


Abstract
Synthetic genetic sensors and circuits enable programmable control over the timing and conditions of gene expression. They are being increasingly incorporated into the control of complex, multigene pathways and cellular functions. Here, we propose a design strategy to genetically separate the sensing/circuitry functions from the pathway to be controlled. This separation is achieved by having the output of the circuit drive the expression of a polymerase, which then activates the pathway from polymerase-specific promoters. The sensors, circuits and polymerase are encoded together on a ‘controller’ plasmid. Variants of T7 RNA polymerase that reduce toxicity were constructed and used as scaffolds for the construction of four orthogonal polymerases identified via part mining that bind to unique promoter sequences. This set is highly orthogonal and induces cognate promoters by 8- to 75-fold more than off-target promoters. These orthogonal polymerases enable four independent channels linking the outputs of circuits to the control of different cellular functions. As a demonstration, we constructed a controller plasmid that integrates two inducible systems, implements an AND logic operation and toggles between metabolic pathways that change Escherichia coli green (deoxychromoviridans) and red (lycopene). The advantages of this organization are that (i) the regulation of the pathway can be changed simply by introducing a different controller plasmid, (ii) transcription is orthogonal to host machinery and (iii) the pathway genes are not transcribed in the absence of a controller and are thus more easily carried without invoking evolutionary pressure.
INTRODUCTION
Synthetic genetic circuits have been constructed by engineering biochemical interactions to generate signal processing or dynamic functions (e.g. a logic gate or oscillator) (1–4). These circuits can be connected to each other to implement more intricate computational operations (5,6). Further, genetic sensors, which respond to environmental stimuli, can be connected as inputs to the circuits (7,8). The outputs can be used to control actuators that determine what the cell is making or doing. A number of ‘toy systems’ have been constructed to develop the design principles by which multiple sensors, circuits and actuators can be combined into larger programs. Such programs link cell–cell communication for biofilm formation, enable Escherichia coli to form patterns, and control the direction of chemotaxis (9–12). For practical applications, simple circuits have been used to control metabolic pathways to implement feedback or to turn on at a particular growth phase (13–15). Increasingly sophisticated programs containing many sensors and circuits will be required to integrate multiple cellular processes, implement fine spatial and temporal control over gene expression and move process control algorithms into individual cells. As the programs become larger, it will be more challenging to design and test in isolation before moving to control relevant pathways. Here, we present a design strategy that relies on multiple engineered phage polymerases to link the output of genetic programs with the pathways to be controlled, both of which are carried in distinct genetic locations. This modular design allows the program and pathways to be constructed and tested independently and then combined to create a complete operational system.
When introducing synthetic DNA into a cell, it is desirable that the encoded processes be functionally distinct from host processes. This has been articulated as building a ‘virtual machine’ in cells that make separate transcription/translation machinery and resources available to a synthetic system, thus reducing the load on the host (16). To this end, RNA regulator systems have been engineered to produce orthogonal parts libraries for controlling gene expression (17,18). Similarly, phage polymerases are a means to control orthogonal transcription and are one of the most used tools in genetic engineering. Specifically, T7 RNA polymerase (RNAP) has been shown to function in a variety of hosts, including Gram-negative and -positive bacteria, plant chloroplasts and mammalian cells (19–21). An advantage of T7 polymerase is that its promoters are tightly inactive in the absence of the polymerase, thus reducing the load on the cell when uninduced (20,22). Recent advances in manipulating and minimizing the ribosome and altering the 16S rRNA to bind to alternative Shine-Delgarno sites are important steps towards the ultimate goal of simultaneous orthogonal translation (23,24).
In this article, we present a design strategy to separate sensing/circuitry functions from pathways/actuation (Figure 1A). They are encoded in genetically distinct regions and linked by having the output of the circuits drive the expression of phage polymerases. This enables the ‘controller’ to be optimized independently using fluorescent proteins and then transferred to the control of a pathway. Bioinformatics and protein engineering were applied to build four orthogonal polymerases by swapping the DNA-binding loop from homologues mined from sequence databases. This yielded orthogonal polymerases that do not cross-react with non-cognate promoters and thus can be used to control four distinct pathways or actuators. Promoter libraries for the polymerases were constructed to enable tuneable control over the expression levels. Further, multiple terminators were constructed to enable multicistron systems be controlled by a polymerase without problems due to homologous recombination. Together, this approach decouples the design of a pathway from the regulation used to control it.
Figure 1.

Separation of synthetic regulation onto a genetic controller using orthogonal phage polymerases. (A) The controller contains genetic sensors and circuits, the output of which is the expression of a phage RNAP, which then activates a pathway. The pathways can be programmed to respond to different conditions by swapping the controller without further genetic manipulation. (B) Modifications are shown to reduce the toxicity of T7 RNAP, the details of which are described in the text. (C) Toxicity of T7 RNAP variants is shown. Each variant was co-transformed with a different plasmid bearing a T7 promoter, and cells were grown on plates containing 1 mM IPTG to fully induce RNAP expression from the Ptac promoter. RNAP variants containing different combinations of modifications are shown (v1 = T7 RNAP in pIncW plasmid, v2 = addition of Lon tag to v1, v3 = addition of weak RBS and GTG start codon to v2). The data are shown for the expression of the T7 variant in the absence of a T7 promoter (black, plasmid N23, Supplementary Data) and the T7 promoter driving red fluorescent protein (white, plasmid N155, Supplementary Data). Asterisks indicate that no colonies were observed.
MATERIALS AND METHODS
Strains and media
Escherichia coli strain DH10B was used for routine cloning, plasmid propagation and T7 characterization assays. E. coli strain MG1655 was used for pigment expression. Luria–Bertani (LB)-Miller (Difco) was used for strain propagation and assays. The antibiotics used were 34.4 μg·ml−1 chloramphenicol, 100 μg·ml−1 spectinomycin, 50 μg·ml−1 kanamycin and/or 100 μg·ml−1 ampicillin. The inducers used were isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich) and/or anhydrotetracycline (Sigma-Aldrich).
Computational methods
RNAP homologues were identified using BLAST (http://blast.ncbi.nlm.nih.gov/) (25). The search was run against the non-redundant protein sequence database, with an expect threshold of 10, word size of 3, the BLOSUM62 matrix, gap costs of 11 for existence and 1 for extension and the conditional compositional score matrix adjustment. Protein sequences were aligned using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (26). Pairwise alignment utilized the Gonnet weight matrix, a gap opening penalty of 100 and an extension penalty of 0.1. The multiple sequence alignment also utilized the Gonnet matrix, a gap opening penalty of 10, an extension penalty of 0.2, gap distances of 5 and no end gaps. Phage promoters were identified by scanning phage genomes with the PHIRE package (http://www.biw.kuleuven.be/logt/PHIRE.htm) (27). Default parameters were used (string length 20, degeneracy 4, dominanNum 4 and window size 20).
Fluorescence characterization via flow cytometry
Cells harbouring the appropriate plasmids were inoculated into 5 ml of LB media (supplemented with antibiotics, 37°C, 250 rpm) in 15 ml Falcon tubes and grown for 14 h. The cultures were diluted into fresh 5 ml LB media (supplemented with antibiotics and inducers) to a final OD600 of 0.25 in 15 ml Falcon tubes and were incubated for 6 h (37°C, 250 rpm). To halt the assay, 2 μl of cells were transferred from each tube to a 96-well plate containing phosphate-buffered saline supplemented with 2 mg·ml−1 kanamycin. Fluorescence data were collected using a BD Biosciences LSRII flow cytometer. GFP measurements utilized a 488 nm laser with a 530/30 nm filter, and mRFP measurements utilized a 532 nm laser with 575/26 nm filter. Data were gated by forward and side scatter, and each dataset consisted of at least 25 000 cells. FlowJo was used to calculate the geometric means of the fluorescence distributions and perform compensation. The autofluorescence value of E. coli cells harbouring no plasmid was subtracted to generate the values reported in this study.
RNAP toxicity assay
To assess RNAP toxicity, we measured the impact of RNAP-mediated expression on cell viability. Cells were co-transformed with an RNAP plasmid and one of the three reporter plasmids. The reporter plasmids carried no insert (N23), a wild-type T7 promoter with lacO-binding site and mRFP that produces moderate gene expression (N155) or a wild-type T7 promoter without lacO-binding site and mRFP producing high levels of gene expression (N489). Cells carrying appropriate plasmids were grown as described for the assay to characterize fluorescence. After dilution to an OD600 of 0.25, cells were further diluted by a factor of 106, and 50 μl of cells were plated on LB media containing antibiotics and 100 μM IPTG. After 14 h of incubation at 37°C, colonies that had formed on each plate were counted.
Pigment production and quantification
Strains were grown as in the Fluorescence Characterization assay with an exception. Instead of incubation in inducing media for 6 h, cells were grown 24 h before pigment extraction and quantification. After 24 h of induction, cultures were diluted to an OD600 of 1.0, and 1 ml of diluted culture was centrifuged at 15 000g for 60 sec. Supernatant was removed, cells were resuspended in 200 μl H2O, and re-centrifuged at 15 000g for 60 sec. The pigments were then extracted from cell pellets following one of the two methods. For lycopene extraction, cells were resuspended in 250 μl acetone and incubated at 55°C for 15 min with frequent vortexing. For deoxychromoviridans extraction, after removal of supernatant, cells were resuspended in 50 μl of 10% sodium dodecyl sulfate and incubated at 55°C for 15 min with frequent vortexing. Following, 250 μl of methanol were added to the tubes. The mixture was incubated at 55°C for an additional 15 min with frequent vortexing. Pigment extraction mixtures (both lycopene and deoxychromoviridans) were then centrifuged at 15 000g for 60 sec and the supernatant was transferred to a 96-well plate for quantification. Absorbance spectra from 350 to 700 nm were collected using a Tecan Safire plate spectrophotometer. Lycopene and deoxychromoviridans were quantified using measurements at 470 and 650 nm, respectively. Data are reported as the percent of max absorbance observed after subtraction of the absorbance of wild-type E. coli MG1655 cell extracts.
Creation of synthetic parts
T7 promoters of varying strength were generated from a random library and cloned in front of mRFP (28). The promoter was randomized in the following way: TAATACGACTCACTANNNNNAGA. The library was screened by picking 36 random colonies and growing overnight in 5 ml LB media containing 1 mM IPTG. Fluorescence was measured by flow cytometry, and four representative clones that exhibited diverse strengths were selected for further analysis. These four clones were sequenced and characterized. T7 terminators were generated from a random library and inserted into the characterization vector N292 (SBa_000566). The T7 terminator library was diversified as follows: TANNNAACCSSWWSSNSSSSTCWWWCGSSSSSSWWSSGTTT. The terminator library was co-transformed with T7* to screen for active terminators. Thirty-six colonies were selected and grown overnight in 5 ml LB media containing 1 mM IPTG. Fluorescence was measured by flow cytometry, and eight clones that exhibited highest termination were selected for further analysis. These eight clones were sequenced and characterized. The RBS Calculator (‘Forward Engineering’ mode, ‘ACCTCCTTA’ as the 16S RNA sequence) was used to generate RBSs of 25 000 AU for each gene in the lycopene biosynthetic pathway. Insulator sequences were designed using the Random DNA Generator using a random GC content of 50% (http://www.faculty.ucr.edu/~mmaduro/random.htm).
Part sequences are listed in Supplementary Table S1, and plasmid maps are shown in Supplementary Figure S10.
Engineering phage polymerases
Each orthogonal polymerase was constructed in the T7* backbone. The specificity loop for the N4 subfamily was inserted in T7*, and the gene was cloned into pIncW without further modification. This construct is denoted T7*(N4). The K1F and T3 specificity loops were cloned into T7*, combined with the Lon-mediated N-terminal degradation tag (LFIKPADLREIVTFPLFSDLVQCGFPSPAADYVEQRIDL) and inserted in pIncW to produce T7*(K1F) and T7*(T3).
RESULTS AND DISCUSSION
A practical challenge in using phage polymerases is that they can exhibit toxicity (20,29,30). To overcome this, we constructed a variant of T7 RNAP with reduced toxicity by combining mutations to lower its concentration and activity. The wild-type gene was placed under control of an IPTG-inducible promoter in a low copy plasmid and co-transformed with a second plasmid containing a T7 promoter and fluorescent reporter (Figure 1B). Toxicity was assessed by plating strains on inducing media and counting colonies after 24 h of growth. Significant toxicity was observed when T7 RNAP is highly expressed, even in the absence of a T7 promoter (Figure 1C, v1). A Lon-mediated N-terminal degradation tag from the umuD gene in E. coli (31) was added to limit polymerase concentration (Figure 1C, v2). This resulted in a slightly lower toxicity. Next, RNAP expression was tightly controlled using a weak ribosome-binding site and GTG start codon. Finally, during cloning, a spontaneous mutation in the polymerase active site (R632S) arose. This mutation was found to significantly reduce host toxicity while maintaining activity (Figure 1C, v3). It is interesting to note that the previous studies have reported that mutations to this region of the polymerase can reduce its processivity (32–34).
Once the toxicity was reduced, we sought to expand the number of polymerases that are orthogonal and can be used simultaneously in a cell. In the context of a controller, this would increase the number of circuit outputs that could be linked to different pathways (Figure 1A). Orthogonality can be achieved by engineering polymerases to bind to different promoters and not cross-react. One approach would be to apply part mining (35), where homologous phage polymerases are identified from the sequence databases, constructed using DNA synthesis and screened for activity and orthogonality. However, we did not want to repeat the process of reducing the toxicity for each polymerase. To avoid this, we used the set of T7 backbones (v1 to v3) as scaffolds into which we inserted the peptide loop responsible for promoter recognition from different phage polymerases identified in the NCBI database (Figure 2A). These chimeras were then tested for activity and orthogonality.
Figure 2.

Design of orthogonal T7 RNAPs and promoters. (A) Sequences of the specificity loop and promoter are shown for each orthogonal polymerase. (B) The interaction between the T7 RNAP and T7 promoter is shown (36). The β-hairpin ‘specificity’ loop (red) interacts with the 5′ end of the promoter in the region marked ‘binding’. (C) The orthogonality between the engineered RNAP promoter pairs is shown. Each RNAP was co-transformed individually with each promoter cloned into the base plasmid N155 (Supplementary Data) and expressed by 1 mM IPTG induction. The data represent the average of three experiments performed on different days after subtraction of the autofluorescence of Escherichia coli harbouring no plasmid. The standard deviation of the experiments is reported in Supplementary Figure S4 and representative cytometry distributions are shown in Supplementary Figure S7.
The DNA sequence to which T7 RNAP binds is determined by a β-hairpin, known as the specificity loop, which contacts the −12 to −7 bp region of the promoter (36). Changes to the specificity loop confer the ability to recognize different promoter sequences (37,38). Remarkably, it had been shown that a single mutation (N748D) switches T7 RNAP to preferentially bind a T3-like phage promoter (39). Thus, this is a good region to target mutagenesis; however, random mutations may be disruptive and it could require too many simultaneous mutations to generate orthogonality (40). Instead, we took the approach of exchanging the entire β-hairpin identified in RNAP from homologous phages. This is related to previous work, where orthogonal transcription factors were made by using information from a sequence database to guide mutations to the protein-DNA interface (39).
Each phage in the T7 family contains an RNAP and 10–20 promoters that provide a wealth of information about the interaction of the polymerase with DNA. To identify β-hairpins that confer different binding specificities, we identified homologues of T7 RNAP and computationally determined their DNA-binding preferences. First, 43 T7 RNAP homologues were identified from NCBI via a protein–protein BLAST against non-redundant protein sequences (25). RNAP with an E-value less than 1−100 and for which a fully sequenced phage genome exists were selected for further analysis. A multiple sequence alignment of the RNAP amino acid sequences was performed using ClustalW (26) to identify and extract the β-hairpin in each RNAP corresponding to T7 amino acids G732-P780. Three RNAPs (RSB2, W5455 and ϕIBB-PF7A) were eliminated due to significant differences in length of the β-hairpins. A second multiple sequence alignment was performed with only the β-hairpin sequences, and 13 RNAP subfamilies were identified (distance between members < 0.1 in the ClustalW guide tree). Putative promoters were identified from each phage genome using PHIRE, software that scans genomes for regulatory elements by identifying conserved sequences with a limited number of user-defined degeneracies (27). WebLogo was used to determine the consensus sequence for each phage RNAP (41). Remarkably, for each β-hairpin subfamily, the binding region of the consensus promoter is identical (Supplementary Figure S1).
Novel RNAPs were generated by swapping the β-hairpin from T7 RNAP (Q744 to I761) with the equivalent region from each subfamily (Figure 2B). The corresponding binding region of the T7 promoter (−12C to −7C) was replaced with the promoter subfamily consensus sequences (Figure 2A). The resulting RNAPs were screened for activity against their predicted promoters. Four RNAPs exhibited strong activity (42-, 12-, 17- and 40-fold induction by T7*, T7*(T3), T7*(K1F) and T7*(N4), respectively) (Supplementary Figure S4) and similar temporal induction (Supplementary Figure S6). The activity of these RNAPs against non-cognate promoters was then characterized. Each RNAP is highly orthogonal, even after significant mutations to both the specificity loop and promoter (Figure 2C).
It is valuable to be able to tune the strength of a promoter to achieve varied levels of transcription. The T7 promoter has been shown to be modular, consisting of a 6 bp RNAP-binding region and a 5 bp strength-determining region (36,42,43). Mutations to the strength-determining region should alter promoter strength without affecting RNAP specificity (44,45). We created a promoter library by randomizing the wild-type T7 promoter from −2bp to +3 bp. The library was screened using flow cytometry, and a set of representative promoters was identified that includes a broad range of expression levels spanning two orders of magnitude. These promoters were sequenced to determine the mutations to the strength-determining region (Figure 3A). These regions were then combined with a different specificity-determining region that is specific for T7*(T3). The rank order for strength persists with fair correlation between strengths of individual promoters (Figure 3B and Supplementary Figure S5). Orthogonality is retained for the hybrid promoters, where T7* only transcribes those promoters containing the cognate-binding sequence and vice versa for T7*(T3).
Figure 3.
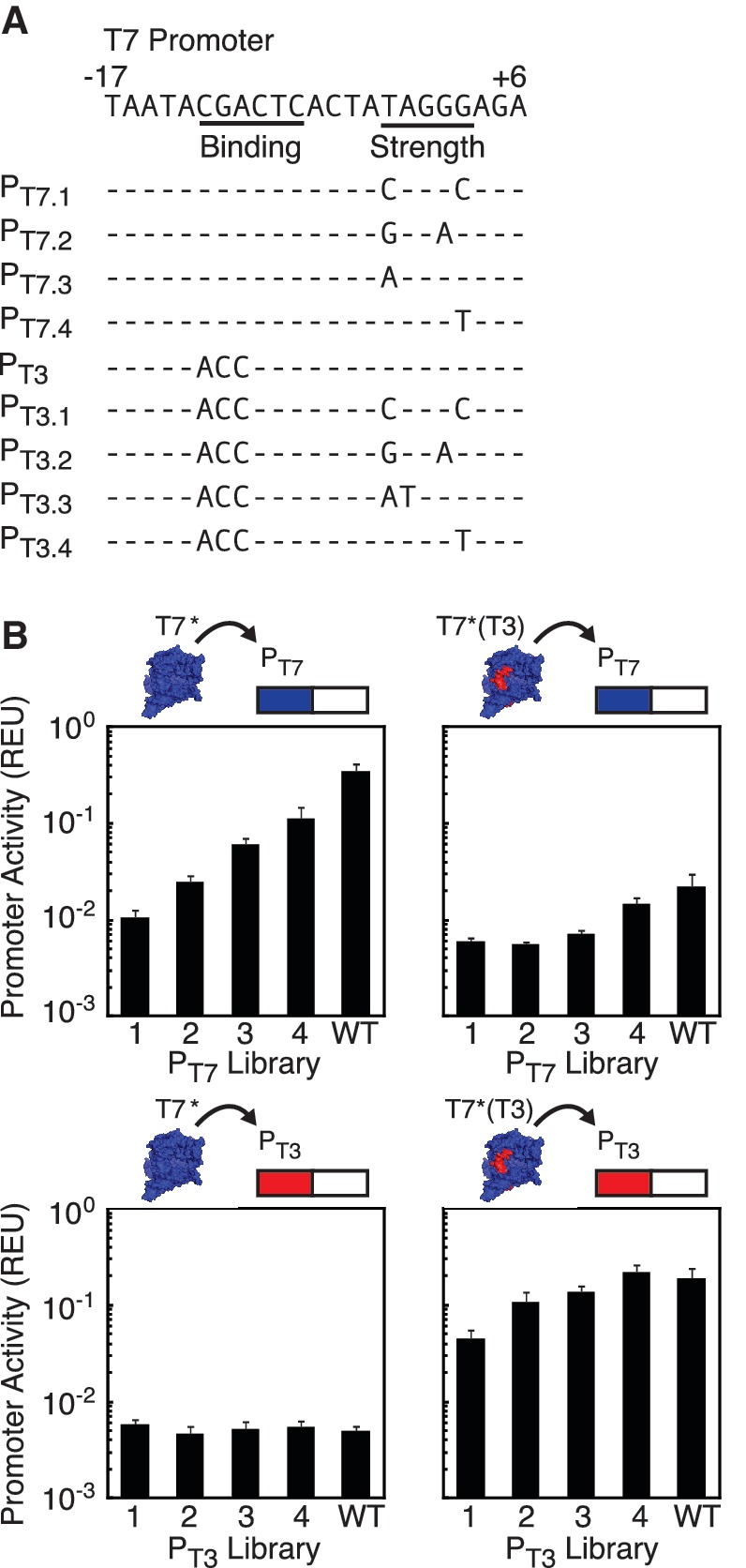
Modularity of T7 promoters. (A) A library of pT7 promoters of differing strengths was created by mutating the strength-determining region (−2bp to +3bp) and cloned into plasmid N155 (Supplementary Data). Synonymous mutations were also made to the pT3 promoter. Sequences are shown for the two libraries. (B) The promoter libraries were used to control fluorescent reporter proteins, and each library was co-transformed with both T7* and T7*(T3). Activity of co-transformants under 1 mM IPTG induction was characterized by flow cytometry to study cognate interactions (T7*-PT7, top left and T7*(T3)-PT3, bottom right) and non-cognate interactions (T7*-PT3, bottom left and T7*(T3)-PT7, top right). Error bars represent the standard deviation of three experiments on different days.
The set of orthogonal RNAPs enables the independent control of multiple pathways by a genetic program encoded on a controller. The modularity of the controller allows it to be tested using fluorescent reporters before implementing it to control metabolic pathways or difficult to assay cellular functions. To demonstrate this, we constructed a simple genetic program whose output is two RNAPs (T7* and T7*(K1F)) under the control of multiple inducing signals and a logic gate (Figure 4). T7* is expressed from the Ptac promoter by IPTG induction (characterized in Supplementary Figure S3), whereas T7*(K1F) is controlled by an AND gate that is active only in the presence of both IPTG and anhydrotetracycline (aTc). The AND function is achieved by placing a lacO-binding site after the transcription start site of the Ptet promoter. Such promoter engineering has been applied previously to build gates (46,47), and it is similar to that used to build an edge detector program (11). We characterized the performance of the genetic program using reporter plasmids for each RNAP and verified that it produced the expected circuit logic (Figure 4B). The reporters are placed under the control of promoters responsive to each RNAP in the same genetic context as the genes ultimately to be controlled. The T7* RNAP exhibits 7.8-fold induction, whereas the T7*(K1F) RNAP is induced 9.2-fold when tested using red fluorescent protein and a low copy pSC101* origin.
Figure 4.

Design and characterization of a two-input genetic program. (A) A genetic program is shown that controls two orthogonal polymerases via different logic. The activity of T7* was measured by co-transformation with a PT7 reporter plasmid (N489, Supplementary Data) with different combinations of inducer (1 mM IPTG and/or 10 ng/ml aTc). (B) K1F* activity was measured using a PK1F fluorescent protein reporter (THSS40, Supplementary Data, 1 mM IPTG and/or 10 ng/ml aTc). Error bars represent the standard deviation of three experiments of different days.
Once the controller is verified, it can then be co-transformed with the target pathways, which are carried on a second plasmid. We applied the genetic program to control the expression of two small molecule pigments, lycopene (red) and deoxychromoviridans (green). Escherichia coli produces small amounts of lycopene through the 1-deoxy-d-xylulose-5-phosphate (DXP) pathway following the introduction of the carotenoid genes crtEBI (Figure 5B) (48). Lycopene production can be improved by overexpressing two genes, dxs and idi (49,50). Deoxychromoviridans is synthesized from l-tryptophan by the genes vioABE (51,52).
Figure 5.

Control of multiple pathways using a genetic controller and orthogonal polymerases. (A) The controller described from Fig. 4 is utilized to control production of two pigments (lycopene and deoxychromoviridans). Operons are drawn using SBOLv symbols (55): RBSs (half circles), insulators (crossed circles) and terminators (T). SynBERC Registry part numbers (registry.synberc.org) are shown for each part, and nucleotide numbers for the plasmids are indicated. T7 promoter strength is reported as the fluorescence of a RFP reporter from plasmid N155 (Supplementary Data) and presented in relative expression units (REU, Supplementary Data, Supplementary Figure S2). (B) Biosynthetic pathways for lycopene and deoxychromoviridans are shown. Intermediate products are shown in black, and genes expressed by the engineered system are shown in italics. DMAPP, dimethylallyl diphosphate; DXP, 1-deoxy-d-xylulose 5-phosphate; FPP, farnesyl diphosphate; GGPP, geranylgeranyl pyrophosphate; G3P, glyceraldehyde 3-phosphate; HMBPP, (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate; IPA, indole-3-pyruvic acid; IPP, isopentenyl diphosphate. (C) Pigment production follows the logic encoded by the controller. Escherichia coli strain MG1655 was co-transformed with plasmids encoding the genetic program and both pathways. Cells were grown overnight in non-inducing conditions, diluted to an OD600 of 1.0, and spotted onto plates containing inducers. In the presence of IPTG and aTc, both pigments are synthesized. The combination of green and red pigments appears brownish purple. Photographs of representative colonies are shown. (D) Quantitative assessment of pigment production. Cells were grown for 24 h in LB media containing the indicated inducers. Lycopene and deoxychromoviridans were extracted, and absorbance was measured (lycopene, 580 nm; deoxychromoviridans, 650 nm). Data are reported as the fraction of maximum absorbance observed. Error bars represent standard deviation of three experiments of different days.
Each gene from both pathways was placed in a cistron comprising an insulator, T7 or K1F promoter, synthetic ribosome-binding site, the gene of interest and a T7 terminator (Figure 5A). A library of T7-derived terminators was created to avoid homologous recombination between cistrons (Supplementary Data, Supplementary Figures S8 and S9). Synthetic cistrons were assembled into either a lycopene operon or a deoxychromoviridans operon using the Gibson assembly method (53). This method enabled us to introduce a library of T7 promoters into each lycopene cistron, co-transform the library with T7*, and screen for efficient producers under inducing conditions. We identified and sequenced a clone that exhibited excellent pigment production and contrast (T7 promoters indicated in Figure 5A). For deoxychromoviridans, we obtained sufficient expression using wild-type promoters but found that clones were not stable when vioE was expressed as a cistron. Expressing vioB and vioE from a single K1F promoter is stable and eliminates leaky expression of deoxychromoviridans.
We then assessed the feasibility of connecting the genetic program that was tuned and characterized in isolation to the multigene pigment pathways. The genetic program was co-transformed with the two biosynthetic pathways, and cells were plated on varying inducer combinations (Figure 5C). Lycopene is synthesized in the presence of IPTG and is not affected by the presence of aTc. Deoxychromoviridans, in contrast, is only synthesized when both inducing molecules are present. Further quantification was performed by extracting pigments and measuring relative absorbance under the different inducing states (50,54). We measured a 7.9-fold induction of lycopene expression between the absence of inducers and the addition of IPTG (Figure 5D). Lycopene production in the presence of both IPTG and aTc yields a 5.6-fold induction, and induction of deoxychromoviridans is 3.3-fold. The circuit performance obtained using the fluorescent reporters (Figure 4) closely matches with that obtained using the more complex pathways (Figure 5D).
Genetic engineering is becoming increasingly complex, requiring integrated control over multiple many-gene pathways. Ultimately, it is envisioned that synthetic systems could approach the size and complexity of complete genomes. Our ability to engineer systems at this scale is going to require modularization in their design and testing. To this end, we present an approach to decouple the genetic regulation controlling the conditions and dynamics of gene expression from the pathways that are being controlled. The sensors and circuits can be constructed and tuned using fluorescent proteins, and the engineered pathways can be optimized under simplified inducing conditions. Because the output channels are orthogonal polymerases, the ‘controller’ can switch from testing to implementation simply by co-transforming it with the more complex pathways. This has several advantages. First, the pathways may be challenging to assay and inappropriate for the characterization of circuit dynamics. Second, this approach enables the future development of highly integrated genetic programs linking dozens of sensors and circuits that all can be encoded in the controller. Thus, the modularization of genetic programming enables it to be abstracted from the idiosyncrasies of the biology being controlled.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–10 and Supplementary References [56–63].
FUNDING
Office of Naval Research [N00014-10-1-0245 to C.A.V.]; NSF [CCF-0943385 to C.A.V.]; National Institutes of Health [AI067699 and AI067699 to C.A.V.]; NSF graduate student fellowships (to K.T. and F.M.) and NDSEG and Hertz graduate fellowships to (T.H.S.S.). K.T., R.H., T.H.S.S., F.M., and C.A.V. are part of the NSF Synthetic Biology Engineering Research Center (SynBERC). Funding for open access charge: National Institutes of Health [AI067699 and AI067699 to C.A.V.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank George Church and Harris Wang for plasmid pAC-LYC containing lycopene biosynthesis genes.
REFERENCES
- 1.Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- 2.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 3.Rinaudo K, Bleris L, Maddamsetti R, Subramanian S, Weiss R, Benenson Y. A universal RNAi-based logic evaluator that operates in mammalian cells. Nat. Biotechnol. 2007;25:795–801. doi: 10.1038/nbt1307. [DOI] [PubMed] [Google Scholar]
- 4.Tamsir A, Tabor JJ, Voigt CA. Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature. 2011;469:212–215. doi: 10.1038/nature09565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber W, Stelling J, Rimann M, Keller B, Daoud-El Baba M, Weber CC, Aubel D, Fussenegger M. A synthetic time-delay circuit in mammalian cells and mice. Proc. Natl Acad. Sci. USA. 2007;104:2643–2648. doi: 10.1073/pnas.0606398104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sohka T, Heins RA, Phelan RM, Greisler JM, Townsend CA, Ostermeier M. An externally tunable bacterial band-pass filter. Proc. Natl Acad. Sci. USA. 2009;106:10135–10140. doi: 10.1073/pnas.0901246106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prindle A, Samayoa P, Razinkov I, Danino T, Tsimring LS, Hasty J. A sensing array of radically coupled genetic ‘biopixels’. Nature. 2011;481:39–44. doi: 10.1038/nature10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science. 2011;333:1307–1311. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- 9.Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature. 2005;434:1130–1134. doi: 10.1038/nature03461. [DOI] [PubMed] [Google Scholar]
- 10.Moon TS, Clarke EJ, Groban ES, Tamsir A, Clark RM, Eames M, Kortemme T, Voigt CA. Construction of a genetic multiplexer to toggle between chemosensory pathways in Escherichia coli. J. Mol. Biol. 2011;406:215–227. doi: 10.1016/j.jmb.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabor JJ, Salis HM, Simpson ZB, Chevalier AA, Levskaya A, Marcotte EM, Voigt CA, Ellington AD. A synthetic genetic edge detection program. Cell. 2009;137:1272–1281. doi: 10.1016/j.cell.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner K, Karig DK, Weiss R, Arnold FH. Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium. Proc. Natl Acad. Sci. USA. 2007;104:17300–17304. doi: 10.1073/pnas.0704256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carothers JM, Goler JA, Juminaga D, Keasling JD. Model-driven engineering of RNA devices to quantitatively program gene expression. Science. 2011;334:1716–1719. doi: 10.1126/science.1212209. [DOI] [PubMed] [Google Scholar]
- 14.Fung E, Wong WW, Suen JK, Bulter T, Lee SG, Liao JC. A synthetic gene-metabolic oscillator. Nature. 2005;435:118–122. doi: 10.1038/nature03508. [DOI] [PubMed] [Google Scholar]
- 15.Holtz WJ, Keasling JD. Engineering static and dynamic control of synthetic pathways. Cell. 2010;140:19–23. doi: 10.1016/j.cell.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 16.Canton B. 2008 Ph.D., Massachusetts Institute of Technology, Biological Engineering Division. [Google Scholar]
- 17.Isaacs FJ, Dwyer DJ, Ding C, Pervouchine DD, Cantor CR, Collins JJ. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 2004;22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 18.Lucks JB, Qi L, Mutalik VK, Wang D, Arkin AP. Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc. Natl Acad. Sci. USA. 2011;108:8617–8622. doi: 10.1073/pnas.1015741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride KE, Schaaf DJ, Daley M, Stalker DM. Controlled expression of plastid transgenes in plants based on a nuclear DNA-encoded and plastid-targeted T7 RNA polymerase. Proc. Natl Acad. Sci. USA. 1994;91:7301–7305. doi: 10.1073/pnas.91.15.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 21.Wyatt LS, Moss B, Rozenblatt S. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology. 1995;210:202–205. doi: 10.1006/viro.1995.1332. [DOI] [PubMed] [Google Scholar]
- 22.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. Designing and engineering evolutionary robust genetic circuits. J. Biol. Eng. 2010;4:12. doi: 10.1186/1754-1611-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rackham O, Chin JW. Cellular logic with orthogonal ribosomes. J. Am. Chem. Soc. 2005;127:17584–17585. doi: 10.1021/ja055338d. [DOI] [PubMed] [Google Scholar]
- 24.Wang K, Neumann H, Peak-Chew SY, Chin JW. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007;25:770–777. doi: 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- 25.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 27.Lavigne R, Sun WD, Volckaert G. PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics. 2004;20:629–635. doi: 10.1093/bioinformatics/btg456. [DOI] [PubMed] [Google Scholar]
- 28.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson JC, Voigt CA, Arkin AP. Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 2007;3:133. doi: 10.1038/msb4100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Temiakov D, Patlan V, Anikin M, McAllister WT, Yokoyama S, Vassylyev DG. Structural basis for substrate selection by t7 RNA polymerase. Cell. 2004;116:381–391. doi: 10.1016/s0092-8674(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy WP, Momand JR, Yin YW. Mechanism for de novo RNA synthesis and initiating nucleotide specificity by t7 RNA polymerase. J. Mol. Biol. 2007;370:256–268. doi: 10.1016/j.jmb.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 34.Bonner G, Lafer EM, Sousa R. Characterization of a set of T7 RNA polymerase active site mutants. J. Biol. Chem. 1994;269:25120–25128. [PubMed] [Google Scholar]
- 35.Bayer TS, Widmaier DM, Temme K, Mirsky EA, Santi DV, Voigt CA. Synthesis of methyl halides from biomass using engineered microbes. J. Am. Chem. Soc. 2009;131:6508–6515. doi: 10.1021/ja809461u. [DOI] [PubMed] [Google Scholar]
- 36.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399:80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 37.Chelliserrykattil J, Cai G, Ellington AD. A combined in vitro/in vivo selection for polymerases with novel promoter specificities. BMC Biotechnol. 2001;1:13. doi: 10.1186/1472-6750-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desai TA, Rodionov DA, Gelfand MS, Alm EJ, Rao CV. Engineering transcription factors with novel DNA-binding specificity using comparative genomics. Nucleic Acids Res. 2009;37:2493–2503. doi: 10.1093/nar/gkp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolon DN, Voigt CA, Mayo SL. De novo design of biocatalysts. Curr Opin Chem Biol. 2002;6:125–129. doi: 10.1016/s1367-5931(02)00303-4. [DOI] [PubMed] [Google Scholar]
- 41.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bandwar RP, Jia Y, Stano NM, Patel SS. Kinetic and thermodynamic basis of promoter strength: multiple steps of transcription initiation by T7 RNA polymerase are modulated by the promoter sequence. Biochemistry. 2002;41:3586–3595. doi: 10.1021/bi0158472. [DOI] [PubMed] [Google Scholar]
- 43.Rong M, He B, McAllister WT, Durbin RK. Promoter specificity determinants of T7 RNA polymerase. Proc. Natl Acad. Sci. USA. 1998;95:515–519. doi: 10.1073/pnas.95.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davidson EA, VAN Blarcom T, Levy M, Ellington AD. Emulsion based selection of t7 promoters of varying activity. Pac. Symp. Biocomput. 2010:433–443. doi: 10.1142/9789814295291_0045. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda RA, Warshamana GS, Chang LL. In vivo and in vitro activities of point mutants of the bacteriophage T7 RNA polymerase promoter. Biochemistry. 1992;31:9073–9080. doi: 10.1021/bi00152a051. [DOI] [PubMed] [Google Scholar]
- 46.Cox RS, III, Surette MG, Elowitz MB. Programming gene expression with combinatorial promoters. Mol. Syst. Biol. 2007;3:145. doi: 10.1038/msb4100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramalingam KI, Tomshine JR, Maynard JA, Kaznessis YN. Forward engineering of synthetic bio-logical AND gates. Biochem. Eng. J. 2009;47:38–47. [Google Scholar]
- 48.Cunningham FX, Jr, Sun Z, Chamovitz D, Hirschberg J, Gantt E. Molecular structure and enzymatic function of lycopene cyclase from the cyanobacterium Synechococcus sp strain PCC7942. Plant Cell. 1994;6:1107–1121. doi: 10.1105/tpc.6.8.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc. Natl Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balibar CJ, Walsh CT. In vitro biosynthesis of violacein from L-tryptophan by the enzymes VioA-E from Chromobacterium violaceum. Biochemistry. 2006;45:15444–15457. doi: 10.1021/bi061998z. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez C, Brana AF, Mendez C, Salas JA. Reevaluation of the violacein biosynthetic pathway and its relationship to indolocarbazole biosynthesis. Chembiochem. 2006;7:1231–1240. doi: 10.1002/cbic.200600029. [DOI] [PubMed] [Google Scholar]
- 53.Ramon A, Smith HO. Single-step linker-based combinatorial assembly of promoter and gene cassettes for pathway engineering. Biotechnol. Lett. 2011;33:549–555. doi: 10.1007/s10529-010-0455-x. [DOI] [PubMed] [Google Scholar]
- 54.Ahmetagic A, Pemberton JM. Stable high level expression of the violacein indolocarbazole anti-tumour gene cluster and the Streptomyces lividans amyA gene in E. coli K12. Plasmid. 2010;63:79–85. doi: 10.1016/j.plasmid.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez C, Bartram S, Ramasubramanian A, Endy D. 2009 BBF RFC 16: Synthetic Biology Open Language Visual (SBOLv) Specification. http://www.sbolstandard.org/ (25 June 2012, date last accessed) [Google Scholar]
- 56.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, de Mora K, Glieberman AL, Monie DD, Endy D. Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Temme K, Zhao D, Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl Acad. Sci. USA. 2012;109:7085–7090. doi: 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shetty RP, Endy D, Knight TF., Jr Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyazaki K. Creating random mutagenesis libraries by megaprimer PCR of whole plasmid (MEGAWHOP) Methods Mol. Biol. 2003;231:23–28. doi: 10.1385/1-59259-395-X:23. [DOI] [PubMed] [Google Scholar]
- 61.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 62.Dykxhoorn DM, St Pierre R, Linn T. A set of compatible tac promoter expression vectors. Gene. 1996;177:133–136. doi: 10.1016/0378-1119(96)00289-2. [DOI] [PubMed] [Google Scholar]
- 63.August PR, Grossman TH, Minor C, Draper MP, MacNeil IA, Pemberton JM, Call KM, Holt D, Osburne MS. Sequence analysis and functional characterization of the violacein biosynthetic pathway from Chromobacterium violaceum. J. Mol. Microbiol. Biotechnol. 2000;2:513–519. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.