

Abstract
α-Fluorinated-1,1-bisphosphonic acids derived from fatty acids were designed, synthesized and biologically evaluated against Trypanosoma cruzi, the etiologic agent of Chagas disease and against Toxoplasma gondii, the responsible agent of toxoplasmosis and also towards the target parasitic enzymes farnesyl pyrophosphate synthase of T. cruzi (TcFPPS) and T gondii (TgFPPS), respectively. Interestingly, 1-fluorononylidene-1,1-bisphosphonic acid (compound 43) has proven to be an extremely potent inhibitor of the enzymatic activity of TgFPPS at the low nanomolar range exhibiting an IC50 of 30 nM. This compound was two-fold more potent than risedronate (IC50 = 74 nM) taken as a positive control. This enzymatic activity was associated to a strong cell growth inhibition against tachyzoites of T. gondii having an IC50 value of 2.7 μM.
Introduction
Bisphosphonates of general formula 1 are metabolically stable pyrophosphate (2) analogues in which a methylene group replaces the oxygen atom bridge between the two phosphorus atoms of the pyrophosphate moiety. Substitution at the carbon atom with different side chains has generated a large family of compounds.1 Several bisphosphonates such as pamidronate (3), alendronate (4), risedronate (5), and ibandronate (6) are in clinical use for the treatment and prevention of osteoclast-mediated bone resorption associated with osteoporosis, Paget’s disease, hypercalcemia, tumor bone metastases, and other bone diseases (Chart 1).2–4 Bisphosphonates became compounds of pharmacological importance since calcification studies were done more than 40 years ago.5–7
Chart 1.

General formula and chemical structure of representative FDA-approved bisphosphonates clinically employed for different bone disorders.
Selective action on bone is based on the binding of the bisphosphonate moiety to the bone mineral.8 It has been postulated that the acidocalcisomes are equivalent in composition to the bone mineral and that accumulation of bisphosphonates in these organelles, as they do in bone mineral, assists their antiparasitic action.8 Bisphosphonates act by a mechanism that lead to osteoclast apoptosis.9 The site of action of aminobisphosphonates has been narrowed down to the isoprenoid pathway and, more specifically, to an inhibition of protein prenylation.10 Farnesyl pyrophosphate synthase (FPPS) constitutes the principal target of bisphosphonates.11–15 This enzyme catalyzes the two mandatory biosynthetic steps to form farnesyl pyrophosphate as indicated briefly in Scheme 1. Inhibition of the enzymatic activity of FPPS blocks farnesyl pyrophosphate and geranylgeranyl pyrophosphate formation that are required for the post-translational prenylation of small GTP-binding proteins, which are also GTPases such as Rab, Rho and Rac within osteoclasts.16
Scheme 1.

Isoprenoid biosynthesis in trypanosomatids and apicomplexan parasites.
Besides their use in long-term treatments for different bone disorders, bisphosphonates additionally exhibit a wide range of biological actions such as stimulation of γδ T cells of the immune system,17 antibacterial action,18 herbicidal properties,19 anticancer action,20–23 as potent and selective inhibitors of the enzymatic acitivity of acid sphingomyelinase,24 and, particularly, as antiparasitic agents.25–29 Certainly, at the beginning, aminobisphosphonates have proven to be effective growth inhibitors of T. cruzi in in vitro and in vivo assays without toxicity to the host cells.8 Inspired on this work, different bisphosphonates were found to be potent inhibitors of the proliferation of pathogenic trypanosomatids other than T. cruzi, such as T. brucei rhodesiense, Leishmania donovani, and L. mexicana and apicomplexan parasites such as Toxoplasma gondii and Plasmodium falciparum.27,28,30–36 In vivo assays of bisphosphonates have shown that risedronate can significantly increase survival of T. cruzi-infected mice.37 In view of the above results, it is possible to assume that bisphosphonates are potential candidates for chemotherapy of neglected infectious diseases. In addition, bisphosphonates have the advantage that their synthesis is straightforward and relatively inexpensive. It is reasonable to assume a low toxicity for bisphosphonate-containing drugs bearing in mind that many bisphosphonates are FDA-approved drugs for the long-term treatment of different bone disorders.38
Bisphosphonates derived from fatty acids have become interesting putative antiparasitic agents, especially 2-alkylaminoethyl derivatives, which were shown to be potent growth inhibitors against the clinically more relevant form of T. cruzi possessing IC50 values at the nanomolar range against the target enzyme.31,32 Compounds 10–12 arise as representative members of this type of bisphosphonates, which have proven to be by far more efficient than their parent drugs 1-hydroxy-, 1-alkyl-, and 1-amino-bisphosphonates as growth inhibitors of trypanosomatids (Chart 2).33–35
Chart 2.
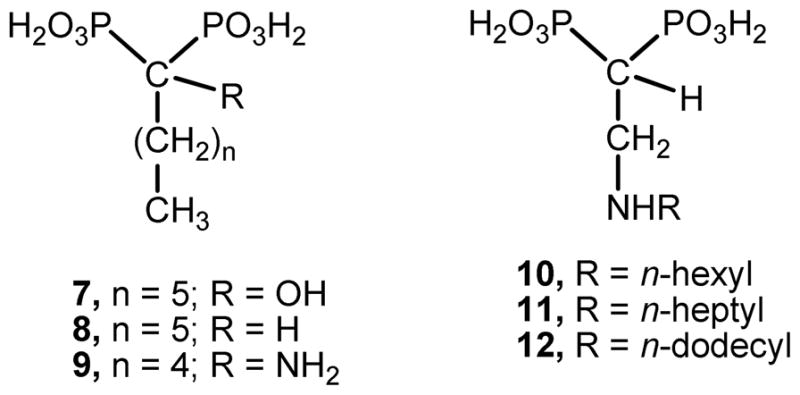
Chemical structure of representative members of bisphosphonic acids derived from fatty acids.
T. cruzi, which is the etiologic agent of American trypanosomiasis, has a complex life cycle involving blood-sucking Reduviid insects and mammals.39,40 It multiplies in the insect gut as an epimastigote form and is spread as a non-dividing metacyclic trypomastigote from the insect feces by contamination of intact mucosa or wounds produced by the blood-sucking activity of the vector. In the mammalian host, the parasite proliferates intracellularly in the amastigote form and is subsequently released into the blood stream as a non-dividing trypomastigote.40 On the other hand, the opportunistic pathogen T. gondii causes a broad spectrum of disease but most infections are asymptomatic.41 This apicomplexan parasite has adopted an essential intracellular life style. The parasite actively penetrates host cells, sets up a privileged compartment in which it replicates and finally kills the cell.42 There are two asexual forms that can cause disease in humans. The tachyzoite form can invade all types of cells and proliferate leading to host cell death. The bradyzoite form divides slowly and forms cysts in muscle and brain. The sexual cycle occurs in the superficial epithelium of the small intestine of members of the cat family. Oocysts, which are shed in feces of recently infected cats, remain in the upper soil horizon, where they may contaminate skin and may be ingested by hand-to-mouth transmission or on natural vegetables. Oocysts require at least 12 hours in order to complete sporulation, afterward they are infectious by mouth.43–46 Chemotherapy for these neglected diseases is still deficient and based on old and empirically discovered drugs.47,48 Therefore, there is a critical need to develop new safe drugs based on the knowledge of the biochemistry and physiology of these microorganisms.
Rationale
The precise mechanism of action by which bisphophonates inhibit the enzymatic activity of the target enzyme remains unsolved. The main members of 2-alkylaminoethyl bisphosphonates family were originally designed in order to maintain the ability to coordinate Mg2+ in a tridentate manner as 1-hydroxy- and 1-amino- derivatives do.32 However, preliminary studies on the interaction of inhibitor 11 (IC50 = 58 nM) with TcFPPS based on the X-ray crystallographic structure of 11–TcFPPS have indicated that the nitrogen atom did not coordinate49 the Mg2+ present at the active site of the target enzyme.50,51 The tridentate coordination structure is circumvented to the hydroxyl groups bonded to the phosphorus atoms either for 2-alkylaminoethyl- or 1-hydroxy-1,1-bisphosphonates.52,53 In addition, the X-ray structure of the complex risedronate–TcFPPS indicated that the residue Asp250 forms a hydrogen bond with the hydroxyl group at the C-1 position of risedronate, event not possible with the 2-aminoalkyl derivatives.31,32,54 In connection with the above ideas, it has been postulated that the presence of an electron withdrawing group at C-1 would enhance the ability to coordinate Ca2+ or Mg2+ in a tridentate manner.55–57 Derivatives where the hydroxyl group is absent, that is, replaced by a hydrogen atom would coordinate in a bidentate manner. In fact, we have found that these analogues exhibited less potency as inhibitors towards the target enzyme than 1-hydroxy,34 or 1-amino derivatives.35 In addition, the influence of the hydroxyl group at C-1 on biological action in many bisphosphonates remains unclear; as a matter of fact there exits strong evidence available showing that the hydroxyl group at C-1 does not interact with Mg2+ at the active site of FPPS suggesting that this group would influence the capacity of the adjacent bisphosphonic moiety to chelate Mg2+ and to increase the pKa of the gem-phopshonate functionality as well.
The introduction of a fluorine atom at the C-1 position of an 1-alkyl-1,1-bisphosphonate derivative such as 8 would seem of interest for a number of reasons: (a) isosteric replacement of hydroxyl groups or hydrogen atoms by fluorine atoms is usually employed in drug design based on the stability of the carbon–fluorine bond and the small atomic size that is benefit for molecular recognition;58 (b) the presence of a fluorine atom would increase metabolic stability; (c) 1-fluoro-1,1-bisphosphonic acids would mimic quite well pKa values of inorganic pyrophophoric acid;59–62 (d) these fluorine-containing bisphosphonic acid derivatives would increase acidity in at least one order of magnitude compared to conventional ones;63 (e) it has been recently depicted that some fluorine-containing bisphosphonate having an aromatic ring at the side chain proven to be inhibitors of the enzymatic activity of human FPPS.64
Results and Discussion
Formation of a carbon–fluorine bond can be envisioned through a reaction between an appropriate carbanion and an electrophilic fluorine donor such as Selectfluor (1-chloromethyl-4-fluoro-1,4-diazobicyclo[2.2.2]octane bis(tetrafluoroborate), F-TEDABF4).65,66 Sodium hydride has proven to be a good base to form a carbanion at the carbon bridge of a gem-phosphonate unit to react with Selectfluor™ to afford the desired monofluoro bisphosphonate derivatives.59,67,68 There were two synthetic approaches to follow in order to access to our title compounds (α-fluorinated bisphosphonates derived from fatty acids) taken 7–12 as reference structures (Chart 2): (a) to employ the already depicted tetraethyl 1-(fluoromethylidene)-1,1-bisphosphonate (compound 14) as a common synthetic intermediate.67 Alkylation of the respective enolate-type carbanion of 1467 by treatment with different alkyl halides followed by hydrolysis of the alkylated α-fluoro-tetraethyl esters would yield the title compounds; (b) fluorination of the already depicted tetraethyl 1-alkyl-1,1-bisphosphonates34 followed by hydrolysis of the resulting products would be the latter approach. Although it would be possible to obtain the corresponding precursors of the title compounds from the simplest fluorine-containg intermediate (compound 14) via an alkylation reaction as recently described;68 however, the first route resulted to be impractical from the synthetic point of view, especially, this transformation was always associated to low reaction yields. The presence of a fluorine atom would increase the acidity of the hydrogen atom bonded at C-1 resulting in a weak and less reactive conjugate base. We succeeded in preparing the title drugs by using the second approach. Fluorination of compound 13 gave rise to 14 in 50% yield according to the literature.67 We were able to fluorinate all the tetraethyl 1-alkyl-1,1-bisphosphonates in moderate but reproducible yields. Precursors (16–25) were obtained via either a hydrogenation reaction or a Michael addition on the already depicted tetraethyl ethenylidene bisphosphonate 15.69 Hydrolysis by treatment with bromotrimethylsilane in anhydrous methylene chloride followed by digestion with methanol afforded the desired 1-fluoro-1,1-bisphosphonic acids 35–44 (Scheme 2).
Scheme 2.

Reagents and Condition for the Preparation of the Title Compounds.
It is worth mentioning the difficulty to monitore the course of the reaction by t.l.c. These reactions, which involved compounds without chromophore groups and without sensitivity for universal revelators, should be followed by NMR.
Biological evaluation of the title compounds 35–44 was very surprising. All of these compounds were almost devoid of antiparasitic activity against the amastigote form of T. cruzi. Only compound 43 exhibited a marginal activity against intracellular amastigotes with 13% of growth inhibition at a concentration of 10 μM. In addition, these results were consistent with inhibition studies towards the enzymatic activity of FPPS. Then, compounds 35–44 were devoid of activity against T. cruzi FPPS. These data were quite unexpected bearing in mind the inhibition action exhibited by lineal structurally related bisphosphonates.31–36 On the other hand, these fluorine-containing bisphosphonic acids exhibited an extremely potent activity as inhibitors of the enzymatic activity of T. gondii FPPS.70 In fact, relatively aliphatic long chain derivatives such as 40–44 showed an inhibitory action at the nanomolar level. Compounds 43 and 44 arose as main member of these drugs exhibiting an unusual potency against TgFPPS possessing IC50 values of 35 nM and 60 nM, respectively, and were more effective than risedronate (IC50 = 74 nM) taken as positive control. This high selectively towards TgFPPS versus TcFPPS might be understood by comparison of the amino acid sequence between both of these enzymes having less than 50% of identity.71 Compound 43 was able to control growth of tachyzoites of T. gondii exhibiting an IC50 value of 2.67 μM (Table 1).
Table 1.
| Comp | IC50 (μM) | IC50 (μM) | IC50 (μM) | IC50 (μM) |
|---|---|---|---|---|
|
| ||||
| TcFPPS | TgFPPS | T. cruzi amastigotes | T. gondii tachyzoites | |
| 35 | >1 | >10 | >10 | >10 |
| 36 | >1 | >10 | >10 | >10 |
| 37 | >1 | >10 | >10 | >10 |
| 38 | >1 | >10 | >10 | >10 |
| 39 | >1 | 3.42 ± 1.67 | >10 | >10 |
| 40 | >1 | 0.160 ± 0.078 | >10 | >10 |
| 41 | >1 | 0.241 ± 0.120 | >10 | >10 |
| 42 | >1 | 0.616 ± 0.259 | >10 | >10 |
| 43 | >1 | 0.035 ± 0.019 | 13% at 10 μM | 2.67 |
| 44 | >1 | 0.060 ± 0.006 | >10 | >10 |
| Risedronate | 0.027 ± 0.003 | 0.074 ± 0.017 | - | |
| benznidazole | - | 2.768 ± 0.488 | ||
In summary, lineal α-fluoro-1,1-bisphosphonic acids represent an interesting new family of bisphosphonates that was able to efficiently and selectively inhibit the enzymatic activity of T. gondii farnesyl pyrophosphate synthase. Particularly, compound 43, which contains nine carbon atoms in the aliphatic chain, exhibited superior activity against T. gondii FPPS. As a result of this enzymatic action, this compound was able to impair tachyzoites of T. grondii growth. Structural variation in the aliphatic chain that includes the addition of conformational restriction tools will be considered in the near future in order to optimize its structure. Work aimed at exploiting the potential usefulness of these drugs is currently being pursued in our laboratory.
Experimental Section
The glassware used in air and/or moisture sensitive reactions was flame-dried and carried out under a dry argon atmosphere. Unless otherwise noted, chemicals were commercially available and used without further purification. Solvents were distilled before use. Tetrahydrofuran and ethyl ether were distilled from sodium/benzophenone ketyl. Anhydrous N, N-dimethylformamide was used as supplied from Aldrich. Tetraethyl methylenebisphosphonate and Selectfluor™ were purchased from Aldrich.
Nuclear magnetic resonance spectra were recorded using a Bruker AM-500 MHz or a Bruker AC-200 MHz spectrometers. Chemical shifts are reported in parts per million (δ) relative to tetramethylsilane. Coupling constants are reported in Hertz. 13C-NMR spectra were fully decoupled. 31P-NMR spectra were fully decoupled. Chemical shifts are reported in parts per million relative to phosphoric acid. Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet.
High resolution mass spectra were conducted using a Bruker micrOTOF-Q II spectrometer, which is a hybrid quadrupole time of flight mass spectrometer with MS/MS capability. Low-resolution mass spectra were obtained on a VG TRIO 2 or on a VG ZAB2-SEQ instruments in electron impact mode at 70 eV (direct inlet).
Melting points were determined using a Fisher-Johns apparatus and are uncorrected. IR spectra were recorded using a Nicolet Magna 550 spectrometer.
Column chromatography was performed with E. Merck silica gel (Kieselgel 60, 230–400 mesh). Analytical thin layer chromatography was performed employing 0.2 mmcoated commercial silica gel plates (E. Merck, DC-Aluminum sheets, Kieselgel 60 F254).
Elemental analyses were performed by Atlantic Microlab, Norcross, Georgia, USA.
As judged from the homogeneity of the 1H, 13C, 31P NMR spectra of the title compounds 35–44 and HPLC analyses of the committed intermediates 14, 16–18, 20–25, 26–34, employing a Beckmann Ultrasphere ODS-2 column 5 μM, 250 × 10 mm eluting with acetonitrile–water (1:1) at 3.00 mL/min with a refractive index detector indicated a purity >97%.
Tetraethyl Ethylidene-1,1-bisphosphonate (16)
A solution of 15 (200 mg, 0.66 mmol) in ethyl acetate (10 mL) in the presence of 10% palladium on charcoal (30 mg) was treated with hydrogen under 3 atm at room temperature for 4 h. Then the reaction mixture was filtered and the solvent was evaporated to afford 190 mg (95% yield) of compound 16 as a colorless oil: Rf 0.20 (EtOAc); IR (film, cm−1) 2985, 1645, 1251, 1022, 968, 819; 1H NMR (200.13 MHz, CDCl3) δ 1.34 (t, J = 7.1 Hz, 12H, H-2′), 1.46 (dt, J = 17.4, 7.3 Hz, 3H, H-2), 2.39 (tq, J = 30.8, 7.2 Hz, 1H, H-1), 4.18 (m, 8H, H-1′); 13C NMR (50.33 MHz, CDCl3) δ 10.1 (t, J = 6.1 Hz, C-2), 16.3 (d, J = 5.4 Hz, C-2′), 31.0 (t, J = 136.3 Hz, C-1), 62.46 (d, J = 6.8, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 21.47; MS (m/z, relative intensity) 302 (M+, 1), 275 (5), 247 (6), 191 (13), 165 (100).
Tetraethyl n-Propylidene-1,1-bisphosphonate (17)
A solution of 15 (200 mg, 0.66 mmol) in anhydrous tetrahydrofuran (10 mL) cooled at −78 °C under argon atmosphere was treated with 1.6 M of methyllithium in diethyl ether (0.4 mL). The reaction mixture was stirred at 0 °C for 2 h. the reaction was worked up by addition of an aqueous saturated solution of ammonium chloride (10 mL). The aqueous phase was extracted with chloroform (3 × 10 mL). The combined organic layers were dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAct (9:1) as eluent to give 83.2 mg (40% yield) of 17 as a colorless oil: Rf 0.67 (CH2Cl2–iPrOH, 19:1); IR (film, cm−1) 3664, 2983, 1246, 1024, 962; 1H NMR (200.13 MHz, CDCl3) δ 1.15 (t, J = 7.3 Hz, 3H, H-3), 1.33 (t, J = 7.1 Hz, 12H, H-2′), 1.94 (m, 2H, H-2), 2.20 (tt, J = 23.9, 5.8 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 13.9 (dd, J = 6.3, 3.6 Hz, C-2), 16.4 (dd, J = 6.8, 3.4 Hz, C-2′), 19.1 (t, J = 5.5 Hz, C-3), 38.2 (t, J = 133.5 Hz, C-1), 62.7 (dd, J = 15.4, 6.4 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) 23.84; MS (m/z, relative intensity) 316 (M+, 1), 301 (11), 288 (17), 261 (14), 233 (15), 179 (100), 123 (45).
Tetraethyl n-Butylidene-1,1-bisphosphonate (18)
A solution of ethyl iodide (1.0 mL, 8.3 mmol) in anhydrous ethyl ether (10 mL) was added to magnesium turnings (204.8 mg, 8.3 mmol) in the presence of iodine (30 mg) under argon atmosphere. The reaction mixture was stirred at room temperature for 2 h. The resulting dark gray mixture was added slowly via cannula to a solution of 15 (500 mg, 1.6 mmol) in ethyl ether (10 mL) cooled at 0 °C. The mixture was stirred for 2 h and the reaction was quenched as depicted for the preparation of 17. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAc (4:1) to afford 450 mg (85% yield) of 18 as a colorless oil: Rf 0.72 (AcOEt–iPrOH–H2O, 6:3:1); IR (film, cm−1) 2980, 1394, 1245, 1165, 1033, 969; 1H NMR (200.13 MHz, CDCl3) δ 0.89 (t, J = 7.3 Hz, 3H, H-4), 1,31 (t, J = 6.9 Hz, 12H, H-2′), 1.55 (m, 2H, H-3), 1.86 (m, 2H, H-2), 2.25 (tt, J = 24.1, 5.8 Hz, 1H, H-1), 4.14 (m, 8H, H-1′); 13C NMR (50.33 MHz, CDCl3) δ 13.7 (C-4), 16.3 (d, J = 6.8 Hz, C-2′), 26.3 (t, J = 6.8 Hz, C-2), 27.5 (t, J = 4.7 Hz, C-3), 36.5 (t, J = 133.6 Hz, C-1), 62.4 (d, J = 7.5 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.15; MS (m/z, relative intensity) 331 ([M+1]+, 11), 301 (34), 288 (69), 261 (38), 193 (100), 165 (53), 152 (67), 137 (81). Anal. Calcd for C12H28O6P2.H2O: C 41.38, H 8.68; Found C 41.64, H 8.78.
Tetraethyl n-Pent-4-enylidene-1,1-bisphosphonate (19)
A solution of allyl chloride (0.4 mL, 5.0 mmol) in anhydrous tetrahydrofuran (10 mL) was added to magnesium turnings (121.5 mg, 5.0 mmol) in the presence of iodine (20 mg) under argon atmosphere. The mixture was stirred at room temperature for 2 h. Then the mixture was cooled at 0 °C and a solution of 15 (415.7 mg, 1.38 mmol) in tetrahydrofuran (5 mL) was added. The reaction mixture was treated as described for the preparation of 18. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAc (1:1) to afford 150 mg (31% yield) of 19 as a colorless oil: Rf 0.75 (AcOEt–iPrOH–H2O, 6:3:1); IR (film, cm−1) 2983, 2933, 2910, 1641, 1456, 1392, 1247, 1165, 1024, 972; 1H NMR (200.13 MHz, CDCl3) δ 1.35 (t, J = 7.1 Hz, 12H, H-2′), 2.19 (m, 5H, H-1, H-2, H-3); 4.19 (m, 8H, H-1′), 5.07 (m, 2H, H-5), 5.77 (ddt, J = 17.1, 10.2, 6.7 Hz, 1H, H-4); 13C NMR (50.33 MHz, CDCl3) δ 16.3 (t, J = 6.8 Hz, C-2′), 24.6 (t, J = 4.7 Hz, C-2), 32.7 (t, J = 6.1 Hz, C-3), 35.6 (t, J = 133.6, C-1), 62.5 (t, J = 6.8 Hz, C-1′), 116.0 (C-5), 137.1 (C-4); 31P NMR δ (202.45 MHz, CDCl3) 24.13; MS (m/z, relative intensity) 343 ([M+1]+, 5), 288 (70), 261 (51), 233 (45), 205 (100), 152 (92), 109 (50).
Tetraethyl n-Pentylidene-1,1-bisphosphonate (20)
A solution of 19 (100 mg, 0.29 mmol) in ethyl acetate (10 mL) in the presence of 10% palladium on charcoal (30 mg) was treated as depicted for the preparation of compound 16 to give 85.1 mg (85% yield) of 20 as a colorless oil: IR (film, cm−1) 2960, 1662, 1471, 1247, 1024, 970; 1H NMR (500.13 MHz, CDCl3) δ 0.91 (t, J = 7.1 Hz, 3H, H-5), 1.35 (t, J = 7.1, 12H, H-2′), 1.73 (m, 6H, -CH2-), 2.28 (tt, J = 24.1, 6.0 Hz, 1H, H-1) 4.17 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 13.7 (C-5), 16.3 (dd, J = 6.4, 2.3 Hz, C-2′), 22.4 (C-4), 25.2 (t, J = 5.0 Hz, C-3), 31.2 (t, J = 6.4 Hz, C-2), 36.7 (t, J = 133.4 Hz, C-1), 62.4 (dd, J = 18.5, 6.7 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.14; MS (m/z, relative intensity) 344 (M+, 5), 301 (12), 288 (28), 207 (21), 152 (27), 57 (46), 41 (100).
Tetraethyl n-Hexylidene-1,1-bisphosphonate (21)
A solution of butyl chloride (0.42 mL, 4.3 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with magnesium turnings (100 mg, 4.3 mmol) as depicted for the preparation of 19. Purification by column chromatography (silica gel) eluting with hexane–EtOAc (4:1) afforded 48 mg (33% yield) of 21 as a colorless oil: Rf 0.20 (AcOEt–iPrOH, 19:1); IR (film, cm−1) 2983, 1662, 1458, 1394, 1247, 1165, 1026, 972, 804; 1H NMR (200.13 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 3H, H-6), 1.33 (t, J = 7.1 Hz, 12H, H-2′), 1,25-1,4 (m, 4H, H-5, H-4) 1.59 (m, 2H, H-3), 1.91 (m, 2H, H-2), 2.26 (tt, J = 24.1, 5.8 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (50.33 MHz, CDCl3) δ 13.9 (C-6), 16.3 (d, J = 5.4 Hz, C-2′), 22.2 (C-5), 25.5 (t, J = 4.8 Hz, C-3), 28.8 (t, J = 6.1 Hz, C-2), 31.5 (C-4), 36.8 (t, J = 132.9 Hz, C-1), 62.4 (t, J = 6.8 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.16; MS (m/z, relative intensity) 359 ([M+1]+, 2), 301 (39), 288 (97), 261 (47), 233 (32), 221 (100), 165 (55), 152 (88). Anal. Calcd for C14H32O6P2.H2O: C 44.68, H 9.11; Found C 44.97, H 8.71.
Tetraethyl n-Heptylidene-1,1-bisphosphonate (22)
A solution of n-pentyl bromide (0.62 mL, 5,0 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with magnesium turnings (121.5 mg, 5.0 mmol) as described for the preparation of 19. The residue was purified by column chromatography (silica gel) eluting with hexane–EtOAc (4:1) to yield 63.2 mg (17% yield) of 22 as a colorless oil: Rf 0.23 (AcOEt–iPrOH, 19:1); IR (film, cm−1) 2981, 2929, 2860, 1647, 1471, 1394, 1249, 1165, 1026, 970, 839, 792; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H, H-7), 1.34 (t, J = 7.1 Hz, 12H, H-2′), 1.30 (m, 6H, -CH2-), 1.55 (p, J = 7.4 Hz, 2H, H-3), 1.91 (m, 2H, H-2), 2.27 (tt, J = 24.2, 6.0 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 14.0 (C-7), 16.4 (dd, J = 6.6, 3.0 Hz, C-2′), 22.5 (C-6), 25.5 (t, J = 5.5 Hz, C-3), 28.9 (C-4), 29.1 (t, J = 6.4 Hz, C-2), 31.4 (C-5), 36.7 (t, J = 133.2 Hz, C-1), 62.4 (dd, J = 18.5, 6.8 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.16; MS (m/z, relative intensity) 373 ([M+1]+, 10), 301 (39), 288 (90), 261 (40), 235 (100), 207 (24), 189 (15), 179 (42), 165 (24), 152 (72), 137 (27), 125 (21). Anal. Calcd for C15H34O6P2: C 46.92, H 9.00; Found C 47.41, H 9.13.
Tetraethyl n-Octylidene-1,1-bisphosphonate (23)
A solution of n-hexyl bromide (0.70 mL, 5.0 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with magnesium turnings (121.5 mg, 5.0 mmol) as depicted for the preparation of 19. The product was purified by column chromatography (silica gel) eluting with hexane–EtOAc (2:3) to afford 112.6 mg (30% yield) of 23 as a colorless oil: Rf 0.22 (AcOEt–iPrOH, 19:1); IR (film, cm−1) 2927, 2856, 1652, 1465, 1394, 1247, 1165, 1026, 970, 856, 800; 1H NMR (500.13 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H, H-8), 1.32 (m, 20H, -CH2-, H-2′), 1.55 (m, 2H, H-3), 1.92 (m, 2H, H-2), 2.27 (tt, J = 24.2, 6.1 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 14.1 (C-8), 16.4 (dd, J = 5.9, 2.9 Hz, C-2′), 22.6 (C-7), 25.5 (t, J = 5.4 Hz, C-3), 28.9 (C-4), 29.1 (t, J = 6.3 Hz, C-2), 29.3 (C-5), 31.7 (C-6), 36.8 (t, J = 133.5 Hz, C-1) 62.4 (dd, J = 18.8, 6.7 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.15; MS (m/z, relative intensity) 387 ([M+1]+, 3), 301 (42), 288 (100), 261 (37), 249 (96), 193 (30), 152 (67). Anal. Calcd for C16H36O6P2.0.2H2O: C 49.27, H 9.41; Found C 49.21, H 9.61.
Tetraethyl n-Nonylidene-1,1-bisphosphonate (24)
A solution of n-heptyl chloride (0.76 mL, 5.0 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with magnesium turnings (121.5 mg, 5.0 mmol) as described for the preparation of 19. Column chromatography purification eluting with hexane–EtOAc (9:1) afforded 160 mg (40% yield) of 24 as a colorless oil: Rf 0.28 (AcOEt–iPrOH, 19:1); IR (film, cm−1) 2927, 2856, 1647, 1467, 1444, 1392, 1251, 1165, 1024, 970, 835, 794; 1H NMR (200.13 MHz, CDCl3) δ 0.87 (t, J = 6.4 Hz, 3H, H-9), 1.32 (m, 10H, -CH2-), 1.34 (t, J = 7.1 Hz, 12H, H-2′), 1.56 (m, 2H, H-3), 1.90 (m, 2H, H-2), 2.26 (tt, J = 23.9, 6.0 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (50.33 MHz, CDCl3) δ 14.0 (C-9), 16.4 (d, J = 6.8 Hz, C-2′), 22.6 (C-8), 25.5 (t, J = 5.4 Hz, C-3), 25.7 (C-4), 29.1 (t, J = 6.5 Hz, C-2), 29.2 (C-5),* 29.3 (C-6),* 31.8 (C-7), 36.7 (t, J = 133.6 Hz, C-1), 62.4 (t, J = 7.5 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.15; MS (m/z, relative intensity) 401 ([M+1]+, 28), 301 (38), 288 (100), 263 (90), 261 (35), 152 (58). Anal. Calcd for C17H38O6P2.H2O: C 48.80, H 9.64; Found C 48.48, H 9.57.
Tetraethyl n-Decylidene-1,1-bisphosphonate (25)
A solution of n-octyl chloride (0.85 mL, 5.0 mmol) in anhydrous tetrahydrofuran (10 mL) was treated with magnesium turnings (121.5 mg, 5.0 mmol) as described for the preparation of 19. The product was purified by column chromatography (silica gel) eluting with hexane–EtOAc (19:1) to afford 186.3 mg (45% yield) of 25 as a colorless oil: Rf 0.12 (hexane–EtOAc, 4:1); IR (film, cm−1): 2979, 2929, 2873, 1458, 1392, 1251, 1165, 1026, 968, 813; 1H NMR (200.13 MHz, CDCl3) δ 0.89 (t, J = 6.7 Hz, 3H, H-10), 1.28 (m, 12H, -CH2-), 1.34 (t, J = 6.7 Hz, 12H, H-2′), 1.51 (m, 2H, H-3), 1.89 (m, 2H, H-2), 2.22 (tt, J = 24.3, 5.8 Hz, 1H, H-1), 4.17 (m, 8H, H-1′); 13C NMR (50.33 MHz, CDCl3) δ 14.0 (C-10), 16.3 (d, J = 6.8 Hz, C-2′), 23.0 (C-9), 21.7 (t, J = 5.4 Hz, C-3), 29.1 (t, J = 6.2 Hz, C-2), 29.5 (C-4, C-7), 28,7 (C-5, C-6), 32.4 (C-8), 37.0 (t, J = 133.6 Hz, C-1), 62.3 (t, J = 6.1 Hz, C-1′); 31P NMR (202.45 MHz, CDCl3) δ 24.15; MS (m/z, relative intensity) 415 ([M+1]+, 20), 385 (5), 371 (5), 357 (6), 315 (10), 301 (28), 288 (100), 277 (67), 261 (32), 165 (23), 152 (49). Anal. Calcd for C18H40O6P2.H2O: C 49.99, H 9.79; Found C 50.39, H 9.76.
Fluorination Reaction. General procedure
Sodium hydride (10.5 mmol, 60% in mineral oil) was washed with anhydrous hexane under an argon atmosphere, and then anhydrous tetrahydrofuran (190 mL) was added. The suspension was cooled at 0 °C, and, in independent experiments, each tetraethyl 1-alkyl-1,1-bisphosphonates such as 14, 16–18, 20–25 (10.0 mmol) in anhydrous tetrahydrofuran (10 mL) were added. The solution was stirred at 0 °C for 15 min, then, the mixture was allowed to reach room temperature and was stirred for 60 min. The mixture was cooled at 0 °C and Selectfluor™ (12.5 mmol) was added in one portion followed by addition of anhydrous N, N-dimethylformamide (3.5 mL). The reaction mixture was allowed to reach room temperature and was stirred for 4 h. The mixture was partitioned between methylene chloride (30 mL) and an aqueous saturated solution of ammonium chloride (30 mL). The aqueous phase was extracted with methylene chloride (2 × 30 mL). The combined organic layers were washed with water (2 × 30 mL), dried (MgSO4), and the solvent was evaporated. The residue was purified by column chromatography eluting with mixtures of (EtOAc–CH3OH) in a ratio indicated in each case to afford the corresponding 1-fluoro tetraethyl ester derivative (compounds 14, 26–34).
Tetraethyl 1-(Fluoromethylidene)-1,1-bisphosphonate (14)
50% Yield; Colorless oil; IR (film, cm−1) 2987, 2935, 2914, 1479, 1444, 1394, 1371, 1261, 1164, 1028, 981, 869, 794; 1H NMR (500.13 MHz, CDCl3) δ 1.38 (dt, J = 7.1, 1.4 Hz, 12H, H-2′), 4.29 (m, 8H, H-1′), 5.01 (dt, J = 45.9, 13.9 Hz, H-1); 13C NMR (50.3 MHz, CDCl3) δ 15.8 (C-2′), 63.9 (dt, J = 17.7, 2.7 Hz, Hz, C-1′), 82.78 (dt, J = 192.0, 157.1 Hz, C-1), 31P NMR (202.46 MHz, CDCl3) δ 11.08 (d, J = 74.6 Hz); HRMS (ESI) Calcd. for (C9H21O6P2F) [M+Na]+: 329.0695; found 329.0697.
Tetraethyl 1-(Fluoroethylidene)-1,1-bisphosphonate (26)
43% Yield; Colorless oil; IR (film, cm−1) 2984, 2935, 2874, 1445, 1393, 1369, 1256, 1165, 1024, 970, 845, 816, 793, 536; 1H NMR (500.13 MHz, CDCl3) δ 1.37 (d, J = 7.1 Hz, 12H, H-2′), 1.83 (dt, J = 25.7, 15.4 Hz, 3H, H-2), 4.28 (m, 8H, H-1′); 13 C NMR (125 MHz, CDCl3) δ 16.4 (dd, J = 7.1, 2.9 Hz, C-2), 19.1 (d, J = 20.9 Hz, C-2′), 64.0 (dt, J = 28.0, 3.2 Hz, C-1′), 93.0 (dt, J = 183.9, 160.5 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 14.92 (d, J = 72.1 Hz). HRMS (ESI) Calcd. for (C10H24O6P2F) [M+H]+: 321.1032; found 321.1022.
Tetraethyl 1-(Fluoro-n-propylidene)-1,1-bisphosphonate (27)
36% Yield; Colorless oil; IR (film, cm−1) 2981, 2933, 1444, 1394, 1369, 1263, 1164, 1022, 972, 784, 769; 1H NMR (500.13 MHz, CDCl3) δ 1.17 (t, J = Hz, 3H, H-3), 1.37 (t, J = Hz, 12H, H-2′), 2.24 (m, 2H, H-2), 4.29 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 7.8 (dd, J = 12.7, 4.6 Hz,), 16.4 (dt, J = 5.9, 3.0 Hz), 26.5 (d, J = 20.9 Hz, C-2′), 63.8 (dt, J = 28.2, 3.2 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 15.00 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C11H25FO6P2Na) [M+Na]+: 357.1008; Found 357.1013.
Tetraethyl 1-(Fluoro-n-butylidene)-1,1-bisphosphonate (28)
47% Yield; Colorless oil; IR (film, cm−1) 2981, 2935, 2875, 1649, 1444, 1392, 1369, 1261, 1164, 1022, 972, 794, 746; 1H NMR (500.13 MHz, CDCl3) δ 0.97 (t, J = 7.3 Hz, 3H, H-4), 1.38 (t, J = 7.0 Hz, 12H, H-2′), 1.68 (m, 2H, H-3), 2.14 (dtt, J = 47.0, 15.1, 8.6 Hz, 2H, H-2), 4.25 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 14.52 (C-4), 16.43 (p, J = 2.9 Hz, C-2′), 16.54 (q, J = 5.4 Hz), 35.29 (d, J = 20.0 Hz), 63.87 (dt, J = 27.4, 2.4 Hz, C-1′), 95.86 (dt, J = 187.3, 156.2 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 14.52 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C12H28FO6P2) [M+H]+: 349.1345; Found: 349.1348.
Tetraethyl 1-(Fluoro-n-pentylidene)-1,1-bisphosphonate (29)
49% Yield; Colorless oil; IR (film, cm−1) 2964, 2934, 2874, 1445, 1393, 1261, 1165, 1024, 975, 797, 540; 1H NMR (500.13 MHz, CDCl3) δ 0.93 (t, J = 7.4 Hz, 3H, H-5), 1.37 (t, J = 7.1 Hz, 12H, H-2′), 1.70 (br s, 2H), 2.17 (m, 2H, H-2), 4.27 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 13.76, 14.43 (q, J = 2.9 Hz, C-2′), 23.09, 25.04 (t, J = 5.5. Hz), 32.99 (d, J = 20.1 Hz), 63.86 (dt, J = 27.2, 3.2 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 15.79 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C13H30O6P2F) [M+H]+: 363.1502; found 363.1508.
Tetraethyl 1-(Fluoro-n-hexylidene)-1,1-bisphosphonate (30)
57% Yield; Colorless oil; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 7.1 Hz, 3H, H-5), 1.30 (m, 8H, H-4, H-5, H-6, H-7), 1.37 (t, J = 7.1 Hz, 12H, H-2′),1.64 (p, J = 7.2 Hz, 2H, H-3), 2.15 (dt, J = 22.3, 15.3 Hz, H-2), 4.28 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 13.88 (C-6), 16.39 (p, J = 3.0 Hz, C-2′), 22.23 (C-5), 22.57 (q, J = 5.4 Hz, C-3), 32.09 (C-4), 33.17 (d, J = 20.0 Hz, C-2), 63.81 (dt, J = 27.2, 3.2 Hz, C-1′), 95.85 (dt, J = 187.1, 156.2 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 15.02 (d, J = 74.6 Hz).
Tetraethyl 1-(Fluoro-n-heptylidene)-1,1-bisphosphonate (31)
35% Yield; Colorless oil; IR (film, cm−1) 2984, 2961, 2932, 2872, 2858, 1456, 1393, 1369, 1261, 1164, 1022, 976, 794, 576; 1H NMR (500.13 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H, H-7), 1.30 (m, 6H, H-4, H-5, H-6), 1.36 (t, J = 7.1 Hz, 12H, H-2′), 1.64 (m, 2H, H-3), 2.15 (m, 2H, H-2), 4.26 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 13.98 (C-7), 16.38 (p, J = 2.7 Hz, C-2′), 22.45 (C-6), 22.84 (q, J = 5.4 Hz, C-3), 29.59 (C-5), 31.38 (C-4), 33.21 (d, J = 20.1 Hz, C-2), 63.78 (dt, J = 27.2, 3.2 Hz, C-1′), 95.84 (dt, J = 188.0, 156.2 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 14.76 (d, J = 75.1 Hz). Calcd. for (C15H34FO6P2) [M+Na]+: 413.1634; found: 413.1635.
Tetraethyl 1-(Fluoro-n-octylidene)-1,1-bisphosphonate (32)
48% Yield; Colorless oil; IR (film, cm−1) 2982, 2959, 2930, 2858, 2358, 1452, 1393, 1258, 1165, 1026, 970, 797, 580, 538; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H, H-8), 1.30 (t, J = 7.1 Hz, 12H, H-2′), 2.15 (dtt, J = 23.8, 15.3, 8.4 Hz, 2H, H-2), 4.27 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 14.06 (C-8), 16.46 (p, J = 3.1 Hz, C-2′), 22.59 (C-7), 22.96 (q, J = 5.3 Hz, C-3), 28.90 (C-6), 29.93 (C-5), 31.70 (C-4), 33.27 (d, J = 19.8 Hz, C-2), 63.87 (dt, J = 27.3, 2.8 Hz, C-1′), 95.84 (dt, J = 188.0, 156.2 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 14.58 (d, J = 74.6 Hz). (F8OEt) HRMS (ESI) Calcd. for (C16H36O6P2F) [M+H]+: 405.1971; found 405.1977.
Tetraethyl 1-(Fluoro-n-nonylidene)-1,1-bisphosphonate (33)
24% Yield; Colorless oil; IR (film, cm−1) 2981, 2959, 2928, 2856, 1467, 1392, 1369, 1261, 1163, 1024, 974, 797, 663, 577, 540; 1H NMR (500.13 MHz, D2O) δ 0.88 (t, J = 7.0 Hz, 3H, H-9), 1.30 (m, 10H), 1.37 (t, J = 7.1 Hz, 12H, H-2′), 1.64 (p, J = 7.3 Hz, 2H, H-3), 2.15 (dtt, J = 47.3, 15.3, 8.3 Hz, 2H, H-2), 4.27 (m, 8H, H-1′); 13C NMR (125.77 MHz, CDCl3) δ 14.06 (C-9), 16.43 (p, J = 2.7 Hz, C-2′), 22.62 (C-8), 22.93 (q, J = 5.4 Hz, C-3), 29.15 (C-7), 29.19 (C-6), 29.98 (C-5), 31.80 (C-4), 33.25 (d, J = 20.1 Hz, C-2), 63.83 (dt, J = 27.3, 2.7 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 15.01 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C17H38O6P2F) [M+H]+: 419.2128; found 419.2131.
Tetraethyl 1-(Fluoro-n-decylidene)-1,1-bisphosphonate (34)
33% Yield; colorless oil; IR (film, cm−1) 2982, 2961, 2926, 2856, 1468, 1450, 1393, 1369, 1261, 1165, 1024, 976, 797, 663, 540; 1H NMR (500.13 MHz, CDCl3) δ 0.90 (t, J = 6.9 Hz, 3H, H-10), 1.32 (m, 6H, CH2), 1.39 (t, J = 7.1 Hz, 8H, H-2′), 1.63 (m, 2H, H-3), 2.12 (dtt, J = 47.4, 15.4, 8.4 Hz, 2H, H-2), 4.26 (m, 8H, H-1′); 13 C NMR (125.77 MHz, CDCl3) δ 14.1 (C-10), 16.5 (p, J =2.9 Hz, C-2′), 22.6 (C-9), 23.0 (q, J = 5.5 Hz, C-3), 29.2 (C-6), 29.5 (C-7), 30.0 (C-5), 31.9 (C-4), 33.3 (d, J = 19.6 Hz, C-2), 63.9 (dt, J = 27.9, 3.2 Hz, C-1′); 31P NMR (202.46 MHz, CDCl3) δ 14.78 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C10H39O6P2FNa) [M+Na]+: 455.2104; found 455.2105.
Silylation/Methanolysis. General procedure
To a solution of the resulting tetraethyl ester (14, 26–34; 1 equivalent) in anhydrous methylene chloride was added dropwise trimethylsilyl bromide (10 equivalents) in an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h. After cooling at 0 °C, anhydrous methanol (10 mL) was added, and the resulting mixture was allowed to reach room temperature. The solution was then concentrated under reduced pressure. The residue was dissolved in dry methanol (10 mL) and subsequently concentrated under reduced pressure twice. The solvent was evaporated and the residue was purified by reverse phase column chromatography or crystallized from ethanol–water to afford the title compounds 35–44.
1-(Fluoromethylene)-1,1-bisphosphonic acid (35)
93% Yield; syrup; 1H NMR (500.13 MHz, D2O) δ 4.78 (dt, J = 45.1, 12.7 Hz, H-1); 13C NMR (50.3 MHz, DMSO-d6) δ 86.2 (dt, J = 185.3, 148.4 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 10.06 (d, J = 64.7 Hz); HRMS (ESI) Calcd. for (CH5O6P2F) [M+Na]+: 216.9443; found 216.9438.
1-(Fluoroethylidene)-1,1-bisphosphonic acid (36)
96% Yield; syrup; IR (KBr, cm−1) 3406, 2985, 2929, 2866, 1731, 1681, 1400, 1220, 1024, 935, 825, 767, 628, 518; 1H NMR (500.13 MHz, D2O) δ 1.39 (dt, J = 27.3, 14.8 Hz, H-2); 13C NMR (125 MHz, CD3OD) δ 19.8 (d, J = 20.9 Hz, C-2), 94.21 (dt, J = 180.3, 152.8 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 12.24 (d, J = 69.7 Hz). HRMS (ESI) Calcd. for (C2H8O6P2F) [M+H]+: 208.9780; found 208.9780.
1-(Fluoro-n-propylidene)-1,1-bisphosphophonic acid (37)
96% Yield; syrup; IR (KBr, cm−1) 3604, 2995, 2950, 2931, 2314, 1728, 1703, 1467, 1205, 1022, 956, 763, 626; 1H NMR (500.13 MHz, D2O) δ 1.01 (t, J = 7.5 Hz, 3H, H-3), 2.07 (dtt, J = 45.7, 15.0, 7.7 Hz, 2H, H-2); 13C NMR (125.77 MHz, DMSO-d6) δ 9.41 (d, J = 5.4 Hz, C-3), 27.18 (d, J = 20.9 Hz, C-2), 97.86 (dt, J = 180.7, 141.2 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 14.16 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C3H9FO6P2Na) [M+Na]+: 244.9756; Found: 244.9748.
1-(Fluoro-n-butylidene)-1,1-bisphosphonic acid (38)
90% Yield; syrup; 1H NMR (500.13 MHz, CDCl3) δ 0.85 (t, J = 7.3 Hz, 3H, H-4), 1.55 (m, 2H, H-3), 1.96 (m, 2H, H-2); 13C NMR (125.77 MHz, CDCl3) δ 14.63, 16.62 (q, J = 5.6 Hz), 34.51 (d, J = 19.5 Hz), 94.73 (dt, J = 184.1, 146.7 Hz, C-1); 31P NMR (202.46 MHz, CDCl3) δ 13.20 (d, J = 72.1 Hz); HRMS (ESI) Calcd. for (C4H11O6P2F) [M+Na]+: 258.9913; found 258.9903.
1-(Fluoro-n-pentylidene)-1,1-bisphosphonic (39)
93% Yield; syrup; (KBr, cm−1) 3155, 2962, 2875, 1653, 1468, 1402, 1213, 1022, 943, 660, 520; 1H NMR (500.13 MHz, D2O) δ 0.94 (t, J = 7.3 Hz, 3H, H-5), 1.36 (1.35 (sext, J = 7.4 Hz, 2H, H-4), 1.68 (m, 2H, H-3), 2.17 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.01 (C-5), 22.59 (C-4), 25.19 (q, J = 5.1 Hz, C-3), 32.13 (d, J = 19.1 Hz, C-2) HRMS (ESI) Calcd. for (C5H13O6P2FNa) [M+Na]+: 273.0069; found 273.0069.
1-(Fluoro-n-hexylidene)-1,1-bisphosphonic acid (40)
95% Yield; Syrup; (KBr, cm−1) 3612, 2961, 2924, 2872, 1685, 1462, 1198, 1177, 1140, 1028, 1020, 943, 659, 513; 1H NMR (500.13 MHz, CD3OD) δ 0.90 (t, J = 6.9 Hz, 3H, H-6), 1.34 (m, 4H, H-4, H-5), 1.68 (m, 2H, H-3), 2.15 (m, 2H, H-2); 13C NMR (125.77 MHz, CD3OD) δ 14.39 (C-6), 23.40 (C-5), 24.07 (q, J = 4.5 Hz, C-3), 33.49 (C-4), 33.91 (d, J = 20.0 Hz, C-2); 31P NMR (202.46 MHz, CD3OD) δ 14.55 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C6H16O6P2F) [M+H]+: 265.0406; found 265.0401.
1-(Fluoro-n-heptylidene)-1,1-bisphosphonic acid (41)
99% Yield; syrup; IR (KBr, cm−1) 3611, 2951, 2937, 2916, 2856, 2246, 1712, 1470, 1200, 1120, 1049, 955, 895, 878, 717, 667, 561, 460; 1H NMR (500.13 MHz, D2O) δ 0.77 (t, J = 7.1 Hz, 3H, H-7), 1.34 (m, 6H, H-4, H-5, H-6), 1.51 (p, J = 7.6 Hz, 2H, H-3), 2.15 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.30 (C-7), 21.86.45 (C-6), 22.98 (q, J = 5.5 Hz, C-3), 29.00 (C-5), 30.73 (C-4), 33.42 (d, J = 19.7 Hz, C-2), 95.89 (dt, J = 181.4, 144.5 Hz, C-1); 31P NMR (202.46 MHz, D2O) δ 14.76 (d, J = 72.15 Hz).HRMS (ESI) Calcd. for (C7H17O6P2FNa) [M+Na]+: 301.0382; found 301.0390.
1-(Fluoro-n-octylidene)-1,1-bisphosphonic acid (42)
97% Yield; syrup; IR (KBr, cm−1) 3570, 2959, 2928, 2858, 1695, 1468, 1211, 1016, 960, 662, 567, 513; 1H NMR (500.13 MHz, D2O) δ 0.76 (t, J = 6.9 Hz, 3H, H-8), 1.20 (m, 8H, H-4, H-5, H-6, H-7), 1.51 (p, J = 7.9 Hz, 2H, H-3), 2.01 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.35 (C-8), 21.95 (C-7), 23.07 (q, J = 5.3 Hz, C-3), 28.13 (C-6), 29.32 (C-5), 31.00 (C-4), 33.42 (d, J = 18.8 Hz, C-2); 31P NMR (202.46 MHz, D2O) δ 14.2 (d, J = 74.6 Hz). HRMS (ESI) Calcd. for (C8H20O6P2F) [M+H]+: 293.0719; found 293.0720.
1-(Fluoro-n-nonylidene)-1,1-bisphosphonic acid (43)
96% Yield; white solid; mp 119 °C; IR (KBr, cm−1) 3608, 2950, 2916, 2852, 1469, 1201, 1126, 1053, 1028, 956, 667, 561, 462; 1H NMR (500.13 MHz, D2O) δ 0.74 (t, J = 6.9 Hz, 3H, H-9), 1.18 (m, 10H, H-4, H-5, H-6, H-7, H-8), 1.49 (m, 2H, H-3), 1.98 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.4 (C-9), 22.0 (C-8), 28.4 (C-7), 28.5 (C-6), 29.5 (C-5), 31.1 (C-4), 32.4 (d, J = 20.4 Hz, C-3), 34.9 (C-2); 31P NMR (202.46 MHz, D2O) δ 13.95 (d, J = 69.8 Hz). HRMS (ESI) Calcd. for (C9H21O6P2FNa) [M+Na]+: 329.0695; found 329.0686.
1-(Fluoro-n-decylidene)-1,1-bisphosphonic acid (44)
94% Yield; white solid; mp 58–59 °C; IR (KBr, cm−1) 3626, 2920, 28532, 1470, 1195, 1144, 1080, 1032, 947, 717, 665, 563, 514, 457; 1H NMR (500.13 MHz, D2O) δ 0.70 (t, J = 7.0 Hz, 3H, H-10), 1.13 (m, 8H, CH2), 1.17 (m, 4H, CH2), 1.48 (p, J = 7.1 Hz, 2H, H-3), 2.01 (m, 2H, H-2); 13C NMR (125.77 MHz, D2O) δ 13.37 (C-10), 22.02 (C-9), 23.14 (q, J = 5.4 Hz, C-3), 28.42 (C-8), 28.46 (C-7), 28.66 (C-6), 29.39 (C-5), 31.13 (C-4), 33.41 (d, J = 20.0 Hz, C-2); 31P NMR (202.46 MHz, D2O) δ 14.22 (d, J = 71.6 Hz). HRMS (ESI) Calcd. for (C10H24O6P2F) [M+H]+: 321.1032; found 321.1032.
Drug Screening
T. cruzi amastigotes assays
Gamma-irradiated (2,000 Rads) Vero cells (3.4 × 104 cells/well) were seeded in 96 well plates (black, clear bottom plates from Greiner Bio-One) in 100 μL RPMI media (Sigma) with 10 % FBS. Plates were incubated overnight at 35 °C and 7 % CO2. After overnight incubation, Vero cells were challenged with 3.4 × 105 trypomastigotes/well (CL strain overexpressing a tdTomato red fluorescent protein) in 50 μL volume and incubated for 5 h at 35 °C and 7 % CO2. After infection, cells were washed once with Hanks solution (150 μL/well) to eliminate any extracellular parasites and compounds were added in serial dilutions in RPMI media in 150 μL volumes. Each dilution was tested in quadruplicate. Each plate also contained controls with host cells and no parasites (for background check), controls with two representative drug dilutions and no parasites (for cytotoxicity assays), and controls with parasites and no drugs (positive control). For each plate, benznidazole was also used as a positive control at 3.5 and 1.5 μM. After drug addition, plates were incubated at 35 °C and 7 % CO2. At day 3 post-infection, plates were assayed for fluorescence.72 IC50 values were determined by non-linear regression analysis using SigmaPlot.
T. gondii tachyzoites assays
Experiments on T. gondii tachyzoites were carried out as described previously73 using T.gondii tachyzoites expressing red fluorescence protein.74 Cells were routinely maintained in hTerT cells grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% fetal bovine serum, 2 mM glutamine, 1 mM pyruvate, at 37 °C in a humid 5% CO2 atmosphere. Confluent monolayers grown in 96-well black plates with optical bottoms (Falcon/Becton-Dickinson, Franklin Lakes, NJ) were used and drugs dissolved in the same medium and serially diluted in the plates. Freshly isolated tachyzoites were filtered through a 3 μm filter and passed through a 22 gauge needle, before use. The cultures were inoculated with 104 tachyzoites/well in the same media. The plates were incubated at 37 °C and read daily in a Molecular Devices fluorescence plate reader. To preserve sterility the plates were read with covered lids, and both excitation (510 nm) and emission (540 nm) were read from the bottom.75 For the calculation of the IC50, the percent of growth inhibition was plotted as a function of drug concentration by fitting the values to the function: I = Imax C/(IC50 + C), where I is the percent inhibition, Imax = 100% inhibition, C is the concentration of the inhibitor, and IC50 is the concentration for 50% growth inhibition.
TcFPPS and TgFPPS Assays and Product Analysis
For TcFPPS76–78 100 μL of assay buffer (10 mM Hepes, pH 7.4, 1 mM MgCl2, 2 mM dithiothreitol, 4.7 μM [4-14C]IPP (10 μCi/μmol)), and 55 μM DMAPP were prewarmed to 37 °C. The assay was initiated by the addition of recombinant protein (10–20 ng). The assay was allowed to proceed for 30 min at 37 °C and was quenched by the addition of 6 M HCl (10 μL). The reactions were made alkaline with 6.0 M NaOH (15 μL), diluted in water (0.7 mL), and extracted with hexane (1 mL). The hexane solution was washed with water and transferred to a scintillation vial for counting. One unit of enzyme activity was defined as the activity required to incorporate 1 nmol of [4-14C]IPP into [14-14C]FPP in 1 min.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Research Council of Argentina (PIP 1888), ANPCyT (PICT 2008 #1690), and the Universidad de Buenos Aires (X-191) to J.B.R., and Bunge & Born Foundation to S. H. S., and the U.S. National Institutes of Health to R. D. (AI-082542) and S. N. J. M. (AI-068467). V. S. R. was supported in part by a training grant of the Ellison Medical Foundation to the Center for Tropical and Emerging Global Diseases.
References
- 1.Roelofs AJ, Thompson K, Ebetino FH, Rogers MJ, Coxon FP. Curr Pharm Des. 2010;16:2950–2960. doi: 10.2174/138161210793563635. [DOI] [PubMed] [Google Scholar]
- 2.Reszka AA, Rodan GA. Mini-Rev Med Chem. 2004;4:711–717. [PubMed] [Google Scholar]
- 3.Russell RGG, Rogers MJ. Bone. 1999;25:97–106. doi: 10.1016/s8756-3282(99)00116-7. [DOI] [PubMed] [Google Scholar]
- 4.Reszka AA, Rodan GA. Curr Osteoporos Rep. 2003;1:45–52. doi: 10.1007/s11914-003-0008-5. [DOI] [PubMed] [Google Scholar]
- 5.Fleisch H, Russell RGG, Straumann F. Nature. 1966;212:901–903. doi: 10.1038/212901a0. [DOI] [PubMed] [Google Scholar]
- 6.Fleisch H, Russell RGG, Francis MD. Science. 1969;165:1262–1264. doi: 10.1126/science.165.3899.1262. [DOI] [PubMed] [Google Scholar]
- 7.Francis MD, Russell RGG, Fleisch H. Science. 1969;165:1264–1266. doi: 10.1126/science.165.3899.1264. [DOI] [PubMed] [Google Scholar]
- 8.Urbina JA, Moreno B, Vierkotter S, Oldfield E, Payares G, Sanoja C, Bailey BN, Yan W, Scott DA, Moreno SNJ, Docampo R. J Biol Chem. 1999;274:33609–33615. doi: 10.1074/jbc.274.47.33609. [DOI] [PubMed] [Google Scholar]
- 9.Hughes DE, Wright KR, Uy HL, Sasaki A, Yoneda T, Roodman GD, Mundy GR, Boyce BF. J Bone Miner Res. 1995;10:1478–1487. doi: 10.1002/jbmr.5650101008. [DOI] [PubMed] [Google Scholar]
- 10.Rogers MJ, Frith JC, Luckman SP, Coxon FP, Benford HL, Mönkkönen J, Auriola S, Chilton KM, Russell RGG. Bone. 1999;24:73S–79S. doi: 10.1016/s8756-3282(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 11.Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RGG, Oppermann U. Proc Natl Acad Sci. 2006;103:7829–7834. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J Biol Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 13.Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. ChemMedChem. 2006;1:267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 14.Cheng F, Oldfield E. J Med Chem. 2004;47:5149–5158. doi: 10.1021/jm040036s. [DOI] [PubMed] [Google Scholar]
- 15.Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ. J Pharmacol Exp Ther. 2001;296:235–242. [PubMed] [Google Scholar]
- 16.Coxon FP, Thompson K, Rogers MJ. Curr Op Pharmacol. 2006;6:307–312. doi: 10.1016/j.coph.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Sanders JM, Ghosh S, Chan JMW, Meints G, Wang H, Raker AM, Song Y, Colantino A, Burzynska A, Kafarski P, Morita CT, Oldfield E. J Med Chem. 2004;47:375–384. doi: 10.1021/jm0303709. [DOI] [PubMed] [Google Scholar]
- 18.Reddy R, Dietrich E, Lafontaine Y, Houghton TJ, Belanger O, Dubois A, Arhin FF, Sarmiento I, Fadhil I, Laquerre K, Ostiguy V, Lehoux D, Moeck G, Parr TR, Jr, Rafai Far A. ChemMedChem. 2008;3:1863–1868. doi: 10.1002/cmdc.200800255. [DOI] [PubMed] [Google Scholar]
- 19.Forlani G, Giberti S, Berlicki Ł, Petrollino D, Kafarski P. J Agric Food Chem. 2007;55:4340–4347. doi: 10.1021/jf0701032. [DOI] [PubMed] [Google Scholar]
- 20.Clézardin P, Massaia M. Curr Pharm Des. 2010;16:3007–3014. doi: 10.2174/138161210793563545. [DOI] [PubMed] [Google Scholar]
- 21.Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. Angew Chem Int Ed. 2009;48:2949–2954. doi: 10.1002/anie.200805133. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Cao R, Yin F, Hudock MP, Guo R–T, Krysiak K, Mukherjee S, Gao Y–G, Robinson H, Song Y, No JH, Kyle Bergan K, Leon A, Cass L, Goddard A, Chang T–K, Lin F–Y, Van Beek E, Papapoulos S, Wang AH–J, Kubo T, Ochi M, Mukkamala D, Oldfield E. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coleman RE. British J Cancer. 2008;98:1736–1740. doi: 10.1038/sj.bjc.6604382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth AG, Drescher D, Yang Y, Redmer S, Uhlig S, Arenz C. Angew Chem Int Ed. 2009;48:7560–7563. doi: 10.1002/anie.200903288. [DOI] [PubMed] [Google Scholar]
- 25.Docampo R, Moreno SNJ. Curr Pharm Des. 2008;14:882–888. doi: 10.2174/138161208784041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oldfield E. Acc Chem Res. 2010;43:1216–1226. doi: 10.1021/ar100026v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh S, Chan JMW, Lea CR, Meints GA, Lewis JC, Tovian ZS, Flessner RM, Loftus TC, Bruchhaus I, Kendrick H, Croft SL, Kemp RG, Kobayashi S, Nozaki T, Oldfield E. J Med Chem. 2004;47:175–187. doi: 10.1021/jm030084x. [DOI] [PubMed] [Google Scholar]
- 28.Martin MB, Grimley JS, Lewis JC, Heath HT, III, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SNJ, Docampo R, Croft SL, Oldfield E. J Med Chem. 2001;44:909–916. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 29.Yardley V, Khan AA, Martin MB, Slifer TR, Araujo Fausto G, Moreno SNJ, Docampo R, Croft SL, Oldfield E. Antimicrob Agents Chemother. 2002;46:929–931. doi: 10.1128/AAC.46.3.929-931.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin MB, Sanders JM, Kendrick H, De Luca-Fradley K, Lewis JC, Grimley JS, Van Brussel EM, Olsen JR, Meints GA, Burzynska A, Kafarski P, Croft SL, Oldfield E. J Med Chem. 2002;45:2904–2914. doi: 10.1021/jm0102809. [DOI] [PubMed] [Google Scholar]
- 31.Rosso VS, Szajnman SH, Malayil L, Galizzi M, Moreno SNJ, Docampo R, Rodriguez JB. Bioorg Med Chem. 2011;19:2211–2217. doi: 10.1016/j.bmc.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szajnman SH, García Liñares GE, Li Z–H, Galizzi M, Jiang C, Bontempi E, Ferella M, Moreno SNJ, Docampo R, Rodriguez JB. Bioorg Med Chem. 2008;16:3283–3290. doi: 10.1016/j.bmc.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szajnman SH, Bailey BN, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2001;11:789–792. doi: 10.1016/s0960-894x(01)00057-9. [DOI] [PubMed] [Google Scholar]
- 34.Szajnman SH, Montalvetti A, Wang Y, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2003;13:3231–3235. doi: 10.1016/s0960-894x(03)00663-2. [DOI] [PubMed] [Google Scholar]
- 35.Szajnman SH, Ravaschino EL, Docampo R, Rodriguez JB. Bioorg Med Chem Lett. 2005;15:4685–4690. doi: 10.1016/j.bmcl.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 36.Ling Y, Sahota G, Odeh S, Chan JMW, Araujo FG, Moreno SNJ, Oldfield E. J Med Chem. 2005;48:3130–3140. doi: 10.1021/jm040132t. [DOI] [PubMed] [Google Scholar]
- 37.Bouzahzah B, Jelicks LA, Morris SA, Weiss LM, Tanowitz HB. Parasitol Res. 2005;96:184–187. doi: 10.1007/s00436-005-1331-9. [DOI] [PubMed] [Google Scholar]
- 38.García Liñares G, Ravaschino EL, Rodriguez JB. Curr Med Chem. 2006;13:335–360. doi: 10.2174/092986706775476043. [DOI] [PubMed] [Google Scholar]
- 39.Urbina JA, Docampo R. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Brener Z. Annu Rev Microbiol. 1973;27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 41.Moreno SNJ, Li Z-H. Expert Opinion Ther Targets. 2008;12:253–263. doi: 10.1517/14728222.12.3.253. [DOI] [PubMed] [Google Scholar]
- 42.Levine ND, Corliss JO, Cox FEG, Deroux G, Grain J, Honigberg BM, Leedale GF, Loeblich AR, 3rd, Lom J, Lynn D, Merinfeld EG, Page FC, Poliansky G, Sprague V, Vavra J, Wallace FG. J Protozool. 1980;27:37–58. doi: 10.1111/j.1550-7408.1980.tb04228.x. [DOI] [PubMed] [Google Scholar]
- 43.Carruthers V, Boothroyd JC. Curr Opin Microbiol. 2007;10:83–89. doi: 10.1016/j.mib.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 44.Fichera ME, Roos DS. Nature. 1997;390:407–409. doi: 10.1038/37132. [DOI] [PubMed] [Google Scholar]
- 45.Stokkermans TJW, Schwartzman JD, Keenan K, Morrissette NS, Tilney LG, Roos DS. Exp Parasitol. 1996;84:355–370. doi: 10.1006/expr.1996.0124. [DOI] [PubMed] [Google Scholar]
- 46.Roberts F, Roberts CW, Johnson JJ, Kyle DE, Krell T, Coggins JR, Coombs GH, Milhous WK, Tzipori S, Ferguson DJ, Chakrabarti D, McLeod R. Nature. 1998;393:801–805. doi: 10.1038/31723. [DOI] [PubMed] [Google Scholar]
- 47.Urbina JA. Acta Tropica. 2010;115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 48.Urbina JA. Drugs Fut. 2010;35:409–420. [Google Scholar]
- 49.Amzel LM. Personal communication.
- 50.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson R, Finn J. J Biol Chem. 2003;278:18401–18407. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 51.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Proc Natl Acad Sci USA. 1996;93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang C-H, Gabelli SB, Oldfield E, Amzel ML. Proteins. 2010;78:888–899. doi: 10.1002/prot.22614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao R, Chen CK-M, Guo R-T, Wang AH, Oldfield E. Proteins. 2008;73:431–439. doi: 10.1002/prot.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel ML. Proteins. 2006;62:80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 55.Van Beek E, Lowik C, Queans I, Papapoulos S. J Bone Miner Res. 1996;11:1492–1497. doi: 10.1002/jbmr.5650111016. [DOI] [PubMed] [Google Scholar]
- 56.Ebetino FH, Francis MD, Rogers MJ, Russell RGG. Rev Contemp Pharmacother. 1998;9:233–243. [Google Scholar]
- 57.Jung A, Bisaz S, Fleisch H. Calcif Tissue Res. 1973;11:269–280. doi: 10.1007/BF02547227. [DOI] [PubMed] [Google Scholar]
- 58.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 59.Marma MS, Xia Z, Stewart C, Coxon F, Dunford JE, Baron R, Kashemirov BA, Ebetino FH, Triffit JT, Russell RGG, McKenna CE. J Med Chem. 2007;50:5967–5975. doi: 10.1021/jm0702884. [DOI] [PubMed] [Google Scholar]
- 60.McKenna CE, Shen P-D. J Org Chem. 1981;46:4573–4576. [Google Scholar]
- 61.Berkowitz DB, Bose M. J Fluorine Chem. 2001;112:13–33. [Google Scholar]
- 62.Blackburn GM, England DA, Kolkmann F. Chem Commun. 1981:930–932. [Google Scholar]
- 63.Romanenko VD, Kukhar VP. Chem Rev. 2006;106:3868–3935. doi: 10.1021/cr051000q. [DOI] [PubMed] [Google Scholar]
- 64.De Schutter JW, Zaretsky S, Welbourn S, Pause A, Tsantrizos YS. Bioorg Med Chem Lett. 2010;20:5781–5786. doi: 10.1016/j.bmcl.2010.07.133. [DOI] [PubMed] [Google Scholar]
- 65.Lal GS. J Org Chem. 1993;57:4676–4683. [Google Scholar]
- 66.Lal GS, Pez GP, Syvret RG. Chem Rev. 1996;96:1737–1755. doi: 10.1021/cr941145p. [DOI] [PubMed] [Google Scholar]
- 67.Xu Y, Qian YL, Prestwich GD. Org Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- 68.Beier P, Opekar S, Mikhail Zibinsky M, Inessa Bychinskaya I, Prakash GKS. Org Biomol Chem. 2011;9:4035–4038. doi: 10.1039/c1ob05095h. [DOI] [PubMed] [Google Scholar]
- 69.Degenhardt CR, Burdsall DC. J Org Chem. 1986;51:3488–3490. [Google Scholar]
- 70.Ling Y, Li Z-H, Miranda K, Oldfield E, Moreno SNJ. J Biol Chem. 2007;282:30804–30816. doi: 10.1074/jbc.M703178200. [DOI] [PubMed] [Google Scholar]
- 71.The amino acid sequence of Proteins was obtained from the protein database from NCBI http://www.ncbi.nlm.nih.gov/protein (T. cruzi, 1YHK_A; T. brucei, gi:30522953; L. donovani, gi:112363472; T. gondii, gi:108745448; Human, 2F7M_F). Sequence alignments were created with ClustalX: Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Trends Biochem Sciences. 1998;23:403–405. doi: 10.1016/s0968-0004(98)01285-7.
- 72.Canavaci AM, Bustamante JM, Padilla AM, Pereza Brandan CM, Simpson LJ, Xu D, Boehlke CL, Tarleton RL. PLOS Negl Trop Dis. 2010;4:e740. doi: 10.1371/journal.pntd.0000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gubbels MJ, Li C, Striepen B. Antimicrob Agents Chemother. 2003;43:309–316. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Agrawal S, van Dooren GG, Beatty WL, Striepen B. J Biol Chem. 2009;284:33683–33691. doi: 10.1074/jbc.M109.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ravaschino EL, Docampo R, Rodriguez JB. J Med Chem. 2006;49:426–435. doi: 10.1021/jm050922i. [DOI] [PubMed] [Google Scholar]
- 76.Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R. J Biol Chem. 2001;276:33930–33937. doi: 10.1074/jbc.M103950200. [DOI] [PubMed] [Google Scholar]
- 77.Montalvetti A, Fernandez A, Sanders JM, Ghosh S, Van Brussel E, Oldfield E, Docampo R. J Biol Chem. 2003;278:17075–17083. doi: 10.1074/jbc.M210467200. [DOI] [PubMed] [Google Scholar]
- 78.Ogura K, Nishino T, Shinka T, Seto S. Methods Enzymol. 1985;110:167–171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.