


Abstract
The essential DNA helicase, PcrA, regulates recombination by displacing the recombinase RecA from the DNA. The nucleotide-bound state of RecA determines the stability of its nucleoprotein filaments. Using single-molecule fluorescence approaches, we demonstrate that RecA displacement by a translocating PcrA requires the ATPase activity of the recombinase. We also show that in a ‘head-on collision’ between a polymerizing RecA filament and a translocating PcrA, the RecA K72R ATPase mutant, but not wild-type RecA, arrests helicase translocation. Our findings demonstrate that translocation of PcrA is not sufficient to displace RecA from the DNA and assigns an essential role for the ATPase activity of RecA in helicase-mediated disruption of its filaments.
INTRODUCTION
Helicases play essential roles in DNA replication, repair, recombination and transcription (1,2). Whereas the major replicative helicase in bacteria, DnaB, actively converts double-stranded DNA (dsDNA) at the replication fork into single-stranded DNA (ssDNA) for use as a template by the DNA polymerase, other helicases such as PcrA and UvrD facilitate replication by clearing various protein barriers from the DNA (3–12). One such barrier is the RecA nucleoprotein filament, which is formed during recombination or when RecA binds to ssDNA gaps to facilitate DNA repair. If these filaments persist on the DNA, they can impede an advancing replisome (7,8,11,12). Therefore, unregulated RecA nucleoprotein filaments can be detrimental to a cell and mechanisms must exist to regulate RecA function.
One such mechanism involves disruption of RecA filaments by DNA helicases such as PcrA present in Gram-positive bacteria (3,13). The homologous UvrD helicase from the Gram-negative Escherichia coli also disrupts RecA filaments (12,14). This regulation of RecA function likely supports bacterial growth and survival since suppressors of a pcrA knock-out have been isolated in the recFOR genes that are involved in RecA-mediated recombination in Bacillus subtilis (11). Moreover, conditional pcrA mutants of B. subtilis exhibit a hyper-recombinogenic phenotype and E. coli uvrD mutants are rescued by pcrA (10,11). However, it is known that the ATPase and helicase activities of Staphylococcus aureus PcrA and Mycobacterium tuberculosis UvrD are not essential for inhibiting RecA functions (3,15,16) Therefore, it is likely that helicases utilize multiple mechanisms to regulate RecA function.
The RecA recombinase promotes ATP-dependent joint-molecule formation and the exchange of DNA strands between homologous DNA molecules (17). RecA binds to ssDNA in the presence of ATP to form helical nucleoprotein filaments which serve as the scaffolding upon which homologous recombination occurs (18–20). ATP binding, hydrolysis and product release play critical roles in RecA filament stability and function (21). The high-affinity RecA-ATP ssDNA-binding state is required for DNA strand exchange and other DNA-repair related functions (22) In contrast, the low-affinity RecA-ADP ssDNA-binding state induced by the ATPase activity of RecA is required for its dissociation during the subsequent stages of recombination. Since the nucleotide-bound state of RecA determines its affinity for ssDNA, we hypothesized that the ATPase activity of RecA may play a major role in the disruption of nucleoprotein filaments by PcrA.
Here, we use the Geobacillus stearothermophilus PcrA and single-molecule fluorescence approaches to demonstrate that ATP hydrolysis by RecA is essential for PcrA-mediated displacement of the recombinase from ssDNA. Our results show that the conversion of RecA from its high-affinity DNA binding state (ATP-bound) to the low-affinity DNA binding state (ADP-bound) is required for the disruption of the RecA filament by a translocating PcrA.
MATERIALS AND METHODS
Purification of PcrA, RecA and RecA K72R proteins
The G. stearothermophilus pcrA clone was obtained from Dale Wigley, and sub-cloned into the pET-14b plasmid with an N-terminal His6 tag. It was purified as described earlier after confirming the DNA sequence (23). The RecA K72R overexpression clone was obtained from Michael Cox. Overexpression and purification of RecA and the RecA K72R mutant was carried out as reported earlier for wild-type RecA (24).
Oligonucleotide substrates
Fluorescently labeled oligonucleotides were purchased from Integrated DNA Technologies (IDT) and were based on previously published sequences (13). The sequences of the oligonucleotides are 5′(dT)40 GCC TCG CTG CCG TCG CCA-3′ and 5′-TGG CGA CGG CAG CGA GGC-3′.
Single-molecule experiments
All single-molecule fluorescence resonance energy transfer (smFRET) experiments were carried out on a prism-based total internal reflection microscope that has been previously described (25). The buffer conditions were used as previously described for studying RecA displacement by PcrA (13). Incubations were performed at room temperature in a buffer containing 20 mM Tris–HCl (pH 8.0), 10 mM KCl, and 5 mM MgCl2 and an oxygen scavenging system (1 mg/ml glucose oxidase, 0.4% (w/v) D-glucose, 0.04 mg/ml catalase and 2 mM Trolox). In order to maintain the RecA concentration in the flow-cell during the addition of PcrA and ATP, 1 μM RecA or RecA K72R was always added along with PcrA for the displacement experiments. Alternating laser excitation at 10 Hz using a 532 nm diode laser (typically 7 mW; CrystaLaser) and a 642 nm diode laser (typically 6 mW; Melles Griot) allowed us to analyze only molecules with exactly one donor and one acceptor dye molecule. A 610 nm dichroic filter was used to split the fluorescence signal into donor and acceptor signal paths, which were further filtered by either a 580/40 nm bandpass filter or a 660 nm long-pass filter (all filters from Chroma Technology) and then imaged onto separate halves of an EMCCD (Andor iXon). Images were collected at 100 ms time resolution.
Data analysis
Fluorescence signals for individual molecules were background corrected for each frame (25) and FRET was calculated using FRET = IA/(ID + IA), where IA and ID are the intensities of the acceptor and donor dyes respectively. Normalized histograms were calculated from molecules acquired from multiple regions of a given flow cell/experiment (150 ≤ n ≤ 1000), and all histograms employed consistent bin widths (0.02 EFRET units/65 bins total) and range (EFRET = −0.2–1.2). The disruption bar graph (Figure 3B) was generated by summing the histogram data over all bins < EFRET = 0.43. The percentage of disruption was determined using percentage disruption = (r − p)/r, where r is the sum of histogram data from an experiment including RecA, but prior to the addition of PcrA, and p is the same sum after the addition of PcrA. The percentage of repetitive looping bar graph (Figure 3C) was generated by summing histogram data over all bins > EFRET = 0.61 and calculating (p − r). These experiments were repeated three times for each concentration of PcrA. Error bars in the graphs indicate the standard deviations for these triplicate measurements. All experiments were repeated three times, and representative histograms and single-molecule traces are shown.
Figure 3.

Nucleoprotein filaments formed by RecA K72R are disrupted by PcrA with a reduced efficiency compared with wild-type. (A) FRET histograms showing PcrA concentration-dependent displacement of 1 μM RecA and 1 μM K72R, respectively. (B) Comparison of the efficiency with which PcrA disrupts K72R (unshaded) and wild-type RecA (gray) filaments. Data from FRET≤ 0.43 from histograms in (A) were used to calculate clearing efficiency. (C) Comparison of repetitive looping efficiencies of PcrA in the presence of K72R (black) or RecA (gray). Data from FRET ≥ 0.61 from histograms in (A) were used to calculate the looping efficiency.
RESULTS
Selective inhibition of the ATPase activity of RecA prevents its displacement from ssDNA by PcrA in the presence of ATP
We used a smFRET assay to measure ssDNA translocation of PcrA as well as RecA displacement (13). In this assay, the DNA substrate is a single-stranded tailed duplex that is fluorescently labeled with a Cy3 donor at the 5′-end of the ssDNA and a Cy5 acceptor at the ss–dsDNA junction (Figure 1A). The substrate molecules are tethered to the surface of polyethylene glycol treated flow-cells using biotin–streptavidin linkages. Fluorescence measurements were carried out using a custom-built total internal reflection fluorescence (TIRF) microscope (25). Changes in distance between the donor and acceptor molecules resulting from RecA filament formation or its disruption by a translocating PcrA were reflected by changes in fluorescence intensities and the corresponding FRET values of individual DNA molecules.
Figure 1.

Translocating PcrA disrupts preformed RecA filaments that hydrolyze ATP. (A) Model showing smFRET assay for ATP hydrolysis-dependent RecA displacement from ssDNA by PcrA. The DNA substrate is a tailed duplex labeled with Cy3 (donor) at the 5′-end of the ssDNA tail and Cy5 (acceptor) at the ss/ds junction. Blue, gray and red spheres indicate PcrA, RecA-ATP and RecA-ADP, respectively. RecA filament formation holds the ssDNA tail of the substrate in a stretched state and decreases its FRET. RecA displacement by PcrA leads to an increase in FRET. Repetitive looping of ssDNA by PcrA that remains bound to the ss/ds junction prevents the reassembly of RecA filaments on ssDNA and leads to alternating mid- (free ssDNA) and high-FRET (looped ssDNA) states. (B) FRET histograms of the smFRET DNA substrate (black; indicated as a line for clarity) in the presence of RecA-ATP before (blue) and after (green) the addition of 20 nM PcrA, 1 μM RecA and 1 mM ATP. (C) ATP concentration-dependent repetitive looping of ssDNA by PcrA. Representative graphs of fluorescence intensities of Cy3 (green) and Cy5 (red) and corresponding FRET values (blue) obtained for the smFRET substrate depicted in the presence of 1 nM PcrA and different concentrations of ATP as indicated. (D) Representative graphs of the fluorescence intensities of Cy3 (green) and Cy5 (red) and the corresponding FRET values (blue) obtained for RecA displacement by PcrA in the presence of 1 nM PcrA and 1 mM ATP.
Assembly of a RecA filament is expected to lower the FRET as the distance between the Cy3 and Cy5 dyes increases due to the ssDNA being held in an extended conformation by the RecA filament [Figure 1A and (13,26)], whereas disruption of a filament by PcrA will increase the FRET. Subsequent to filament disruption, the ATP-hydrolysis fueled translocation of PcrA that remains bound to the ss/ds junction repetitively loops the ssDNA by reeling in the ssDNA tail in the 3′ to 5′ direction (Figure 1A). This brings the donor dye at the 5′-end of the ssDNA tail into close proximity with the acceptor dye at the junction to produce mid- (free DNA) and high-FRET (looped DNA) states in a cyclical manner (in Supplementary Movie S1).
In the absence of PcrA and RecA, the DNA substrate exhibited a peak FRET value of 0.4 (Figure 1B). The assembly of a RecA filament on the ssDNA in the presence of ATP reduced the peak FRET to 0.2. We also observed a second population of molecules with a peak FRET of 0.4, indicating the presence of naked DNA molecules. These results confirm the dynamic assembly and disassembly of RecA on DNA in the presence of ATP as shown previously (13,27,28). The addition of 20 nM PcrA to preformed RecA-ATP filaments reduced the population of low-FRET molecules (FRET≤ 0.2) by 78% based on data in Figure 1B. Two populations of molecules with peak FRET values of 0.5 and 0.8 were observed under these conditions, which reflect the displacement of RecA by PcrA and the subsequent repetitive looping of ssDNA by the helicase. We confirmed the ability of PcrA to repetitively loop the ssDNA tail in an ATP concentration-dependent manner (Figure 1C). In addition, gel-based assays confirmed the 3′ to 5′ unidirectional nature of G. stearothermophilus PcrA (Supplementary Figure S1).
The disruption of preformed RecA filaments was tested in real-time by adding 1 nM PcrA to preformed RecA filaments (Figure 1D). The disruption of RecA filaments was observed as a gradual increase in the fluorescence intensity of the acceptor dye with a corresponding decrease in donor dye intensity followed by the saw-tooth pattern of dye intensities and FRET signal, consistent with the repetitive looping activity of PcrA (Figure 1D). These results demonstrate that under conditions in which PcrA and RecA can both hydrolyze ATP, the helicase readily disrupts the nucleoprotein filaments formed by the recombinase. Since the translocase activity of PcrA continues unabated after a fully formed RecA filament is disrupted, these results also indicate that wild-type RecA is not able to reassemble stable filaments in the presence of the translocating helicase.
ATPγS is a competitive inhibitor of the ATPase activity of RecA and is hydrolyzed at a rate that is three orders of magnitude lower than that of ATP (29). RecA filaments formed in the presence of ATPγS are stable for several hours and do not exchange bound ATPγS for ATP in solution (29). We used these properties to selectively inhibit the ATPase activity of RecA and test its role in nucleoprotein filament disruption by ATP-dependent translocation of PcrA. We first assembled RecA-ATPγS filaments on the ssDNA substrate. RecA-ATPγS filaments had a peak FRET value of 0.2 (Figure 2A). The population of naked DNA molecules observed in the presence of RecA-ATP was absent in the presence of ATPγS (Figures 1B and 2A) These results indicated that RecA assembled stable filaments in the presence of ATPγS and ATP hydrolysis-coupled disassembly was largely non-existent. Next, the flow-cell containing the RecA-ATPγS filaments was washed with a buffer containing ATP to remove any free ATPγS that could inhibit PcrA, followed by the addition of PcrA in the same buffer. The distribution of FRET values remained unchanged, demonstrating that PcrA was unable to disrupt RecA-ATPγS filaments (Figure 2A). We confirmed that residual ATPγS was not responsible for the failure of PcrA to disrupt these filaments by performing a control reaction in which the DNA substrate was treated with ATPγS in the absence of RecA, washed with a buffer containing ATP to remove ATPγS, and subsequently incubated with PcrA and ATP (Figure 2B). The results showed that the substrate molecules exhibited both mid- and high-FRET peaks of 0.5 and 0.8, respectively, consistent with the repetitive looping of ssDNA by PcrA. Taken together, our findings demonstrate that the ATPase activity of RecA is essential for its displacement by PcrA.
Figure 2.
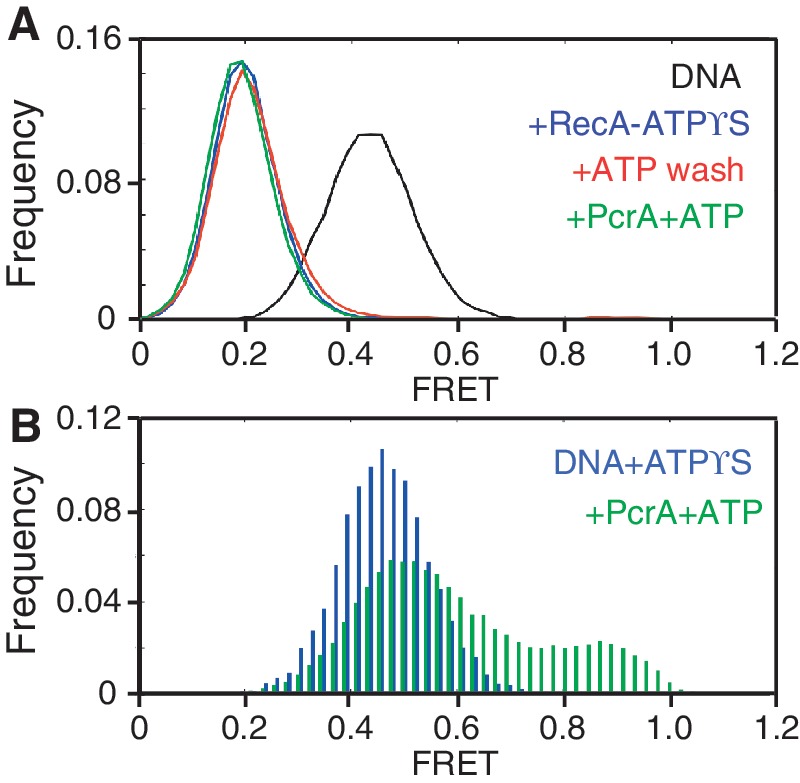
Selective inhibition of the ATPase activity of RecA prevents disruption of RecA filaments by a translocating PcrA. (A) FRET histograms of the smFRET substrate before and after the addition of 1 μM RecA, 1 mM ATPγS, 1 mM ATP and 10 nM PcrA. FRET histograms for each reaction condition that are closely overlapping are shown in different colors as line graphs. (B) FRET histograms of DNA incubated with 1 mM ATPγS before washing with a buffer containing 1 mM ATP and subsequently after the addition of 10 nM PcrA, 1 μM RecA and 1 mM ATP are shown in different colors.
Formation of the RecA-ADP complex, which has low affinity for ssDNA, is essential for its displacement from ssDNA by PcrA
ATP hydrolysis by RecA results in the low-affinity ssDNA binding state of RecA (i.e. the ADP-bound form) (30). To test whether PcrA could displace this form of RecA from the DNA, we used a site-directed mutant of RecA, K72R, which hydrolyses ATP at a significantly reduced rate relative to wild-type RecA (31,32). If ATP hydrolysis is not required for RecA displacement, then the K72R mutant would be displaced as efficiently as the wild-type protein. In contrast, if ATP hydrolysis is required, K72R would be displaced at a much lower efficiency.
After confirming the reduced ATPase activity of the mutant as well as its inability to complete three-strand DNA strand exchange (Supplementary Figure S2), we tested its assembly on ssDNA utilizing the smFRET assay. K72R mutant formed stable filaments on the DNA substrate in the presence of ATP and without residual naked DNA molecules observed with wild-type RecA (Figure 3A). As expected, the mutant also assembled filaments with nearly identical FRET signals in the presence of ATP, dATP, or ATPγS confirming that ATP-hydrolysis by this mutant is significantly lower (Supplementary Figure S3). Furthermore, the peak as well as the narrow distribution of FRET values following the assembly of K72R filaments in the presence of ATP was nearly identical to that of wild-type RecA filaments formed in the presence of ATPγS (Figures 2A and 3A). We observed that individual filaments of K72R were stable up to 2.5 min (Supplementary Figure S4).
To determine whether PcrA can disrupt stable K72R-ATP filaments, we tested the disassembly of these filaments using different concentrations of PcrA (Figure 3A–C). At the lowest concentration of PcrA tested (1 nM), there was little disruption of K72R filaments (4 ± 1%) compared with wild-type RecA (35 ± 5%). Differences in disruption of K72R and wild-type RecA filaments by PcrA were evident even at the highest concentration of PcrA tested (40 nM PcrA; 43 ± 10% versus 62 ± 4%, respectively, for K72R versus wild-type). Concentrations of PcrA > 40 nM were not tested in the displacement assays as previous studies have reported dimerization of PcrA above this concentration, which converts it to a processive helicase (33,34). These results showing that PcrA disrupts K72R filaments at a lower efficiency than those of wild-type RecA suggest that the formation of low-affinity RecA-ADP complex is essential for filament disruption by the helicase.
In addition to the differences in the displacement efficiencies, we noticed a striking difference in the percentage of substrates that exhibited a FRET value ≥0.61 following the addition of PcrA (Figure 3A and C). Depending on the PcrA concentration, there was a 2- to 10-fold decrease in the high-FRET population in the presence of K72R compared with wild-type RecA (Figure 3C). Since the high-FRET population predominantly reflects the looping activity of PcrA, our observations suggest that K72R has an inhibitory effect on the translocase activity of the helicase.
A polymerizing K72R filament impedes helicase translocation
On ssDNA, PcrA translocates in the 3′ to 5′ direction, whereas RecA polymerizes in the 5′ to 3′ direction (35,36). We surmised that in a ‘head-on collision’ between the helicase and a polymerizing filament, the 3′ to 5′ ATP-dependent translocation of PcrA would block the 5′ to 3′ polymerization of K72R, which generates stable nucleoprotein filaments. In contrast, PcrA should be able to use the ATPase activity of wild-type RecA to displace the low-affinity ADP-bound form of the protein.
To test this prediction, we injected either wild-type RecA or K72R into a reaction in which PcrA was already exhibiting repetitive looping of ssDNA in the presence of ATP, as demonstrated by the saw-tooth pattern of changing dye intensities and FRET (Figure 4A). These patterns remained unchanged after the addition of wild-type RecA, indicating that repetitive looping of PcrA continued unabated. In addition, the population of molecules from single-molecule traces failed to show assembly of stable RecA filaments in the presence of translocating PcrA (Figure 4B). It should be noted that in the absence of RecA, the repetitive looping activity of PcrA continues for at least 2.5 min (Supplementary Figure S5). In contrast, following the addition of K72R, the acceptor dye intensity dropped, with a corresponding decrease in FRET indicating the assembly of K72R filaments (Figure 4C and Supplementary Figure S6). The absence of saw-tooth pattern of both FRET and dye intensities after the injection of K72R indicated that PcrA translocation was arrested by the filament formation. Nearly half of the individual K72R filaments were disassembled at various times following their assembly, a behavior not observed in the absence of PcrA (Supplementary Figure S4) indicating that PcrA remained bound to the junction (Figure 4C; Supplementary Figure S6C and S6D). Their rate of disassembly and reassembly were highly variable and ranged from almost instantaneous to several seconds (Figure 4C and Supplementary Figure S6C). The diversity of this behavior probably reflects the diversity in the nucleation sites of K72R on the ssDNA portion of the substrate with respect to the translocating PcrA. The histogram of the distribution of FRET values in the presence of K72R showed an 82% reduction in the population of molecules that exhibited peak FRET values ≥0.8 (Figure 4D). The appearance of a new peak at 0.2 FRET under these conditions confirmed the assembly of stable K72R filaments. These results indicate that repetitive looping of PcrA is inhibited due to the assembly of K72R but not wild-type RecA filaments. Therefore, in a head-on collision between a polymerizing RecA filament and translocating PcrA, K72R RecA impedes helicase translocation. We conclude that PcrA takes advantage of the ATPase activity of RecA to prevent de novo filament assembly.
Figure 4.

Head-on collision between PcrA and RecA arrests translocation of PcrA in the absence of the ATPase activity of RecA. (A) Fluorescence intensities of Cy3 and Cy5 during repetitive looping of ssDNA by 10 nM PcrA on the smFRET substrate before and after the addition of RecA at t = 20 s. Only data up to t = 70 s are shown, although repetitive looping continued for up to 180 s (not shown). Corresponding FRET values are also shown. (B) FRET histogram for experiment shown in (A) before (green histogram) and after (black histogram) the addition of 1 μM RecA at t = 20 s. (C) Fluorescence intensities of Cy3 and Cy5 during an experiment similar to (A) except that 1 μM K72R was added at t = 30 s (black arrow). Corresponding FRET values are also shown. (D) FRET histograms before (green histogram) and after (black histogram) the addition of K72R at t = 30 s for the experiment in (C) showing filament formation by K72R in the presence of translocating PcrA.
RecA in the high-affinity ssDNA binding state resists displacement from ssDNA by PcrA
If formation of the low affinity ssDNA binding state of RecA is essential for its displacement by PcrA, the disruption of RecA-ADP filaments should be prevented in the presence of aluminium fluoride (AlF4). AlF4 can be used to replace the gamma phosphate in RecA prior to ADP release, thereby maintaining the high-affinity ssDNA binding state, or RecA-ADP-AlF4 form (37,38). We first tested the ability of PcrA to disrupt RecA-ADP filaments (Figure 5A). Addition of RecA to the DNA substrate in the presence of ADP reduced the peak FRET in more than one-half of the molecules from 0.4 to 0.2, indicating filament assembly (compare Figure 1B and Figure 5A). Upon adding PcrA and ATP, there was a 30% reduction in the population of molecules with a peak FRET of 0.2 (Figure 5A). The concomitant increase in high-FRET population reflects repetitive looping by PcrA in the presence of RecA and ADP. These results demonstrate that PcrA disrupts low-affinity RecA-ADP filaments, albeit less efficiently than RecA-ATP filaments (Figure 1B). This effect is likely due, at least in part, to the inhibition of the translocase activity of PcrA by the excess of ADP used to form RecA-ADP filaments because both the translocase activity of PcrA as well as the displacement of RecA-ATP filaments decreased in the presence of excess ADP (Supplementary Figure S7). Since the high-FRET population is relatively lower than that measured for PcrA disruption of RecA-ATP filaments, it is possible that the individual looping reactions themselves are incomplete in the presence of ADP. We confirmed that residual ADP did not inhibit PcrA by performing a control reaction in which the DNA substrate was treated with ADP in the absence of RecA, washed with a buffer containing ATP to remove ADP, and subsequently incubated with PcrA and ATP (Supplementary Figure S8A and S8B). The results showed that the substrate molecules exhibited both mid- and high-FRET peaks of 0.5 and 0.8, respectively, consistent with the repetitive looping of ssDNA by PcrA. We also confirmed that in the absence of PcrA, the addition of ATP to RecA-ADP filaments did not cause their disassembly (Supplementary Figure S8C and S8D).
Figure 5.
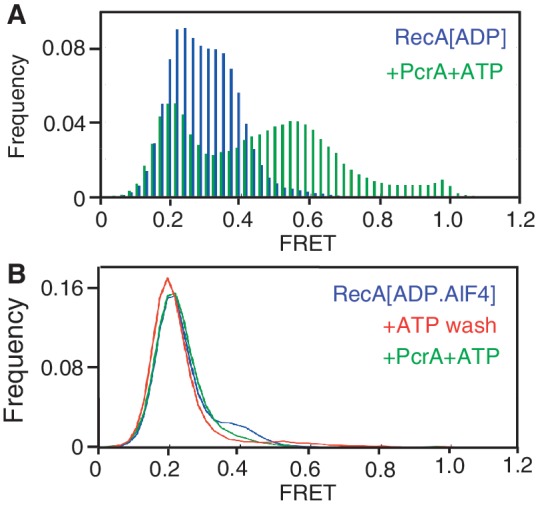
PcrA disrupts low-affinity RecA-ADP filaments but not high-affinity RecA-ADP-AlF4 filaments. (A) FRET histogram of the smFRET substrate in the presence of 1 μM RecA and 5 mM ADP (blue histogram) and after the addition of 10 nM PcrA, 1 μM RecA and 1 mM ATP (green histogram). (B) FRET histograms of the smFRET substrate after the addition of 1 μM RecA, 5 mM ADP-AlF4, 1 mM ATP and 10 nM PcrA. For clarity, histograms of closely overlapping populations are represented as line graphs.
Next, we tested the ability of RecA-ADP-AlF4 filaments to resist displacement by PcrA by adding RecA to the DNA substrate in the presence of ADP and AlF4 (Figure 5B). We observed that the peak FRET value of the substrates was reduced from 0.4 for naked DNA (see Figure 1B) to 0.2 indicating assembly of stable RecA-ADP-AlF4 filaments. The resulting distribution of FRET values is nearly identical to that for RecA-ATPγS and K72R-ATP and without residual naked DNA molecules (see Figures 2B and 3A). We then removed AlF4 from the flow-cell by washing it with a buffer containing ATP. As with ATPγS (see Figure 2B), we observed no change in the distribution of FRET values, as RecA-ADP-AlF4 did not exchange for ATP (Figure 5B). Finally, we treated RecA-ADP-AlF4 filaments with PcrA and ATP and observed that the FRET values of the entire population of molecules remained unchanged with a peak at 0.2, indicating that PcrA was unable to disrupt these filaments (Figure 5B). Based on these results, we conclude that the high-affinity ssDNA binding state of RecA resists displacement by PcrA.
DISCUSSION
In this article, we demonstrate that the ATPase activity of RecA is essential for its displacement from ssDNA by PcrA, thereby identifying a novel component in the regulation of this recombinase by an essential helicase. Our studies show that the translocase activity of PcrA is insufficient to displace RecA from ssDNA, thereby indicating the presence of multiple mechanisms in the regulation of RecA filament formation by PcrA. Additionally, results of the experiment involving a head-on collision between RecA and PcrA on a ssDNA ‘track’ show that the ATPase activity of RecA, which generates the low-affinity ADP-bound form of the protein on ssDNA, is essential for PcrA to remove this barrier, underlining the significance of this enzymatic activity of RecA in its regulation by PcrA. In its absence of its ATPase activity, the RecA filament presents an insurmountable barrier to the translocating helicase. We propose a new model for RecA displacement by PcrA that involves the hydrolysis of ATP to generate the low-affinity ssDNA binding state of RecA (Figure 6 and Supplementary Movie S2).
Figure 6.
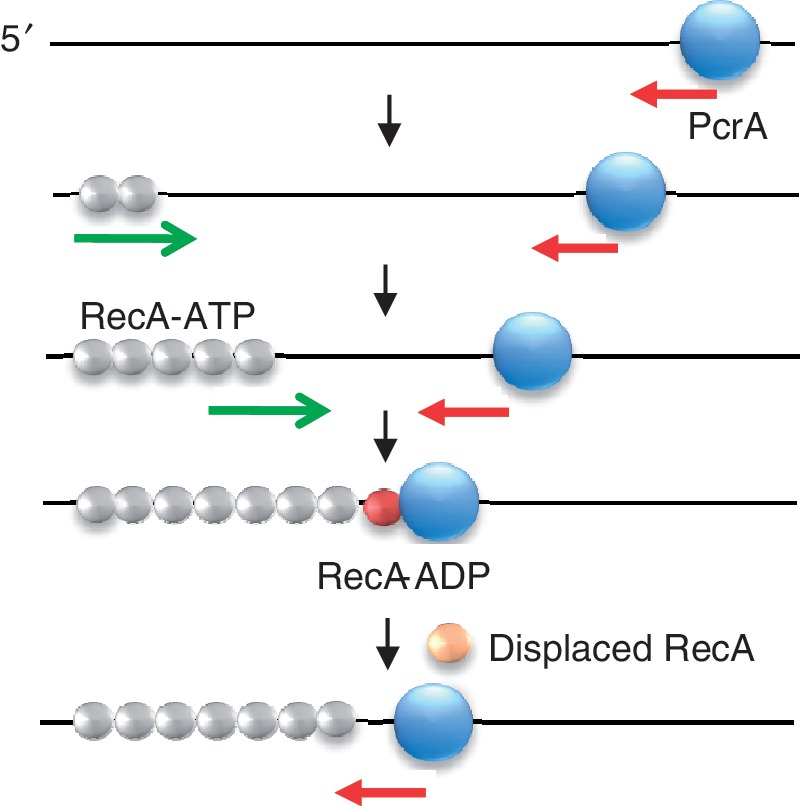
Model for PcrA-mediated disruption of RecA nucleoprotein filaments. Disruption of RecA filaments by a translocating PcrA (blue sphere) requires hydrolysis of ATP by RecA. Red arrow, 3′ to 5′ translocation of PcrA; green arrow, 5′ to 3′ polymerization of RecA. Formation of RecA-ADP, which has lower-affinity for DNA (indicated by the color change of RecA spheres from gray to red) allows PcrA to displace RecA from the DNA and disrupt an entire filament. A monomer of RecA displaced by translocating PcrA is shown as an orange sphere.
Since polymerization of RecA extends the filaments initiating on ssDNA into dsDNA portion of the substrate, it is possible that the inability of PcrA to disrupt RecA-ATPγS and RecA-ADP-AlF4 filaments (Figure 5) could be a result of the non-availability of the ss/ds junction for binding PcrA. However, our head-on collision experiments in which PcrA is prebound to DNA demonstrate that in the absence of the ATPase activity of RecA, the filaments formed by the recombinase present a barrier for PcrA translocation.
It could be argued that the inhibition of translocase activity of PcrA by K72R mutant (Figure 4) is a reflection of few opportunist molecules of PcrA that are able to bind to DNA when RecA has cycled off. Thus, RecA K72R could form such a PcrA-blocking filament at any time a PcrA helicase dissociates, and block PcrA after that. We discount this possibility based on the following observations: First, PcrA remains tightly bound to the junction and exhibits repetitive looping for long periods up to 2.5 min (Supplementary Figure S5). In our reactions we have washed off excess PcrA from the flow cell. Therefore, the repetitive looping activity is the result of a single molecule of PcrA that remains bound to the junction. Second, the individual filaments of K72R are very stable for extended periods of time up to 2.5 min (Supplementary Figure S4) whereas those that are assembled on DNA preincubated with PcrA are frequently disassembled (Figure 4C; Supplementary Figure S6C and S6D). Finally, in these same filaments, we observed repetitive looping of DNA for short durations until the reassembly of K72R filaments that again lead to inhibition of the looping activity of PcrA translocase. Taken together, these observations provide proof that PcrA remains bound to the DNA during the cycling of RecA filaments on DNA.
The ATPase activity of RecA (39,40) is dispensable for the SOS response, as well as for DNA strand exchange, except at stages such as branch migration and bypass of heterologous inserts that require RecA dissociation to generate RecA-ADP through ATP hydrolysis (41–43). Consistent with this, studies have shown that ATPase mutants of RecA such as K72R and E38K/K72R exhibit reduced levels of in vivo recombination and DNA repair following UV treatment (14,31). Taken together, these results suggest that the in vivo defects observed with these ATPase mutants of RecA, could at least partly stem from the inability of DNA helicases to disrupt stable mutant RecA filaments.
In vivo, the regulation of RecA-mediated recombination can occur during any of the stages of recombination including presynapsis, synapsis, the strand exchange reaction itself or the heteroduplex extension phase. The experiments reported in this study address the mechanism by which an essential bacterial helicase regulates the early stages of assembly of the presynaptic RecA filaments on ssDNA. It is known that these RecA filaments can also extend into neighboring dsDNA. Considering the fact that RecA filament stability on dsDNA is also dependent upon the nucleotide-bound state of RecA, it is possible that disruption of RecA filaments on dsDNA also requires the ATPase activity of RecA. Experiments are in progress to test whether this is true.
DNA helicases can remove replication barriers caused by proteins bound to the DNA (1,5–12,44,45). Several helicases have been used to identify the mechanisms involved in protein displacement from the DNA (46–48). These differences may be due to a broad range of mechanisms used to displace proteins bound to the DNA. In the case of RecBCD, it is clear that mechano-chemical forces generated as a result of helicase translocation are sufficient for displacing non-specific proteins bound to the DNA (48). This is also the case for the disruption of biotin–streptavidin interactions by Dda (49). Nevertheless, not all protein displacements by helicases occur solely due to their translocation on the DNA, as translocation by itself probably cannot provide specificity during protein displacement (3,15,50–52).
Our earlier studies have shown that ATPase mutants of S. aureus PcrA can displace RecA from the DNA (3). The inability of S. aureus PcrA to exhibit repetitive looping activity on the smFRET substrate (data not shown) precluded further studies to address the mechanism of helicase/translocase independent displacement of RecA by S. aureus PcrA from the DNA utilizing the smFRET assay reported here. However, since even an actively translocating G. stearothermophilus PcrA is incapable of displacing RecA filaments that do not hydrolyze ATP, it is safe to assume that helicase/translocase independent displacement of RecA by ATPase mutants of PcrA is also likely to involve utilization of the ATPase activity of RecA. Experiments are currently in progress to test this prediction by utilizing ATPase mutants of S. aureus PcrA and M. tuberculosis UvrD that have been reported to inhibit RecA-mediated DNA strand exchange.
Nucleoprotein filaments formed by recombinases such as RecA and Rad51 are major impediments for a translocating helicase. In the absence of ATP hydrolysis, such a filament is an immovable barrier consisting of hundreds of protein monomers bound tightly to the DNA. Upon ATP hydrolysis, the change from high- to low-affinity ssDNA binding state of the recombinase allows the helicase to sequentially displace monomers from these filaments. Our results suggest that PcrA and related helicases have evolved other mechanisms in addition to translocation to efficiently displace protein filaments from the DNA. Similar observations have also been made with the Srs2 helicase, which has been shown to stimulate the ATPase activity of Rad51 during the displacement of preformed Rad51 filaments from the DNA (50). In the absence of ATP hydrolysis, Rad51 filaments present an insurmountable barrier for the translocating Srs2. These findings are consistent with the results presented herein and suggest that the ATPase activity of the recombinase required by DNA helicases for filament disruption is likely to be a conserved theme in both prokaryotes and eukaryotes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online: Supplementary Figures 1–8, Supplementary Movies 1 and 2 and Supplementary References [53,54].
FUNDING
National Institutes of Health (NIH) [R01GM31685 to S.A.K., R01GM077872 to S.H.L]; National Institute of General Medical Sciences fellowship [F32GM082132 to K.R.T.]. Funding for open access charge: NIH [R01GM31685 to S.A.K.; R01GM077872 to S.H.L.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dale Wigley for the PcrA expression clone and Michael Cox for the RecA K72R expression clone. We also thank Ranjith Anand, Laurence Brewer, Michelle Kienholz, Harshad Ghodke and Ben Van Houten for critical reading of the manuscript. M.V.F. collected and analyzed the smFRET data. K.R.T. biochemically assayed PcrA. P.R.B. purified RecA. M.V.F. and S.H.L. designed and built the TIRF microscope. M.V.F., S.P.A., G.D.S., P.R.B., S.A.K. and S.H.L. helped design the experiments and analyzed the data. S.P.A. designed the experimental strategy, purified PcrA and RecAK72R, characterized the K72R mutant, and wrote the manuscript. All authors contributed to the interpretation of results and the final manuscript.
REFERENCES
- 1.Lohman T, Tomko E, Wu C. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 2.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 3.Anand S, Zheng H, Bianco P, Leuba S, Khan S. DNA helicase activity of PcrA is not required for the displacement of RecA protein from DNA or inhibition of RecA-mediated strand exchange. J. Bacteriol. 2007;189:4502–4509. doi: 10.1128/JB.00376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benkovic S, Valentine A, Salinas F. Replisome-mediated DNA replication. Annu. Rev. Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 5.Bidnenko V, Lestini R, Michel B. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol. Microbiol. 2006;62:382–396. doi: 10.1111/j.1365-2958.2006.05382.x. [DOI] [PubMed] [Google Scholar]
- 6.Flores M, Sanchez N, Michel B. A fork-clearing role for UvrD. Mol. Microbiol. 2005;57:1664–1675. doi: 10.1111/j.1365-2958.2005.04753.x. [DOI] [PubMed] [Google Scholar]
- 7.Fonville N, Blankschien M, Magner D, Rosenberg S. RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair (Amst.) 2010;9:403–413. doi: 10.1016/j.dnarep.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lestini R, Michel B. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007;26:3804–3814. doi: 10.1038/sj.emboj.7601804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maples VF, Kushner SR. DNA repair in Escherichia coli: identification of the uvrD gene product. Proc. Natl Acad. Sci. USA. 1982;79:5616–5620. doi: 10.1073/pnas.79.18.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petit M, Dervyn E, Rose M, Entian K, McGovern S, Ehrlich S, Bruand C. PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol. 1998;29:261–273. doi: 10.1046/j.1365-2958.1998.00927.x. [DOI] [PubMed] [Google Scholar]
- 11.Petit M, Ehrlich D. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 2002;21:3137–3147. doi: 10.1093/emboj/cdf317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, Fabre F, Petit MA. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J, Myong S, Niedziela-Majka A, Lee K, Yu J, Lohman T, Ha T. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell. 2010;142:544–555. doi: 10.1016/j.cell.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Centore R, Sandler S. UvrD limits the number and intensities of RecA-green fluorescent protein structures in Escherichia coli K-12. J. Bacteriol. 2007;189:2915–2920. doi: 10.1128/JB.01777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh P, Patil KN, Khanduja JS, Kumar PS, Williams A, Rossi F, Rizzi M, Davis EO, Muniyappa K. Mycobacterium tuberculosis UvrD1 and UvrA proteins suppress DNA strand exchange promoted by cognate and noncognate RecA proteins. Biochemistry. 2010;49:4872–4883. doi: 10.1021/bi902021d. [DOI] [PubMed] [Google Scholar]
- 16.Williams A, Guthlein C, Beresford N, Bottger EC, Springer B, Davis EO. UvrD2 is essential in Mycobacterium tuberculosis, but its helicase activity is not required. J. Bacteriol. 2011;193:4487–4494. doi: 10.1128/JB.00302-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox M. Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell Biol. 2007;8:127–138. doi: 10.1038/nrm2099. [DOI] [PubMed] [Google Scholar]
- 18.Cox M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 19.DasGupta C, Wu A, Kahn R, Cunningham R, Radding C. Concerted strand exchange and formation of Holliday structures by E. coli RecA protein. Cell. 1981;25:507–516. doi: 10.1016/0092-8674(81)90069-6. [DOI] [PubMed] [Google Scholar]
- 20.West SC, Cassuto E, Howard-Flanders P. RecA protein promotes homologous-pairing and strand-exchange reactions between duplex DNA molecules. Proc. Natl Acad. Sci. USA. 1981;78:2100–2104. doi: 10.1073/pnas.78.4.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kowalczykowski S, Eggleston A. Homologous pairing and DNA strand-exchange proteins. Annu. Rev. Biochem. 1994;63:991–1043. doi: 10.1146/annurev.bi.63.070194.005015. [DOI] [PubMed] [Google Scholar]
- 22.Menetski JP, Bear DG, Kowalczykowski SC. Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc. Natl Acad. Sci. USA. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang T, Naqvi A, Anand S, Kramer M, Munshi R, Khan S. Biochemical characterization of the Staphylococcus aureus PcrA helicase and its role in plasmid rolling circle replication. J. Biol. Chem. 2002;277:45880–45886. doi: 10.1074/jbc.M207383200. [DOI] [PubMed] [Google Scholar]
- 24.Bianco P, Weinstock G. Interaction of the RecA protein of Escherichia coli with single-stranded oligodeoxyribonucleotides. Nucleic Acids Res. 1996;24:4933–4939. doi: 10.1093/nar/24.24.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fagerburg MV, Leuba SH. Optimal practices for surface-tethered single molecule total internal reflection fluorescence resonance energy transfer analysis. Methods Mol. Biol. 2011;749:273–289. doi: 10.1007/978-1-61779-142-0_19. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Yang H, Pavletich N. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–484. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- 27.Shan Q, Cox M. RecA protein dynamics in the interior of RecA nucleoprotein filaments. J. Mol. Biol. 1996;257:756–774. doi: 10.1006/jmbi.1996.0200. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Jacobs SA, West SC, Ogawa T, Egelman EH. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl Acad. Sci. USA. 2001;98:8419–8424. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinstock GM, McEntee K, Lehman IR. Interaction of the recA protein of Escherichia coli with adenosine 5′-O-(3-thiotriphosphate) J. Biol. Chem. 1981;256:8850–8855. [PubMed] [Google Scholar]
- 30.Menetski JP, Kowalczykowski SC. Interaction of recA protein with single-stranded DNA. Quantitative aspects of binding affinity modulation by nucleotide cofactors. J. Mol. Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 31.Britt R, Chitteni-Pattu S, Page A, Cox M. RecA K72R filament formation defects reveal an oligomeric RecA species involved in filament extension. J. Biol. Chem. 2011;286:7830–7840. doi: 10.1074/jbc.M110.194407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rehrauer W, Kowalczykowski S. Alteration of the nucleoside triphosphate (NTP) catalytic domain within Escherichia coli recA protein attenuates NTP hydrolysis but not joint molecule formation. J. Biol. Chem. 1993;268:1292–1297. [PubMed] [Google Scholar]
- 33.Niedziela-Majka A, Chesnik M, Tomko E, Lohman T. Bacillus stearothermophilus PcrA monomer is a single-stranded DNA translocase but not a processive helicase in vitro. J. Biol. Chem. 2007;282:27076–27085. doi: 10.1074/jbc.M704399200. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Dou SX, Ren H, Wang PY, Zhang XD, Qian M, Pan BY, Xi XG. Evidence for a functional dimeric form of the PcrA helicase in DNA unwinding. Nucleic Acids Res. 2008;36:1976–1989. doi: 10.1093/nar/gkm1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dillingham M, Wigley D, Webb M. Demonstration of unidirectional single-stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry. 2000;39:205–212. doi: 10.1021/bi992105o. [DOI] [PubMed] [Google Scholar]
- 36.Register J, III, Griffith J. The direction of RecA protein assembly onto single strand DNA is the same as the direction of strand assimilation during strand exchange. J. Biol. Chem. 1985;260:12308–12312. [PubMed] [Google Scholar]
- 37.Kowalczykowski S, Krupp R. DNA-strand exchange promoted by RecA protein in the absence of ATP: implications for the mechanism of energy transduction in protein-promoted nucleic acid transactions. Proc. Natl Acad. Sci. USA. 1995;92:3478–3482. doi: 10.1073/pnas.92.8.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreau PL, Carlier MF. RecA protein-promoted cleavage of LexA repressor in the presence of ADP and structural analogues of inorganic phosphate, the fluoride complexes of aluminum and beryllium. J. Biol. Chem. 1989;264:2302–2306. [PubMed] [Google Scholar]
- 39.Cox J, Tsodikov O, Cox M. Organized unidirectional waves of ATP hydrolysis within a RecA filament. PLoS Biol. 2005;3:231–243. doi: 10.1371/journal.pbio.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox M. Why does RecA protein hydrolyse ATP? Trends Biochem. Sci. 1994;19:217–222. doi: 10.1016/0968-0004(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 41.Cox M, Lehman I. RecA protein of Escherichia coli promotes branch migration, a kinetically distinct phase of DNA strand exchange. Proc. Natl Acad. Sci. USA. 1981;78:3433–3437. doi: 10.1073/pnas.78.6.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahn R, Cunningham R, DasGupta C, Radding C. Polarity of heteroduplex formation promoted by Escherichia coli recA protein. Proc. Natl Acad. Sci. USA. 1981;78:4786–4790. doi: 10.1073/pnas.78.8.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J, Cox M, Inman R. On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. II. Four-strand exchanges. J. Biol. Chem. 1992;267:16444–16449. [PubMed] [Google Scholar]
- 44.Krejci L, Van Komen S, Li Y, Villemain J, Reddy M, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 45.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 46.Yeruva L, Raney KD. Protein displacement by helicases. Methods Mol. Biol. 2010;587:85–98. doi: 10.1007/978-1-60327-355-8_6. [DOI] [PubMed] [Google Scholar]
- 47.Eggleston A, O'Neill T, Bradbury E, Kowalczykowski S. Unwinding of nucleosomal DNA by a DNA helicase. J. Biol. Chem. 1995;270:2024–2031. doi: 10.1074/jbc.270.5.2024. [DOI] [PubMed] [Google Scholar]
- 48.Finkelstein I, Visnapuu M, Greene E. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature. 2010;468:983–987. doi: 10.1038/nature09561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Byrd A, Raney K. Protein displacement by an assembly of helicase molecules aligned along single-stranded DNA. Nat. Struct. Mol. Biol. 2004;11:531–538. doi: 10.1038/nsmb774. [DOI] [PubMed] [Google Scholar]
- 50.Antony E, Tomko E, Xiao Q, Krejci L, Lohman T, Ellenberger T. Srs2 disassembles Rad51 filaments by a protein–protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol. Cell. 2009;35:105–115. doi: 10.1016/j.molcel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward JD, Muzzini DM, Petalcorin MI, Martinez-Perez E, Martin JS, Plevani P, Cassata G, Marini F, Boulton SJ. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol. Cell. 2010;37:259–272. doi: 10.1016/j.molcel.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 52.Williams AB, Michael WM. Eviction notice: new insights into Rad51 removal from DNA during homologous recombination. Mol. Cell. 2010;37:157–158. doi: 10.1016/j.molcel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Anand SP, Khan SA. Structure-specific DNA binding and bipolar helicase activities of PcrA. Nucleic Acids Res. 2004;32:3190–3197. doi: 10.1093/nar/gkh641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell MJ, Davis RW. Toxic mutations in the recA gene of E. coli prevent proper chromosome segregation. J. Mol. Biol. 1999;286:417–435. doi: 10.1006/jmbi.1998.2456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.