secondary to ATP-mediated plasma membrane depolarization [6]. One byproduct of mitochondrial electron transportation is the generation of reactive oxygen species (ROS) [5].
3. Impact of Mitochondrial ROS on β Cell Function
Physiological levels of glucose and fatty acids are essential to normal β cell function. However, continuous overstimulation of β cells by these nutrients may be deleterious to β cell function, a phenomenon referred to as glucolipotoxicity. Accordingly, pancreatic β cells chronically exposed to hyperglycemic and hyperlipidemic conditions steadily undergo deterioration and ultimately failure of insulin secreting capacity [7]. This loss of β cell function has been attributed to a variety of mechanisms, most of which having in common the formation of ROS [8–10]. Elevated ROS affect the function and survival of β cells through a direct oxidation of cellular macromolecules [11, 12] and activation of cellular stress-sensitive signaling pathway [13]. Glucose infusion in rats for 48 h to achieve chronic hyperglycaemia increases mitochondrial islet superoxide and reduces glucose-stimulated insulin secretion [14]. However, it should be noticed that physiological glucose stimulation prevents formation and accumulation of ROS [15, 16]. Increasing glucose usage by pharmacological activation of glucokinase reduces ROS toxicity in insulin-secreting cells [17].
4. Mitochondria Generate ROS
ROS refer to a diverse range of species, such as superoxide (O2 ∙−), hydrogen peroxide (H2O2), and hydroxyl radical. The biological consequences of ROS rely on the specific species being involved and the physiological or pathological context. Superoxide can be converted to less reactive H2O2 by superoxide dismutase (SOD) and then to O2 and H2O by catalase, glutathione peroxidase (GPX), and peroxiredoxin, which constitute antioxidant defenses [18].
Mitochondrial electron transport chain is a potent producer of O2 ∙− within cell. Electrons from sugar, fatty acids, and amino acid catabolism accumulate in the electron carriers of the respiratory chain. O2 ∙− formation is coupled to this electron transportation as a byproduct of normal mitochondrial respiration through one electron reduction of molecular O2 ∙−. Complexes I and III of the respiratory chain are the major sites for O2 ∙− generation [19]. In complex I, electrons carried by NADH are accepted by flavin mononucleotide (FMN) and transferred to mobile electron carrier Coenzyme Q (CoQ), with O2 ∙− formation at FMN. This formation of O2 ∙− requires FMN in a fully reduced form, which is determined by the NADH/NAD+ ratio [20]. Accumulation of NADH and enhanced ROS formation are favored by slowdown of mitochondrial respiration caused by complex I inhibition (e.g., rotenone effect), secondary to damages of respiratory chain, or because of low cellular ATP demand [21, 22].
Reverse electron transport (RET) from complex II to complex I also generates large amounts of O2 ∙−. Under conditions of substantial proton motive force, RET occurs when electron supply induces CoQ reduction driving electrons back to complex I, thereby reducing NAD+ to NADH at the FMN site [23]. Succinate and fatty acid oxidation promote high proton motive force along with electron supply to CoQ, giving rise to O2 ∙− formation under RET. Generation of O2 ∙− by complex I, in particular through RET, is very sensitive to mitochondrial uncoupling because of the required proton motive force [24]. When inhibited by antimycin, complex III can produce significant amounts of O2 ∙−, although its production under physiological conditions is only marginal compared to that of complex I [22].
5. Mitochondrial ROS, Friends or Foes?
ROS impact differently on cell function depending on specific reactive oxygen species, their concentrations, and effectiveness of detoxifying systems; thereby, defining signaling or toxic effects of ROS [25]. In insulin-secreting cells, low concentrations of H2O2 have been reported to contribute to the stimulation of insulin secretion [26]. However, exposure to robust concentrations of H2O2 impairs β cell function [27]. When β cells are continuously overstimulated by nutrients, accumulation of ROS can overwhelm detoxification systems and induce deleterious effects [8]. This is particularly relevant for pancreatic islets because of the shifted redox balance favored by high metabolic rate, in particular under glucolipotoxic conditions, and relatively weak detoxifying systems [28]. Being a major source of ROS, mitochondria are in the eye of the storm. Specifically, H2O2 exposure to insulin-secreting cells inactivates mitochondria, thereby interrupting mitochondrial signals normally linking glucose metabolism to insulin exocytosis [27]. One single oxidative stress applied for just 10 minutes induces β cell dysfunction lasting over days, explained by persistent damages in mitochondrial components and accompanied by subsequent generation of endogenous ROS of mitochondrial origin [29]. In the close vicinity of free radical production, mitochondrial inner membrane components are particularly prone to oxidative injuries, such as subunits of electron transport chain complexes and the adenine nucleotide translocase (ANT) [29, 30]. Moreover, some iron-sulfur centers of mitochondrial matrix proteins, among them aconitase, are susceptible to damages induced by direct reaction with O2 ∙− [31] or nitric oxide [32], leading to impaired mitochondria and β cell dysfunction.
Because of relatively low antioxidant-enzyme activities in pancreatic islets, enhancing expression of corresponding genes in insulin-secreting cells has been foreseen as potential protective intervention. However, contradictory findings were reported in studies testing this promising approach. The concept is favored by some studies, among them: (i) β cell-specific overexpression of cytosolic SOD (SOD1) enhances mouse resistance to alloxan-induced diabetogenesis [33]; (ii) adenovirus-mediated overexpression of mitochondrial SOD (SOD2) in isolated islets extends islet function following transplantation into streptozotocin (STZ)-treated nonobese diabetic (NOD) mice [34]; (iii) β cell-specific overexpression of SOD2 and catalase protects islets from STZ-induced oxidative stress [35]; (iv) β cell-specific overexpression of glutathione peroxidase in db/db mice improves β cell volume and granulation [36]. Tempering these promising results, overexpression of antioxidant enzymes, such as catalase and metallothionein, specifically in β cells of NOD mice increases β cell death and sensitizes islets to cytokine-induced injuries [37]. The latter results suggest that a mild dominance of host ROS over detoxifying systems might exhibit beneficial effects.
6. Mitochondrial Protection against ROS: Role of UCP2
Mitochondrial uncoupling refers to the dissociation of electron-dependent oxygen consumption to ATP generation on the respiratory chain. The most efficient way to induce mitochondrial uncoupling is to allow protons to circulate freely across the inner mitochondrial membrane, in other words to create a proton leak. In this regard, UCP1 is a professional mitochondrial uncoupler by inducing proton leakage. As a result, the energy contributed by electron flow is dissipated as heat instead of ATP generation. UCP1 expression in brown adipose tissue confers to these fat depots highly thermogenic properties [38]. UCP2 was named after its loose homology (59%) with UCP1 and exhibits heterogeneous tissue expression, including in pancreatic islets [39]. Similarly to UCP1, UCP2 was proposed to induce proton leakage and to dissipate proton motive force [40], consequently limiting ATP production and glucose-stimulated insulin secretion in pancreatic β cells [41, 42].
Due to tight dependency of complex I on proton motive force for ROS formation, putative uncoupling effects of UCP2 were suggested to compromise mitochondrial ROS generation and associated cell damages [43, 44]. In insulin-secreting cells, a series of in vitro studies have shown that increasing UCP2 expression attenuates ATP synthesis and insulin secretion in response to glucose [41, 42, 45, 46]. Conversely, UCP2 deficiency enhances glucose-stimulated insulin secretion, as shown in islets isolated from both global [47, 48] and β cell-specific [49] UCP2 knockout mice. In diet-induced T2D mouse model, the lack of UCP2 improves blood glucose levels and insulin secretory capacity [47]. Chronic exposure of INS-1 β cells to fatty acid decreases the secretory response to glucose, along with UCP2 gene induction and partial mitochondrial uncoupling [50].
Over the last decade, UCP2 effects in β cells have been tightly correlated with its presumed uncoupling properties and consequences on ATP synthesis from oxidative phosphorylation [41, 42, 45–48]. However, in β cell-specific UCP2-null mice, the potentiated glucose-stimulated insulin secretion correlates with higher intracellular ROS levels, without any changes on mitochondrial coupling and ATP generation [49]. Moreover, pancreatic islets form global UCP2 knockout mice studied on three congenic backgrounds, as opposed to mixed genetic background [48], exhibit impaired glucose response accompanied by increased ROS production and persistent oxidative stress [51]. Finally, cytokine-induced ROS production is reduced in insulin-secreting cells overexpressing UCP2, independently of uncoupling effects [3]. Collectively, these observations are contradictory regarding putative uncoupling properties of UCP2 and its effects on the secretory function of β cells. Instead, they suggest a role in defense mechanisms against oxidative stress [52], as shown by induction of UCP2 that prevents cytokine-induced β cell death through suppression of ROS production [3, 53].
Because UCP2 might play a protective role against ROS, one can hypothesize cooperation and feedback mechanisms with dedicated antioxidant enzymes. In β cells overexpressing UCP2, the associated sheltering effect against oxidative injuries is not associated with changes in antioxidant enzymes (personal communication from Françoise Assimacopoulos-Jeannet, University of Geneva). Ablation of UCP2 in β cells favors both ROS formation and induction of H2O2-scavenging GPX, but not of superoxide scavenger SODs [49]. In islets lacking SOD1 or GPX1, UCP2 is upregulated as a protective response against excessive cellular ROS [54]. Conversely, UCP2 is downregulated upon induction of GPX1 in mouse islets [55]. Collectively, these data indicate crosstalk between UCP2 and antioxidant enzymes through unidentified mechanisms.
7. Hormesis, a Stress-Induced Protective Response
Theophrastus Bombastus von Hohenheim, a Swiss pharmacist born in 1493 also named Paracelsus, developed this revolutionary idea at the Renaissance period: “Alle Ding' sind Gift, und nichts ohn' Gift; allein die Dosis macht, daß ein Ding kein Gift ist” freely translated to “the dose makes the poison.” Five centuries later, this notion has been extended to the so-called hormesis. Hormesis is a phenomenon whereby exposure of cells or organs to low levels of a given toxin confers resistance to subsequent contacts to higher concentrations [56]. Accordingly, hormesis describes an adaptive response to continuous cellular stresses. Hormesis is well illustrated by ischemic preconditioning, a situation where short ischemic episodes protect brain and heart from prolonged lack of oxygen and nutrients [57, 58]. Regarding pancreatic β cells, emerging concepts suggest that efficiency of hormetic responses to detrimental lifestyle factors might set the level of protection, impacting on the progression of T2D [59].
8. Adaptation and Hormesis in β cells
Obesity is a strong risk factor for T2D, appearing in subjects developing β cell dysfunction and death in response to metabolic and inflammatory stresses [60, 61]. However, about half of obese individuals do not develop diabetes, due to efficient long-term adaptation to insulin resistance by increasing β cell mass and insulin secretion. In these resistant individuals, β cells may develop adaptive stress responses to prevent their loss, at least transiently. Peroxisome proliferator-activated receptor alpha (PPARα) is a transcription factor controlling lipid and glucose homeostasis. PPARα-deficient mice on an obese (ob/ob) background develop β cell dysfunction characterized by reduced islet area and glucose response [62]. Human islets treated with PPARα agonist are protected against fatty acid-induced impairment of glucose-induced insulin secretion and apoptosis [62]. This indicates that PPARα could be an adaptive candidate in β cells under pathological conditions, such as lipid-induced dysfunction [63].
Converging evidence suggest that stresses can induce specific responses rendering β cells more resistant to the stress-molecule, or even to other toxins. Pre-exposure to low dose IL-1β renders β cells less susceptible to toxin-induced cell necrosis and to radical-induced damages, though with a loss of normal phenotype [64]. Moreover, islets from pancreatectomized hyperglycemic rats exhibit reduced sensitivity to STZ, an effect associated with induction of protective antioxidant and antiapoptotic genes during chronic hyperglycemia [65]. Finally, islets from GK/Par rat (non-obese model of T2D) also show strong resistance to toxic effects of exogenous ROS, secondary to an adaptive response to the diabetic milieu [66]. Thus, β cells possess hormetic mechanisms in response to inflammatory and metabolic stresses. Stressors are not merely toxic; they can also prime the stressed cell to future pathogenic challenges by rendering them more resistant.
9. Mitochondrial Adaptation and Hormesis, or Mitohormesis, in β Cells
Mitochondrial adaptation and hormesis, or mitohormesis, originally referred to the hypothetical model of cell preservation in response to ROS-induced stresses originating from mitochondria [67]. The concept was substantiated by findings in C. elegans revealing that glucose restriction activates mitochondria and ROS formation, promoting hormetic extension of life span [68]. In conflict with Harman's free radical theory of aging [69], protective effects depend on mitochondrial ROS formation inducing an adaptive response, in turn conferring increased stress-resistance. This might ultimately give rise to long-term cell preservation. In agreement with this model, calorie restriction extend life span in different organisms by increasing mitochondrial ROS production [70]. In pancreatic β cells, mitohormetic response is suggested by adaptation to dietary fat-induced insulin resistance attributed to increased mitochondrial function [71], an effect correlating with elevated ROS levels secondary to fatty acid treatment of insulin-secreting cells [10].
The nature of mitohormesis in insulin-secreting cells can be studied when cells recover from a single transient exposure to sublethal H2O2. We previously reported that INS-1E β cells and rat islets subjected to a 10 min H2O2 exposure exhibit impaired secretory response associated with interrupted mitochondrial signals measured right after stress [27]. Loss of mitochondrial function occurs within the first minutes of oxidative stress [27], as revealed by collapse of mitochondrial membrane potential (Figure 1(a)). Monitored concomitantly on the same cells, gradual discontinuous mitochondrial network is observed, eventually exhibiting some globular patterns 60 min after stress (Figure 1(b)). These phenomena were not observed in control nonstressed cells (Figures 1(c) and 1(d)). Then, the question is whether such oxidative stress results in prolonged mitochondrial damages, recovery of cell function, or improved resistance to stress. After a 3-day recovery period following the 10 min stress, we observed increased mitochondrial H2O2 formation and persistence of mitochondrial dysfunction altering metabolism-secretion coupling [29]. The ROS-induced endogenous H2O2 generation contributes to prolongation of oxidative attacks days after exposure to exogenous H2O2. This is accompanied by increased expression of genes participating to recovery of mitochondrial function, detoxification, and cell survival; such as subunits of mitochondrial electron transport chain complexes and antioxidant enzymes [29]. Mitochondrial defects induced by acute 10 min oxidant exposure are carried on to daughter cells. These cells ultimately achieve gradual turnover of mitochondrial components enabling recovery of their function in the following weeks of culture period [29]. Three weeks after transient oxidant exposure, those insulin-secreting cells respond normally to physiological stimuli. Remarkably, the recovered cells are more resistant than naïve cells to a new exogenous oxidative stress. This beneficial “memory” of mitochondrial oxidative injury represents mitohormetic property and is associated with a higher UCP2 gene expression 3 weeks post-stress [29], suggesting a protective role for UCP2.
Figure 1.

Simultaneous monitoring of mitochondrial membrane potential and morphology in INS-1E β cell under transient oxidative stress. Real-time imaging of INS-1E cells by simultaneous fluorescence recordings of mitochondrial potential (ΔΨm) by TMRE (a and c) and mitochondrial morphology by ΔΨm-independent mito-eYFP (b and d) as described [2]. (a), Signals recorded before oxidant exposure (before-stress), during the 10 min 200 μM H2O2 exposure (stress), and after neutralization of extracellular H2O2 by the addition of 100 U/mL catalase (after-stress). (b), Corresponding mitochondrial morphology monitored simultaneously with ΔΨm shown in (a). (c) and (d) show control nonstressed cells.
10. Does UCP2 Participate to Mitohormesis in β cell?
As described above, some studies have highlighted UCP2 as a protective element under stress conditions [3, 29, 49, 52], possibly implicated in β cell mitohormetic response. To address this question, INS-1 β cells with doxycycline-inducible overexpression of human UCP2 [3] were challenged with an oxidative stress by exposure to 200 μM H2O2 for 10 min as described [27, 29]. Consistent with previous report [3], increased expression of UCP2 (Figure 2(a)) did not alter mitochondrial coupling (Figures 2(b) and 2(c)). Indeed, INS-1 cells with induced UCP2 overexpression exhibited similar respiration upon glucose stimulation compared to non induced cells (Figure 2(c)). Moreover, state 3 respiration measured on isolated mitochondria stimulated with succinate plus ADP was even slightly higher versus controls (Figure 2(b)). INS-1E cells with basal UCP2 expression are highly sensitive to oxidative stress regarding mitochondrial respiration, exhibiting marked reduction of state 3 (−59% versus control nonstressed cells) 3 days after oxidative stress [29]. On the contrary, cells overexpressing UCP2 did not show any impairment of oxygen consumption at day 3 after stress, as shown both on isolated mitochondria and intact cells (Figures 2(b) and 2(c), resp.). Acute oxidant exposure did not further elevate UCP2 protein levels 3 days after stress in UCP2-induced cells (Figure 2(a)). Collectively, these observations support the concept that UCP2 upregulation observed previously as a mitohormetic response [29] can serve as defense mechanism against mitochondrial oxidative damages (Figure 2).
Figure 2.
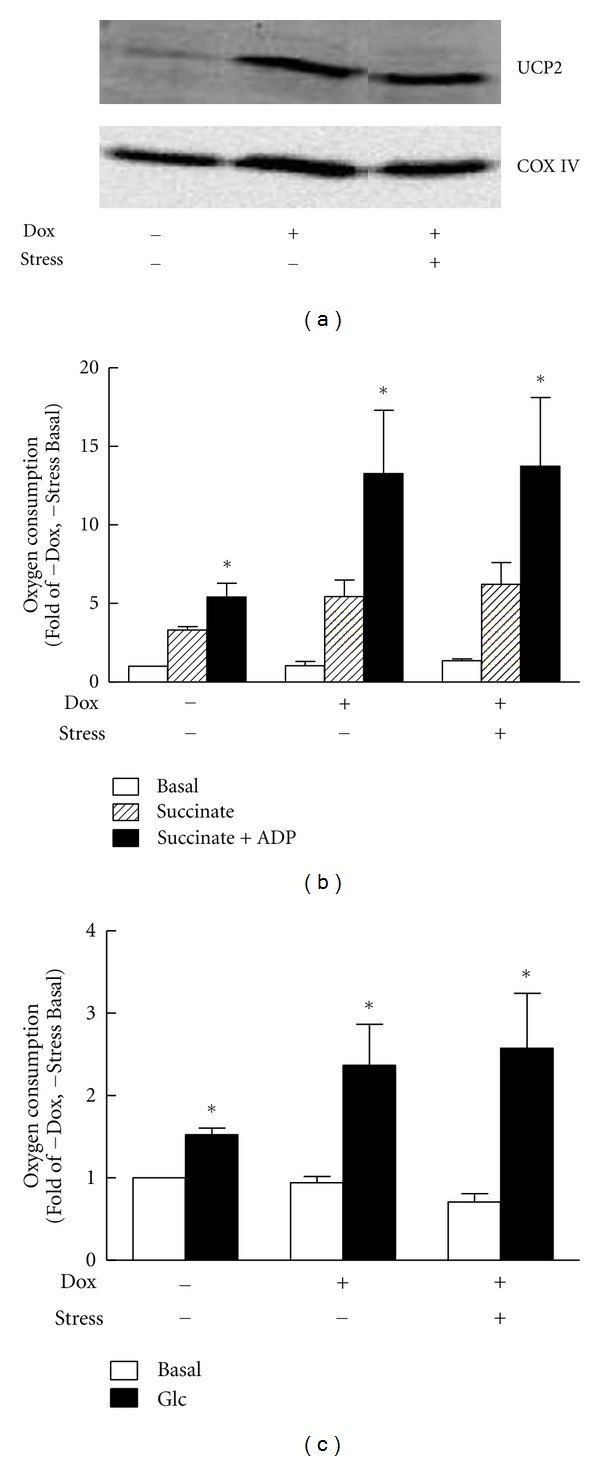
Effects of UCP2 overexpression and oxidative stress on mitochondrial respiration in INS-1 cells. UCP2 was induced in INS-1 cells (hUCP2 INS-1-r9, [3]) by 250 ng/mL doxycycline (+Dox) 2 days before oxidative stress and during stress period. (a) Immunoblotting showing UCP2 protein levels in noninduced (−Dox) versus induced (+Dox) INS-1 cells and nonstressed (−stress) versus stressed cells (+stress, 200 μM H2O2 for 10 min 3 days before analysis). Cytochrome oxidase (COX IV) is shown as control for inner mitochondrial protein. (b) O2 consumption measured on mitochondria isolated from INS-1 cells 3 days after-stress. Respiration was induced by 5 mM succinate (Succinate) followed by addition of 150 μM ADP (Succinate + ADP). (c) O2 consumption measured on intact INS-1 cells stimulated by 15 mM glucose (Glc), compared to basal respiration at 2.5 mM glucose (Basal). Data are means ± SE of 3 independent experiments expressed as nmol O2/min per 100 μg mitochondrial protein (b) or nmol O2/min per 106 cells (c) normalized to basal respiration of controls (no Dox, no stress). *P < 0.05 versus basal O2 consumption of corresponding condition.
11. Conclusions
In pancreatic β cells, stress-response hormesis can develop under different metabolic insults, such as lipotoxicity, cytokines, or ROS. In particular, oxidative stress induces mitohormesis, rendering mitochondria more resistant to oxidative attacks. Various studies in this field reported conflicting results. However, converging evidence points to UCP2 as a marker of mitohormesis, this protein being upregulated following stress conditions. Moreover, overexpression of UCP2 in naïve cells lacking hormesis adaptation is sufficient to confer resistance to oxidative stress (Figure 2). The exact function of UCP2 is still unknown, although its partial homology with uncoupling UCP1 protein suggests a functional link with the electron transport chain. Whether treatment with UCP2 inducers, such as glutamine [72], could promote mitohormesis and protect β cells under metabolic stress remains to be determined.
Acknowledgments
The authors thank Françoise Assimacopoulos-Jeannet (Geneva) for gift of inducible hUCP2 INS-1 cells and for helpful discussions. They are also thankful for the continuous support from the Swiss National Science Foundation and the State of Geneva.
Abbreviations
- ANT:
Adenine nucleotide translocase
- CoQ:
Coenzyme Q
- ER:
endoplasmic reticulum
- FMN:
Flavin mononucleotide
- GPX:
Glutathione peroxide
- GSIS:
Glucose-stimulated insulin secretion
- NADH:
Nicotinamide adenine dinucleotide
- NOD:
Nonobese diabetic
- PPAR:
Peroxisome proliferator-activated receptor
- RET:
Reverse nucleotide transport
- ROS:
Reactive oxygen species
- SOD:
Superoxide dismutase
- STZ:
Streptozotocin
- UCP:
Uncoupling protein.
References
- 1.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. Journal of Neuroscience. 2004;24(24):5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Produit-Zengaffinen N, Davis-Lameloise N, Perreten H, et al. Increasing uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin secretion but decreases production of reactive oxygen species. Diabetologia. 2007;50(1):84–93. doi: 10.1007/s00125-006-0499-6. [DOI] [PubMed] [Google Scholar]
- 4.Maechler P. Mitochondria as the conductor of metabolic signals for insulin exocytosis in pancreatic β-cells. Cellular and Molecular Life Sciences. 2002;59(11):1803–1818. doi: 10.1007/PL00012507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maechler P, Li N, Casimir M, Vetterli L, Frigerio F, Brun T. Role of mitochondria in β-cell function and dysfunction. Advances in Experimental Medicine and Biology. 2010;654:193–216. doi: 10.1007/978-90-481-3271-3_9. [DOI] [PubMed] [Google Scholar]
- 6.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic β-cells. Nature. 2001;414(6865):807–812. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 7.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. Journal of Clinical Investigation. 1992;90(2):320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson RP, Harmon J, Tran POT, Poitout V. β-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(1, supplement):S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 9.Wu L, Nicholson W, Knobel SM, et al. Oxidative stress is a mediator of glucose toxicity in insulin-secreting pancreatic islet cell lines. Journal of Biological Chemistry. 2004;279(13):12126–12134. doi: 10.1074/jbc.M307097200. [DOI] [PubMed] [Google Scholar]
- 10.Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic β-cells exposed to free fatty acids. Journal of Biological Chemistry. 2003;278(22):19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- 11.Dypbukt JM, Ankarcrona M, Burkitt M, et al. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. Journal of Biological Chemistry. 1994;269(48):30553–30560. [PubMed] [Google Scholar]
- 12.Takasu N, Asawa T, Komiya I, Nagasawa Y, Yamada T. Alloxan-induced DNA strand breaks in pancreatic islets: evidence for H2O2 as an intermediate. Journal of Biological Chemistry. 1991;266(4):2112–2114. [PubMed] [Google Scholar]
- 13.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress—activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes. 2003;52(1):1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55(5):1366–1379. doi: 10.1007/s00125-012-2474-8. [DOI] [PubMed] [Google Scholar]
- 15.Martens GA, Cai Y, Hinke S, Stangé G, Van De Casteele M, Pipeleers D. Glucose suppresses superoxide generation in metabolically responsive pancreatic β cells. Journal of Biological Chemistry. 2005;280(21):20389–20396. doi: 10.1074/jbc.M411869200. [DOI] [PubMed] [Google Scholar]
- 16.Sarre A, Gabrielli J, Vial G, Leverve XM, Assimacopoulos-Jeannet F. Reactive oxygen species are produced at low glucose and contribute to the activation of AMPK in insulin-secreting cells. Free Radical Biology and Medicine. 2012;52(1):142–150. doi: 10.1016/j.freeradbiomed.2011.10.437. [DOI] [PubMed] [Google Scholar]
- 17.Futamura M, Yao J, Li X, et al. Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia. 2012;55(4):1071–1080. doi: 10.1007/s00125-011-2439-3. [DOI] [PubMed] [Google Scholar]
- 18.Yu BP. Cellular defenses against damage from reactive oxygen species. Physiological Reviews. 1994;74(1):139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 19.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 20.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochemical Society Transactions. 2008;36(5):976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 21.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochemical Journal. 1972;128(3):617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chance B, Hollunger G. The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate-linked reduction of pyridine nucleotide. The Journal of Biological Chemistry. 1961;236:1534–1543. [PubMed] [Google Scholar]
- 24.Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biology and Medicine. 2004;37(6):755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Murphy MP, Holmgren A, Larsson NG, et al. Unraveling the biological roles of reactive oxygen species. Cell Metabolism. 2011;13(4):361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56(7):1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 27.Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. Journal of Biological Chemistry. 1999;274(39):27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 28.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46(11):1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 29.Li N, Brun T, Cnop M, Cunha DA, Eizirik DL, Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent β-cell dysfunction. Journal of Biological Chemistry. 2009;284(35):23602–23612. doi: 10.1074/jbc.M109.024323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods in Enzymology. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- 32.Corbett JA, Wang JL, Sweetland MA, Lancaster JR, Jr., McDaniel ML. Interleukin 1β induces the formation of nitric oxide by β-cells purified from rodent islets of Langerhans. Evidence for the β-cell as a source and site of action of nitric oxide. Journal of Clinical Investigation. 1992;90(6):2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubisch HM, Wang J, Bray TM, Phillips JP. Targeted overexpression of Cu/Zn superoxide dismutase protects pancreatic β-cells against oxidative stress. Diabetes. 1997;46(10):1563–1566. doi: 10.2337/diabetes.46.10.1563. [DOI] [PubMed] [Google Scholar]
- 34.Bertera S, Crawford ML, Alexander AM, et al. Gene transfer of manganese superoxide dismutase extends islet graft function in a mouse model of autoimmune diabetes. Diabetes. 2003;52(2):387–393. doi: 10.2337/diabetes.52.2.387. [DOI] [PubMed] [Google Scholar]
- 35.Chen H, Li X, Epstein PN. MnSOD and catalase transgenes demonstrate that protection of islets from oxidative stress does not alter cytokine toxicity. Diabetes. 2005;54(5):1437–1446. doi: 10.2337/diabetes.54.5.1437. [DOI] [PubMed] [Google Scholar]
- 36.Harmon JS, Bogdani M, Parazzoli SD, et al. β-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db Mice. Endocrinology. 2009;150(11):4855–4862. doi: 10.1210/en.2009-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Chen H, Epstein PN. Metallothionein and catalase sensitize to diabetes in nonobese diabetic mice: reactive oxygen species may have a protective role in pancreatic β-cells. Diabetes. 2006;55(6):1592–1604. doi: 10.2337/db05-1357. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiological Reviews. 1984;64(1):1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Zhou YT, Shimabukuro M, Koyama K, et al. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(12):6386–6390. doi: 10.1073/pnas.94.12.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleury C, Neverova M, Collins S, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nature Genetics. 1997;15(3):269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 41.Chan CB, De Leo D, Joseph JW, et al. Increased uncoupling protein-2 levels in β-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50(6):1302–1310. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- 42.Chan CB, MacDonald PE, Saleh MC, Johns DC, Marbàn E, Wheeler MB. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes. 1999;48(7):1482–1486. doi: 10.2337/diabetes.48.7.1482. [DOI] [PubMed] [Google Scholar]
- 43.Mattiasson G, Shamloo M, Gido G, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nature Medicine. 2003;9(8):1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- 44.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 45.Hong Y, Fink BD, Dillon JS, Sivitz WI. Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142(1):249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- 46.Wang MY, Shimabukuro M, Lee Y, et al. Adenovirus-mediated overexpression of uncoupling protein-2 in pancreatic islets of Zucker diabetic rats increases oxidative activity and improves β- cell function. Diabetes. 1999;48(5):1020–1025. doi: 10.2337/diabetes.48.5.1020. [DOI] [PubMed] [Google Scholar]
- 47.Joseph JW, Koshkin V, Zhang CY, et al. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002;51(11):3211–3219. doi: 10.2337/diabetes.51.11.3211. [DOI] [PubMed] [Google Scholar]
- 48.Zhang CY, Baffy G, Perret P, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, β cell dysfunction, and type 2 diabetes. Cell. 2001;105(6):745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 49.Robson-Doucette CA, Sultan S, Allister EM, et al. β-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes. 2011;60(11):2710–2719. doi: 10.2337/db11-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lameloise N, Muzzin P, Prentki M, Assimacopoulos-Jeannet F. Uncoupling protein 2: a possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes. 2001;50(4):803–809. doi: 10.2337/diabetes.50.4.803. [DOI] [PubMed] [Google Scholar]
- 51.Pi J, Bai Y, Daniel KW, et al. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic β-cell function. Endocrinology. 2009;150(7):3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li LX, Skorpen F, Egeberg K, Jørgensen IH, Grill V. Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal β-cells. Biochemical and Biophysical Research Communications. 2001;282(1):273–277. doi: 10.1006/bbrc.2001.4577. [DOI] [PubMed] [Google Scholar]
- 53.Zengaffinen N, Perreten H, Lameloise N, Keller P, Muzzin P, Assimacopoulos-Jeannet F. Uncoupling protein 2 overexpression prevents cytokine-induced reactive oxygen species production and apoptosis in pancreatic beta cells. Diabetologia. 2005;48:p. A38. [Google Scholar]
- 54.Wang X, Vatamaniuk MZ, Roneker CA, et al. Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxidants and Redox Signaling. 2011;14(3):391–401. doi: 10.1089/ars.2010.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang XD, Vatamaniuk MZ, Wang SK, Roneker CA, Simmons RA, Lei XG. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia. 2008;51(8):1515–1524. doi: 10.1007/s00125-008-1055-3. [DOI] [PubMed] [Google Scholar]
- 56.Calabrese EJ, Bachmann KA, Bailer AJ, et al. Biological stress response terminology: integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicology and Applied Pharmacology. 2007;222(1):122–128. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 57.Zhang F, Wu Y, Jia J. Exercise preconditioning and brain ischemic tolerance. Neuroscience. 2011;177:170–176. doi: 10.1016/j.neuroscience.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 58.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 59.Kolb H, Eizirik DL. Resistance to type 2 diabetes mellitus: a matter of hormesis? Nature Reviews Endocrinology. 2011;8:183–192. doi: 10.1038/nrendo.2011.158. [DOI] [PubMed] [Google Scholar]
- 60.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(supplement 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 61.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 62.Lalloyer F, Vandewalle B, Percevault F, et al. Peroxisome proliferator-activated receptor α improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes. 2006;55(6):1605–1613. doi: 10.2337/db06-0016. [DOI] [PubMed] [Google Scholar]
- 63.Frigerio F, Brun T, Bartley C, et al. Peroxisome proliferator-activated receptor α (PPARα) protects against oleate-induced INS-1E beta cell dysfunction by preserving carbohydrate metabolism. Diabetologia. 2010;53(2):331–340. doi: 10.1007/s00125-009-1590-6. [DOI] [PubMed] [Google Scholar]
- 64.Ling Z, Van De Casteele M, Eizirik DL, Pipeleers DG. Interleukin-1β-induced alteration in a β-cell phenotype can reduce cellular sensitivity to conditions that cause necrosis but not to cytokine- induced apoptosis. Diabetes. 2000;49(3):340–345. doi: 10.2337/diabetes.49.3.340. [DOI] [PubMed] [Google Scholar]
- 65.Laybutt DR, Kaneto H, Hasenkamp W, et al. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to β-cell survival during chronic hyperglycemia. Diabetes. 2002;51(2):413–423. doi: 10.2337/diabetes.51.2.413. [DOI] [PubMed] [Google Scholar]
- 66.Lacraz G, Figeac F, Movassat J, Kassis N, Portha B. Diabetic GK/Par rat β-cells are spontaneously protected against H2O2-triggered apoptosis. A cAMP-dependent adaptive response. American Journal of Physiology. Endocrinology and Metabolism. 2010;298(1):E17–E27. doi: 10.1152/ajpendo.90871.2008. [DOI] [PubMed] [Google Scholar]
- 67.Tapia PC. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: 'Mitohormesis' for health and vitality. Medical Hypotheses. 2006;66(4):832–843. doi: 10.1016/j.mehy.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 68.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metabolism. 2007;6(4):280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 69.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 70.Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis) Experimental Gerontology. 2010;45(6):410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 71.Fex M, Nitert MD, Wierup N, Sundler F, Ling C, Mulder H. Enhanced mitochondrial metabolism may account for the adaptation to insulin resistance in islets from C57BL/6J mice fed a high-fat diet. Diabetologia. 2007;50(1):74–83. doi: 10.1007/s00125-006-0464-4. [DOI] [PubMed] [Google Scholar]
- 72.Hurtaud C, Gelly C, Chen Z, Lévi-Meyrueis C, Bouillaud F. Glutamine stimulates translation of uncoupling protein 2 mRNA. Cellular and Molecular Life Sciences. 2007;64(14):1853–1860. doi: 10.1007/s00018-007-7039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]