

Abstract
Nowadays, primary hyperparathyroidism (PHPT) is mostly a mild disease. Overt skeletal manifestations are rare but decreased bone mineral density (BMD) can still be demonstrated. Even in mild cases, excess parathyroid hormone (PTH) increases bone turnover leading to bone loss particularly at cortical sites. Conversely, a relative preservation of cancellous bone has been shown by histomorphometric analyses and advanced imaging techniques. An increased fracture rate has been demonstrated in untreated patients with PHPT at peripheral sites and in the spine. Parathyroidectomy (PTx) is the definitive cure for PHPT. With the restoration of normal PTH, bone resorption is quickly tapered down, while bone formation proceeds at the level of bone multicellular units, which were activated prior to PTx. The rapid refilling of the enlarged remodeling space and the subsequent matrix mineralization will result in an increase in BMD at sites rich in trabecular bone, such as lumbar spine and hip, which mainly occurs during the first 6–12 months after PTx. Cortical bone is less responsive to PTX because of the low rate of bone turnover, but sensible increases in BMD at the distal third of the radius can be observed in the long term. PTx seems to decrease the risk of fractures but more data are needed before a definitive conclusion on this important matter can be reached. Treatment with bisphosphonates can be considered for patients with low BMD who do not undergo PTx. Two-year treatment with alendronate has been shown to decrease bone turnover markers and increase BMD at the lumbar spine and hip, but not at the distal radius. Cinacalcet stably decreased serum calcium levels across a broad range of PHPT severity, but no change in BMD occurred in patients treated for up to 5.5 years.
Keywords: bisphosphonates, bone markers, bone mineral density, calcimimetics, histomorphometry, osteitis fibrosa cystica, parathyroidectomy, parathyroid hormone
Introduction
Bone is a classic target organ in primary hyperparathyroidism (PHPT), which is the third most common endocrine disease with the highest incidence in postmenopausal women [Fraser, 2009]. In the past, PHPT was often diagnosed in the context of osteitis fibrosa cystica, a severe skeletal disease characterized by brown tumors, bone cysts and deformities, due to extremely elevated bone resorption elicited by continuously high parathyroid hormone (PTH) levels. Nowadays, PHPT is mostly an asymptomatic, mild disease [Silverberg et al. 2009]. Overt skeletal manifestations are rare but bone involvement can still be demonstrated by means of dual-emission X-ray absorptiometry (DXA). While PHPT is characterized by bone loss in the cortical compartment, histomorphometric analyses of bone biopsies and more advanced imaging techniques have shown a relative preservation of cancellous bone and favorable changes in bone geometry. An increased risk for fracture can be demonstrated in untreated PHPT [Mosekilde, 2008]. Bone loss can be at least partially reversed after parathyroidectomy (PTx), which represents the definitive treatment for this disease. Alternative treatments for PHPT have been proposed to improve bone density and potentially decrease fracture risk [Lewiecki, 2010]. Nonetheless, longitudinal data are still lacking in this field.
Herein, we review the various effects of PTH on skeletal modeling and remodeling and discuss the classic and newly discovered skeletal features in PHPT.
Parathyroid effects on adult bone
PTH is one of the major players in the regulation of mineral homeostasis [Hanley et al. 2008], together with hormones classically involved in calcium and phosphate homeostasis, such as calcitriol [1,25(OH)2 vitamin D] and FGF23, and other hormones targeting bone, such as sex hormones, cortisol, thyroid hormones and growth hormone/insulin-like growth factor 1. Its main role is to maintain serum calcium and phosphate concentrations within the normal range. This is accomplished through a tight, direct regulation of calcium and phosphate fluxes, by increasing renal tubular calcium reabsorption and bone resorption. Moreover, PTH indirectly promotes intestinal calcium absorption by inducing renal activation of vitamin D. PTH secretion is strictly regulated by serum ionized calcium (Ca2+), which interacts with the calcium sensing receptor (CASR) expressed on parathyroid and renal proximal tubule cells. A decrease in Ca2+ stimulates PTH release and, if prolonged, PTH synthesis and parathyroid hyperplasia. At bone level PTH displays direct effects, which will be different according to the intensity and duration of PTH stimulus [Silva et al. 2011].
Catabolic versus anabolic action of parathyroid hormone in bone
The effects of PTH in bone can be either catabolic and/or anabolic (Figure 1). The prevalence of one action over the other will determine the net effect of PTH on bone mass and microarchitecture [Silva et al. 2011]. In the setting of PHPT, a continuous high level of PTH will enhance bone catabolism by a prevalent effect on osteoclast activation. This will lead to a reduced bone mass preferentially at cortical sites and an increased risk of fracture in people with PHPT, as further discussed in this chapter. Conversely, the intermittent administration of full-length PTH (PTH1-84) or biologically active shortened fragment teriparatide (PTH1-34) has been employed as treatment for osteoporosis because of its anabolic properties [Compston, 2007]. Evaluation of markers of bone turnover and histomorphometric analysis of bone biopsies demonstrates that there is an initial uncoupling between bone formation and bone resorption at the beginning of intermittent PTH administration [Dobnig et al. 2005]. In this limited interval, also referred to as the ‘anabolic window’, bone modeling due to PTH effects on osteoblasts and osteocytes overcomes bone remodeling due to PTH effects on osteoclasts. This anabolic effect is typically observed at sites rich in cancellous bone, such as lumbar spine, with improvement in bone volume, microarchitecture and connectivity, as demonstrated by DXA, bone histomorphometry and µCT studies [Rubin and Bilezikian, 2005]. In parallel, cortical bone seems to be minimally affected by intermittent administration of PTH.
Figure 1.

Effects of parathyroid hormone (PTH) on bone cells. The anabolic effects are indicated by the red lines and highlighted by the red shadow, while the catabolic effects are indicated in black and highlighted by the grey shadow (‘+’ = stimulation; ‘–’ = inhibition). See text for details. OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor κB ligand.
Cellular and molecular mechanisms of PTH action in bone
PTH exerts its effects through binding to a specific membrane receptor (PTH1R), a G-protein-coupled receptor. In adult bone PTH1R is expressed in osteoblasts [Datta and Abu-Samra, 2009]. Recent evidence suggests that PTH is also expressed in osteocytes and osteoclasts [Rhee et al. 2011; Dempster et al. 2005]. After binding to its receptor, PTH leads to a Gαs-mediated activation of adenyl cyclase, which in turn activates protein kinase A (PKA), and Gαq-mediated activation of phospholipase C and protein kinase C [Datta and Abu-Samra, 2009].
In coculture experiments, osteoblasts continuously exposed to PTH sustain osteoclastogenesis, mimicking the effect of continuous exposure to high levels of PTH as occurs in patients with PHPT [Silva et al. 2011]. Under these conditions PTH will lead to an increased osteoblast expression of the receptor activator of nuclear factor κB ligand (RANKL). RANKL will bind to its receptor (RANK) expressed on osteoclast precursors, thus stimulating osteoclastogenesis and osteoclast activity [Ma et al. 2001]. In parallel, PTH induces a decrease in osteoprotegerin (OPG), a decoy receptor for RANKL, which prevents RANKL interaction with RANK [Huang et al. 2004]. The increased RANKL/OPG ratio is thought to be the main mechanism by which a sustained high level of PTH elicits bone resorption [Nakchbandi et al. 2008]. Additional mechanisms involve the stimulation of other mediators such as the monocyte chemoattractant protein 1 (MCP-1), which might also contribute to PTH action on osteoclastogenesis [Li et al. 2007].
Recent in vitro evidence suggests that PTH can also directly stimulate osteoclasts since PTH1R is expressed in osteoclast precursors [Dempster et al. 2005]. Peripheral blood monocytes cultured on bone slices and exposed to RANKL and colony-stimulating factor differentiate into fully mature osteoclasts and express PTH1R. At this time, the addition of PTH1-34 results in a two- to threefold increase in bone resorption [Dempster et al. 2005].
Gene expression profiles differ in osteoblasts exposed to intermittent PTH, thus supporting the osteoanabolic action of PTH, which is mainly cyclic adenosine monophosphate mediated and due to PKA activation. PTH induces osteoblast differentiation and decreases osteoblast apoptosis, thus increasing fully mature osteoblast number (Figure 1). PTH induces osteoblast differentiation as demonstrated by increased expression of osteoblast-specific genes such as Runx2, collagen type I α1 in immature osteoblasts and alkaline phosphatase and osteocalcin in mature osteoblasts [Ishizuya et al. 1997]. Along with the prodifferentiative effect, PTH decreases immature osteoblast proliferation, enhancing several cell-cycle inhibitors such as p21 and p27 and reducing expression of cyclin D1, a potent inducer of cell cycle [Datta et al. 2007]. Regarding apoptosis, PTH reduces the expression of proapoptotic genes, such as Bad, and enhances the expression of antiapoptotic proteins, such as Bcl-2 [Bellido et al. 2003]. In addition, PTH promotes DNA repair, thus enhancing cell survival [Schnoke et al. 2009].
A newly identified target cell of PTH is the osteocyte. Sclerostin, the product of Sost gene, is produced by osteocytes and negatively regulates bone formation by inhibiting the Wnt-ß catenin pathway [Poole et al. 2005]. Intermittent PTH directly inhibits sclerostin production by osteocytes, thus enhancing bone formation. As a matter of fact, a transgenic mouse overexpressing PTH1R in the osteocytes displays an enhanced bone mass and a decreased expression of Sost [Rhee et al. 2011]. Thus, PTH anabolic action in bone is sustained not only by the direct actions in osteoblasts but also in osteocytes [Kramer et al. 2010] (Figure 1).
PTH is also directly linked to the development of bone marrow fibrosis in hyperparathyroidism and other conditions such as bone fibrous dysplasia. Mesenchymal and hematopoietic stem cells are indeed regulated by PTH [Ohishi and Schipani, 2011].
Skeletal features of primary hyperparathyroidism
As mentioned before, nowadays most patients with PHPT are diagnosed by routine serum calcium measurement and not by the presence of clinical clues suggesting this diagnosis [Marcocci and Cetani, 2011]. Nevertheless, a bone involvement can still be demonstrated. PHPT is characterized by an increased activation frequency of bone multicellular units (BMUs), resulting in an increased bone remodeling space. In cancellous bone the activation frequency of BMUs and the number of osteoblasts and osteoclasts are increased, but the resorption depth is swallowed and the bone formation period is longer. This can explain the relative preservation of trabecular bone often observed in mild PHPT. Conversely, in the cortical compartment cortical porosity and endocortical bone resportion are enhanced, thus leading to cortical bone loss. These ultrastructural events account for the increase in markers of bone turnover and changes in bone mineral density (BMD).
Untreated primary hyperparathyroidism
Bone turnover markers and vitamin D status
Markers of bone formation activity (bone alkaline phosphatase and osteocalcin) and bone resorption activity (urinary deoxypyridinoline and serum and urinary carboxy-terminal collagen crosslinks of collagen) are normal or in the upper normal range in most patients with mild PHPT, but can be increased in patients with more severe disease [Guo et al. 1996]. Figure 2 depicts data in a consecutive series of patients undergoing PTx.
Figure 2.

Baseline levels of serum bone alkaline phosphatase (BALP) and serum carboxy-terminal collagen crosslinks (S-CTX) in both sexes in a series of patients with primary hyperparathyroidism selected for parathyroidectomy (women in gray, men in black); normal range is indicated between or below the dashed lines.
Serum concentration of 25-hydroxyvitamin D (25OHD) is often below the lower normal limit. The low 25OHD values are likely due either to its increased conversion to 1,25(OH)2 vitamin D and accelerated catabolism (reviewed by Silverberg [2007]). In contrast, the 1,25(OH)2 vitamin D level tends to be in the upper range of normal and elevated in about one-quarter of patients with PHPT. It has been shown that low 25OHD levels are associated with increased indices of disease activity, namely higher serum levels of PTH, accelerated parathyroid tumor growth, increased bone turnover, and more severe bone loss [Rao et al. 2000; Silverberg, 2007].
X-ray evaluation
Severe forms of PHPT are associated with skeletal complications that are radiographically evident [Cope, 1966]. These include, in addition to the classical features of osteitis fibrosa cystica, subperiosteal resorption of the distal phalanges and a ‘salt and pepper’ appearance of the skull (Figure 3). Nowadays, overt bone disease is seen in less than 2% of patients with PHPT [Bilezikian and Silverberg, 2000; Miller and Bilezikian, 2002] and therefore skeletal X-ray evaluation is not routinely performed. However, in some areas of the world, especially where vitamin D deficiency is endemic, symptomatic PHPT still dominates and radiological manifestations of the disease can be detected [Rao et al. 2002; Raef et al. 2004].
Figure 3.

Long bone X-rays of a patient with severe primary hyperparathyroidism and osteitis fibrosa cystica. Arrows indicate brown tumors.
Bone mineral density
BMD has become essential in skeletal evaluation of patients with PHPT. Because of its greater sensitivity compared with conventional X-ray, it can detect bone involvement even in mild disease [Silverberg et al. 1989; Miller and Bilezikian, 2002]. BMD is measured by DXA at lumbar spine, hip and one-third distal radius. Information obtained from BMD measurement is used to make recommendations for parathyroid surgery or surveillance [Bilezikian et al. 2009].
The classical pattern of bone densitometry in PHPT is the preferential reduction in BMD at the distal radius (one-third site), with lower degree of involvement at sites rich in trabecular bone [Silverberg et al. 1989]. Thus, BMD at lumbar spine, which is relatively rich in trabecular bone, generally shows a modest reduction. BMD at the hip, containing an admixture of trabecular and cortical bone, shows intermediate involvement between the lumbar spine and distal radius. The same BMD profile is also seen in postmenopausal women with PHPT, in whom trabecular bone loss would be expected, suggesting that PHPT protects from the bone loss secondary to estrogen deficiency. This typical BMD profile is not always present in patients with PHPT at the time of diagnosis, and 15% of women may have vertebral osteopenia or osteoporosis [Silverberg et al. 1996]. This observation is one of the reasons why the current guidelines for surgery also recommend the measurements of BMD at lumbar spine, in addition to hip and forearm [Bilezikian et al. 2009]. In severe forms of PHPT, reductions in BMD at all three sites can be evident.
Long-term follow-up studies have shown that successful PTx is followed by an increase in BMD [Silverberg et al. 1999; Rubin et al. 2008] (see below). In patients with mild PHPT followed without PTx, BMD remains stable for up to 8 years at all three sites, but afterwards a progressive decrease occurs at cortical sites (distal forearm and hip) [Rubin et al. 2008]. This late bone loss was not predictive of whether patients met or did not meet surgical criteria at initial evaluation.
Quantitative bone ultrasound
Several studies have evaluated bone impairment in patients with PHPT using quantitative ultrasound techniques (QUS), which provide information not only on bone mass but also on bone microarchitecture [Gonnelli et al. 2000; Ingle et al. 2002; Camozzi et al. 2003; Chappard et al. 2006; Cipriani et al. 2009]. QUS measurements have been performed at calcaneus and distal phalanges, sites enriched of trabecular or cortical bone, respectively. All studies showed reduction of most but not all parameters in patients with PHPT compared with controls at the calcaneus and distal phalanges. QUS results were variable but not always correlated with BMD at different skeletal sites. Measurement of QUS parameters at calcaneus and distal phalanges after successful PTx showed a significant increase over 1–2 years [Gonnelli et al. 2000; Ingle et al. 2002].
Histomorphometry and bone structure
Bone histomorphometry has played an important role in clarifying the skeletal effect of mild asymptomatic PHPT. Histomorphometric analysis of the bone biopsy of iliac crest from patients with mild PHPT has demonstrated maintenance of trabecular bone volume, cortical thinning and accelerated bone remodeling [Parisien et al. 1990]. Bone biopsy studies have also shown that in patients with PHPT there is greater cancellous bone volume, increased trabecular number, and preserved trabecular connectivity [Parisien et al. 1990; Dempster et al. 1999]. Preserved trabecular connectivity accounts for the finding that the age-dependent loss of trabecular bone is not seen in postmenopausal women with PHPT. These data suggest that the trabecular plates and their connections are maintained more effectively in patients with PHPT than in normal aging individuals. To further support these findings the same authors compared three groups of postmenopausal women (women with PHPT, healthy women and women with osteoporosis) matched by years since menopause. Trabecular bone volume and indices of connectivity were highest in the patients with PHPT and lowest in those with osteoporosis [Parisien et al. 1995].
Dempster and colleagues confirmed the preservation of trabecular bone in patients with PHPT using microcomputed tomography [Dempster et al. 2007]. In this study, iliac crest bone biopsies were obtained from pre- and postmenopausal women and men with mild PHPT and control women. Preservation of cancellous bone structure and connectivity density and no change in bone surface/total volume was found in postmenopausal women with PHPT compared with healthy women. No difference in any parameters was found between premenopausal women with PHPT and control women or women and men with PHPT.
Fourier transform infrared imaging analysis, a validated spectroscopic technique that measures the ratio of collagen crosslinks in mineralized sections of bone and gives information about the relative maturity of bone, has been used in iliac crest bone biopsies from patients with PHPT [Zoehrer et al. 2008]. Patients with PHPT had a significantly lower collagen crosslinks ratio compared with normal controls. PTx restored this ratio to that observed in normal controls.
Bone mineralization density distribution, an important aspect of bone material quality, has been analyzed in iliac crest bone biopsies from patients with PHPT (16 men and 35 women) by quantitative backscattered electron imaging [Roschger et al. 2007]. This technique allows estimation of the mineralization density (mineral content in bone matrix) and its distribution at the microscopic level. Young bone structural units are less mineralized than older ones. In patients with PHPT the average mineralization density is reduced and there is an increased heterogeneity of the degree of mineralization. A strong correlation was found between these changes and the bone turnover rate. Reduced mineralization density in patients with PHPT would be expected to reduce the stiffness of bone tissue.
Fracture risk
As mentioned above, a variable degree of bone loss occurs in patients with PHPT. Several studies have investigated fracture rates in patients with untreated PHPT. An increased risk of fracture compared with the general population has been reported in most studies, but to what extent the risk of fracture is increased in PHPT is still controversial (for a review, see Mosekilde [2008]). Several factors, such as the retrospective nature of all studies, the selection of the patients and controls, and the methods to define vertebral fractures may account for the discrepant data.
Despite the preferential involvement of cortical bone in PHPT, an increased fracture rate has been reported at peripheral sites (forearm) and in the spine. In evaluating fracture risk in PHPT, other skeletal effects of PTH that contribute to bone quality and strength, besides BMD, should be taken into account. Enlarged cross-sectional diameter of the bone, due to increased periosteal apposition and endosteal resorption, may provide greater biomechanical competence. In addition, preserved cancellous microarchitecture probably adds strength to bone in PHPT [Dempster et al. 2007]. The positive effects of increased cross-sectional diameter and preserved cancellous microstructure should tend to counteract the negative effects of PTH on cortical thickness. However, the increased bone turnover might lead to reduce mineralization, which may increase the risk of fracture.
In a large cohort study performed in Denmark, which mainly included patients who underwent parathyroid surgery, an increase in overall fractures and in spine and forearm fractures was evident in patients with PHPT up to 10 years before the diagnosis [Vestergaard et al. 2000]. Similar data were previously reported by Khosla and colleagues who retrospectively evaluated the prevalence of fracture in cases of mild PHPT followed without PTx [Khosla et al. 1999]. In both studies, men had a lower fracture risk than women and fracture risk increased with advanced age at diagnosis. In a recent prospective case–control study an increased rate of vertebral fractures was found, evaluated by DXA, in postmenopausal women with PHPT who met the criteria for surgery, but not in those who did not [Vignali et al. 2009] (Figure 4). Although lumbar spine BMD predicted fracture rate, it is noteworthy that a substantial percentage of patients fractured with T scores greater than −2.5, whereas this was not seen in the control population. This observation raises the question of whether the cutoff point of −2.5 as a criterion for PTx in patients with asymptomatic PHPT (see below) is as useful as it is in postmenopausal women who do not have PHPT. Our data confirm previous observations of De Geronimo and colleagues who also reported an increased vertebral fracture risk in a case–control study in postmenopausal women [De Geronimo et al. 2006]. However, our findings differ from those of Kaji and colleagues who found a lower vertebral fracture rate in Japanese women with PHPT compared with controls and a lower threshold of BMD for vertebral fractures [Kaji et al. 2005]. Differences in ethnicity and patient characteristics might account for the latter discrepancies.
Figure 4.

Rate of vertebral fractures (evaluated by dual-emission X-ray absorptiometry) in patients with primary hyperparathyroidism and controls. Left panel: symptomatic and asymptomatic patients and controls. Right panel: asymptomatic patients, grouped according to whether they met or did not meet the criteria for surgery established by the 2002 Workshop on Asymptomatic Primary Hyperparathyroidism [Bilezikian et al. 2002], and controls. P value refers to the odds ratio between different groups. (Reproduced from Vignali et al. [2009] with permission, Copyright 2009, The Endocrine Society.)
The risk of hip fracture seems to be only slightly, if at all, increased in patients with PHPT [Mosekilde, 2008]. This is consistent with the finding that PHPT is not a dominant feature in most series of patients with hip fracture [Di Monaco et al. 2004].
Effects of treatment
Surgical treatment is the definitive cure for PHPT. All patients with symptomatic PHPT should be advised to undergo PTx. According to the recently revised surgical guidelines, surgery is also recommended for some patients with asymptomatic PHPT, including those with bone density below −2.5 at any site or a previous fragility fracture [Bilezikian et al. 2009].
Parathyroidectomy
After successful PTx bone turnover rapidly normalizes. With the restoration of normal PTH and calcium levels, bone resorption is quickly tapered down, while bone formation proceeds at the level of bone multicellular units, which were activated prior to PTx [Heaney, 2002]. Markers of bone resorption, such as urinary deoxypyridoline and urinary and serum carboxy-terminal collagen crosslinks of collagen, rapidly decline within the first 1–3 months after PTx, while markers of bone formation, such as bone alkaline phosphatase and osteocalcin, decline more gradually within 6 months [Christiansen et al. 1999]. This postoperative transient phase of uncoupling of the two processes results in an ‘anabolic window’, as previously defined. The rapid refilling of the enlarged remodeling space and the subsequent matrix mineralization will be reflected by an increase (up to 10–12%) in BMD at sites rich in trabecular bone such as lumbar spine and hip, which mainly occurs during the first 6–12 months after PTx [Christiansen et al. 1999]. The extent of postoperative raise in BMD is greater in patients with more severe disease and is positively correlated with preoperative serum calcium, PTH and bone turnover markers. Indeed, an average BMD increase of 20% has been observed in patients with vertebral osteopenia or osteoporosis within 4 years after PTx [Silverberg et al. 1996]. The remineralization process is extremely marked after PTx in patients with severe PHPT. In this setting the rapid bone mineral accrual at the level of the numerous activated bone multicellular units accounts for the so-called hungry bone disease which is characterized by hypocalcemia due to rapid calcium fluxes into the remineralizing bone.
Although the main increase in BMD is generally observed during the first year after PTx, the gain in BMD is sustained over the following 10–15 years, regardless of aging [Silverberg et al. 1999; Rubin et al. 2008]. Cortical bone, as estimated by BMD at the distal one-third of the radius, is less responsive to PTX because of the low rate of bone turnover, but sensible increases can be observed in the long term, with the restoration of the normal secretory patterns of PTH [Rubin et al. 2008].
Patients with asymptomatic PHPT who do not meet the surgical guidelines may also benefit from PTx in terms of BMD, as demonstrated in randomized controlled trials, even if their bones are only partially compromised by the disease [Ambrogini et al. 2007] (Figure 5).
Figure 5.
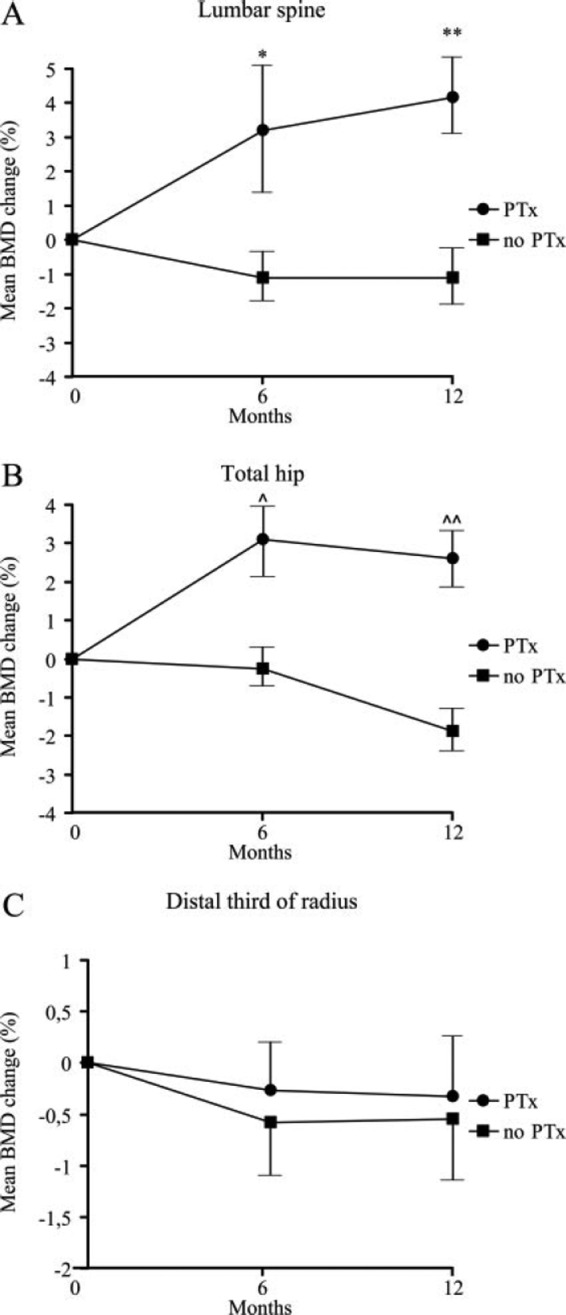
Mean (±SEM) change (% of basal) in lumbar spine (A), total hip (B) and distal one-third of the radius (C) bone mineral density (BMD) in patients with mild primary hyperparathyroidism randomized to parathyroidectomy (Ptx) or followed without intervention (no PTx). *p = 0.0005; **p = 0.0002; ^p = 0.003; ^^p = 0.0001. (Reproduced from Ambrogini et al. [2007] with permission, Copyright 2007, The Endocrine Society.)
The question of whether the observed postoperative improvement in BMD is associated with increased bone strength and decreased fracture risk remains to be clarified. Two cohort studies have evaluated the effect of PTx on fracture risk in PHPT. The first study, which included 674 patients, showed that surgery was followed by a decreased rate of any fracture from 1.8 to 1.0 (p < 0.006) and forearm fracture from 1.9 to 0.7 (p < 0.03), with no change in vertebral and hip fracture rates [Vestergaard et al. 2000]. Ten years after surgery the fracture rate increased again, mainly for an increase in distal forearm fractures. The other study, which included 1934 patients, showed that PTx decreased the risk of all fractures, hip and distal forearm fractures, but not other fractures [Vestergaard and Mosekilde, 2003]. The beneficial effect of surgery was evident in patients who had or not had prevalent fractures before PTx. More data are needed before a definitive conclusion on this important matter can be reached. In addition, changes in bone microarchitecture after PTx could modulate bone strength and in turn influence fracture risk.
Medical therapy
No definitive medical therapy for PHPT is currently available. All patients with PHPT not undergoing PTx should be advised to maintain a normal calcium intake. Vitamin D deficiency, which appears to be associated with a more severe skeletal disease, should be corrected [Rao et al. 2000; Bollerslev et al. 2011]. Treatment options are available for patients not undergoing PTx, but in whom hypercalcemia or reduced BMD are a concern. Beneficial effects have been observed in biochemical parameters or bone density according to different medical treatment. No studies have investigated the effect of medical treatment on fracture rates in patients with PHPT.
Antiresorptive therapy
Antiresorptive drugs would be expected to be of benefit for bone, by decreasing the activation frequency, refilling the remodeling space and increasing mineralization. Indeed, placebo-controlled trials have shown improvement in BMD in postmenopausal women with PHPT and mild hypercalcemia treated with antiresorptive drugs.
Estrogen therapy (conjugated estrogen 0.625 mg/medroxyprogesterone 5 mg daily for 2 years) increased femoral neck and lumbar spine BMD in postmenopausal women with PHPT [Grey et al. 1996]. Long-term estrogen therapy is no longer recommended, but its use for a limited length of time could be considered in early postmenopausal women with mild PHPT and low BMD, who have no contraindications to the use of estrogen.
Bisphosphonate therapy, mainly alendronate, has also been used in PHPT. Rossini and colleagues randomized 26 older women to alendronate (10 mg on alternate days) or no treatment for 2 years [Rossini et al. 2001]. Alendronate was associated with a significant decrease in bone turnover and an increase in BMD over baseline at the lumbar spine (+8.6%), total hip (+4.8%) and total body (+1.2%). Conversely, femoral neck and total body BMD significantly decreased in untreated patients after 2 years. Chow and colleagues evaluated the effect of 10 mg alendronate compared with placebo for 1 year in 48 postmenopausal women [Chow et al. 2003]. Bone turnover markers decreased in patients receiving alendronate and BMD significantly increased in these patients compared with patients given placebo [+3.8% versus 0.2% (p = 0.016) at the lumbar spine and +4.2% versus −0.2% (p = 0.011) at the femoral neck]. Serum calcium slightly decreased with alendronate but not with placebo. In a multicenter trial Khan and colleagues randomized 44 patients with mild, asymptomatic PHPT to treatment with 10 mg daily alendronate or placebo [Khan et al. 2004]. Alendronate therapy was associated with an increase in lumbar spine and total hip BMD (5.3% and 3.7% at year 1 and 6.8% and 4.0% at year 2, respectively), whereas no changes were observed at the distal radius. All three studies showed no consistent changes in serum calcium and PTH levels.
A recent meta-analysis showed that antiresorptive therapies increase BMD to the same extent as PTx in patients with mild PHPT, suggesting that this treatment may be considered in patients with mild hypercalcemia and low BMD [Sankaran et al. 2010].
Calcimimetics
Cinacalcet is an allosteric modulator of the calcium-sensing receptor and acts by sensitizing this receptor to the extracellular calcium [Nemeth, 2002], thus inhibiting the synthesis and secretion of PTH and the renal tubular calcium reabsorption. Cinacalcet was shown to decrease serum calcium levels across a broad range of PHPT severity, whereas PTH levels declined only modestly and generally remained elevated [Peacock et al. 2009, 2011; Marcocci et al. 2009]. No change in BMD was found in patients treated for up to 5.5 years. Cinacalcet is approved in Europe and the USA for the treatment of moderate to severe hypercalcemia in patients with PHPT who are unable to undergo PTx. Its use may be considered for patients in whom BMD is not low, but whose serum calcium level is more than 1 mg/dl above the normal range.
Contributor Information
Claudio Marcocci, Section of Endocrinology and Bone Metabolism, Department of Endocrinology and Metabolism, University of Pisa, Via Paradisa 2, 56124 Pisa, Italy.
Luisella Cianferotti, Section of Endocrinology and Bone Metabolism, Department of Endocrinology and Metabolism, University of Pisa, Pisa, Italy.
Filomena Cetani, Section of Endocrinology and Bone Metabolism, Department of Endocrinology and Metabolism, University of Pisa, Pisa, Italy.
References
- Ambrogini E., Cetani F., Cianferotti L., Vignali E., Banti C., Viccica G., et al. (2007) Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab 92: 3114–3121 [DOI] [PubMed] [Google Scholar]
- Bellido T., Ali A.A., Plotkin L.I., Fu Q., Gubrij I., Roberson P.K., et al. (2003) Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem 278: 50259–50272 [DOI] [PubMed] [Google Scholar]
- Bilezikian J.P., Silverberg S.J. (2000) Clinical spectrum of primary hyperparathyrodiism. Rev Endoc Metab Disord 1: 237–245 [DOI] [PubMed] [Google Scholar]
- Bilezikian J.P., Khan A.A., Potts J.T., Jr on behalf of the Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism (2009) Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Third International Workshop. J Clin Endocrinol Metab 94: 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikian J.P., Potts J.T., Jr, Fuleihan Gel-H., Kleerekoper M., Neer R., Peacock M., et al. (2002) Summary statement from a workshop on asymptomatic primary hyperparathyroidism. A perspective for the 21st century. J Clin Endocrinol Metab 87: 5353–5361 [DOI] [PubMed] [Google Scholar]
- Bollerslev J., Marcocci C., Sosa M., Nordenström J., Bouillon R., Mosekilde L. (2011) Current evidence for recommendation of surgery, medical treatment and vitamin D repletion in mild primary hyperparathyroidism. Eur J Endocrinol 165: 851–864 [DOI] [PubMed] [Google Scholar]
- Camozzi V., Lumachi F., Mantero F., Piccolo M., Luisetto G. (2003) Phalangeal quantitative ultrasound technology and dual energy x-ray densitometry in patients with primary hyperparathyroidism: influence of sex and menopausal status. Osteoporos Int 14: 602–608 [DOI] [PubMed] [Google Scholar]
- Chappard C., Roux C., Laugier P., Paillard M., Houllier P. (2006) Bone status in primary hyperparathyroidism assessed by regional bone mineral density from the whole body scan and QUS imaging at calcaneus. Joint Bone Spine 73: 86–94 [DOI] [PubMed] [Google Scholar]
- Chow C.C., Chan W.B., Li J.K., Chan N.N., Chan M.H., Ko G.T., et al. (2003) Oral alendronate increases bone mineral density in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 88: 581–587 [DOI] [PubMed] [Google Scholar]
- Christiansen P., Steiniche T., Brixen K., Hessov I., Melsen F., Heickendorff L., et al. (1999) Primary hyperparathyroidism: short-term changes in bone remodeling and bone mineral density following parathyroidectomy. Bone 25: 237–244 [DOI] [PubMed] [Google Scholar]
- Cipriani C., Romagnoli E., Scarpiello A., Angelozzi M., Montesano T., Minisola S. (2009) Phalangeal quantitative ultrasound and bone mineral density in evaluating cortical bone loss. A study in postmenopausal women in primary hyperparathyroidism and subclinical iatrogenic hyperthyroidism. J Clin Densitom 12: 456–460 [DOI] [PubMed] [Google Scholar]
- Compston J.E. (2007) Skeletal actions of intermittent parathyroid hormone: effects on bone remodeling and structure. Bone 40: 1447–1452 [DOI] [PubMed] [Google Scholar]
- Cope O. (1966) The study of hyperparathyroidism at the Massachusetts General Hospital. N Eng J Med 26: 1174–1182 [DOI] [PubMed] [Google Scholar]
- Datta N.S., Abou-Samra A.B. (2009) PTH and PTHrP signaling in osteoblasts. Cell Signal 21: 1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta N.S., Pettway G.J., Chen C., Koh A.J., McCauley L.K. (2007) Cyclin D1 as a target for the proliferative effects of PTH and PTHrP in early osteoblastic cells. J Bone Miner Res 22: 951–964 [DOI] [PubMed] [Google Scholar]
- De Geronimo S., Romagnoli E., Diacinti D., D’Erasmo E., Minisola S. (2006) The risk of fractures in postmenopausal women with primary hyperparathyroidism. Eur J Endocrinol 155: 415–420 [DOI] [PubMed] [Google Scholar]
- Dempster D.W., Hughes-Begos C.E., Plavetic-Chee K., Brandao-Burch A., Cosman F., Nieves J., et al. (2005) Normal human osteoclasts formed from peripheral blood monocytes express PTH type 1 receptors and are stimulated by PTH in the absence of osteoblasts. J Cell Biochem 95: 139–148 [DOI] [PubMed] [Google Scholar]
- Dempster D.W., Muller R., Zhou H., Kohler T., Shane E., Parisien M., et al. (2007) Preserved three-dimensional cancellous bone structure in mild primary hyperparathyroidism. Bone 41: 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster D.W., Parisien M., Silverberg S.J., Liang X-G., Schnitzer M., Shen V., et al. (1999) On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism. J Clin Endocrinol Metab 84: 1562–1566 [DOI] [PubMed] [Google Scholar]
- Di Monaco M., Vallero F., Di Monaco R., Mautino F., Cavanna A. (2004) Primary hyperparathyroidism in elderly patients with hip fracture. J Bone Miner Metab 22: 491–495 [DOI] [PubMed] [Google Scholar]
- Dobnig H., Sipos A., Jiang Y., Fahrleitner-Pammer A., Ste-Marie L.G., Gallagher J.C., et al. (2005) Early changes in biochemical markers of bone formation correlate with improvements in bone structure during teriparatide therapy. J Clin Endocrinol Metab 90: 3970–3977 [DOI] [PubMed] [Google Scholar]
- Fraser W.D. (2009) Hyperparathyroidism. Lancet 374: 145–158 [DOI] [PubMed] [Google Scholar]
- Gonnelli S., Montagnani A., Cepollaro C., Monaco R., Gennari L., Rossi B., et al. (2000) Quantitative ultrasound and bone mineral density in patients with primary hyperparathyroidism before and after surgical treatment. Osteoporos Int 11: 255–260 [DOI] [PubMed] [Google Scholar]
- Grey A.B., Stapleton J.P., Evans M.C., Tatnell M., Reid I.R. (1996) Effect of hormone replacement therapy on bone mineral density in postmenopausal women with mild primary hyperparathyroidism. A randomized, controlled trial. Ann Intern Med 125: 360–368 [DOI] [PubMed] [Google Scholar]
- Guo C.Y., Thomas W.E., al-Dehaimi A.W., Assiri A.M., Eastel R. (1996) Longitudinal changes in bone mineral density and bone turnover in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 81: 3487–3491 [DOI] [PubMed] [Google Scholar]
- Hanley D.A., Watson P.H., Hodsman A.B., Dempster D.W. (2008) Pharmacological mechanisms of therapeutics: parathyroid hormone. In Bilezikian J., Raisz L.G., Martin T.J. (eds), Principles of Bone Biology. Elsevier: New York, pp; 1659–1695 [Google Scholar]
- Heaney R.P. (2002) The basis for the post-parathyroidectomy increase in bone mass. J Bone Miner Res 17(Suppl. 2): N154–N157 [PubMed] [Google Scholar]
- Huang J.C., Sakata T., Pfleger L.L., Bencsik M., Halloran B.P., Bikle D.D., et al. (2004) PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res 19: 235–244 [DOI] [PubMed] [Google Scholar]
- Ingle B.M., Thomas W.E.G., Eastell R. (2002) Differential effects of primary hyperparathyroidism on ultrasound properties of bone. Osteoporos Int 13: 572–578 [DOI] [PubMed] [Google Scholar]
- Ishizuya T., Yokose S., Hori M., Noda T., Suda T., Yoshiki S., et al. (1997) Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest 99: 2961–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji H., Yamauchi M., Chihara K., Sugimoto T. (2005) The threshold of bone mineral density for vertebral fractures in female patients with primary hyperparathyroidism. Eur J Endocrinol 153: 373–378 [DOI] [PubMed] [Google Scholar]
- Khan A.A., Bilezikian J.P., Kung A.W., Ahmed M.M., Dubois S.J., Ho A.Y., et al. (2004) Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 89: 3319–3325 [DOI] [PubMed] [Google Scholar]
- Khosla S., Melton L.J., III, Wermers R.A., Crowson C.S., O’Fallon W., Riggs B. (1999) Primary hyperparathyroidism and the risk of fracture: a population-based study. J Bone Miner Res 14: 1700–1707 [DOI] [PubMed] [Google Scholar]
- Kramer I., Keller H., Leupin O., Kneissel M. (2010) Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol Metab 21: 237–244 [DOI] [PubMed] [Google Scholar]
- Lewiecki E.M. (2010) Management of skeletal health in patients with asymptomatic primary hyperparathyroidism. J Clin Densitom 13: 324–334 [DOI] [PubMed] [Google Scholar]
- Li X., Qin L., Bergenstock M., Bevelock L.M., Novack D.V., Partridge N.C. (2007) Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J Biol Chem 282: 33098–33106 [DOI] [PubMed] [Google Scholar]
- Ma Y.L., Cain R.L., Halladay D.L., Yang X., Zeng Q., Miles R.R., et al. (2001) Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene associated bone formation. Endocrinology 142: 4047–4054 [DOI] [PubMed] [Google Scholar]
- Marcocci C., Cetani F. (2011) Primary hyperparathyroidism. N Engl J Med 22: 2389–2397 [DOI] [PubMed] [Google Scholar]
- Marcocci C., Chanson P., Shoback D., Bilezikian J.P., Fernandez-Cruz L., Orgiazzi J., et al. (2009) Cinacalcet reduces serum calcium concentrations in patients with intractable primary hyperparathyroidism. J Clin Endocrinol Metab 94: 2766–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P.D., Bilezikian J.P. (2002) Bone densitometry in asymptomatic primary hyperparathyroidism. J Bone Miner Res 17(Suppl. 2): N98–N102 [PubMed] [Google Scholar]
- Mosekilde L. (2008) Primary hyperparathyroidism and the skeleton. Clin Endocrinol (Oxf) 15: 1–19 [DOI] [PubMed] [Google Scholar]
- Nakchbandi I.A., Lang R., Kinder B., Insogna K.L. (2008) The role of the receptor activator of nuclear factor-kappaB ligand/osteoprotegerin cytokine system in primary hyperparathyroidism. J Clin Endocrinol Metab 93: 967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E.F. (2002) Pharmacological regulation of parathyroid hormone secretion. Curr Pharm Des 8: 2077–2087 [DOI] [PubMed] [Google Scholar]
- Ohishi M., Schipani E. (2011) PTH and stem cells. J Endocrinol Invest 34: 552–556 [DOI] [PubMed] [Google Scholar]
- Parisien M., Cosman F., Mellish R.W., Schnitzer M., Nieves J., Silverberg S.J., et al. (1995) Bone structure in postmenopausal hyperparathyroid, osteoporotic, and normal women. J Bone Miner Res 10: 1393–1399 [DOI] [PubMed] [Google Scholar]
- Parisien M., Mellish R.W., Silverberg S.J., Shane E., Lindsay R., Bilezikian J.P., et al. (1992) Maintenance of cancellous bone connectivity in primary hyperparathyroidism: trabecular strut analysis J Bone Miner Res 7: 913–919 [DOI] [PubMed] [Google Scholar]
- Parisien M., Silverberg S.J., Shane E., de la Cruz L., Lindsay R., Bilezikian J.P., et al. (1990) The histormorphometry of bone in primary hyperparathyroidism: preservation of cancellous bone structure. J Clin Endocrinol Metab 70: 930–938 [DOI] [PubMed] [Google Scholar]
- Peacock M., Bilezikian J.P., Bolognese M.A., Borofsky M., Scumpia S., Sterling L.R., et al. (2011) Cinacalcet HCl reduces hypercalcemia in primary hyperparathyroidism across a wide spectrum of disease severity. J Clin Endocrinol Metab 96: E9–E18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock M., Bolognese M.A., Borofsky M., Scumpia S., Sterling L.R., Cheng S., et al. (2009) Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a 5-year study. J Clin Endocrinol Metab 94: 4860–4867 [DOI] [PubMed] [Google Scholar]
- Poole K.E., van Bezooijen R.L., Loveridge N., Hamersma H., Papapoulos S.E., Löwik C.W., et al. (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19: 1842–1844 [DOI] [PubMed] [Google Scholar]
- Raef H., Ingemansson S., Sobhi S., Sutan A., Ahmed M., Chaudhry M. (2004) The effect of vitamin D status on the severity of bone disease and on the other features of primary hyperparathyroidism in a vitamin D deficient region. J Endocrinol Invest 27: 807–812 [DOI] [PubMed] [Google Scholar]
- Rao D.S., Agarwal G., Talpos G.B., Phillips E.R., Bandeira F., Mishra S.K., et al. (2002) Role of vitamin D and calcium nutrition in disease expression and parathyroid tumor growth in primary hyperparathyroidism: a global perspective. J Bone Miner Res 17(Suppl. 2): N75–N80 [PubMed] [Google Scholar]
- Rao D.S., Honasoge M., Divine G.W., Phillips E.R., Lee M.W., Ansari M.R., et al. (2000) Effect of vitamin D nutrition on parathyroid adenoma weight: patogenetic and clinical implications. J Clin Endocrinol Metab 85: 1054–1058 [DOI] [PubMed] [Google Scholar]
- Rhee Y., Allen M.R., Condon K., Lezcano V., Ronda A.C., Galli C., et al. (2011) PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res 26: 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschger P., Dempster D.W., Zhou H., Paschalis E.P., Silverberg S.J., Shane E., et al. (2007) New observations on bone quality in mild primary hyperparathyroidism as determined by quantitative backscattered electron imaging. J Bone Miner Res 22: 717–723 [DOI] [PubMed] [Google Scholar]
- Rossini M., Gatti D., Isaia G., Sartori L., Braga V., Adami S. (2001) Effects of oral alendronate in elderly patients with osteoporosis and mild primary hyperparathyroidism. J Bone Miner Res 16: 113–119 [DOI] [PubMed] [Google Scholar]
- Rubin M.R., Bilezikian J.P. (2005) Parathyroid hormone as an anabolic skeletal therapy. Drugs 65: 2481–2498 [DOI] [PubMed] [Google Scholar]
- Rubin M.R., Bilezikian J.P., McMahon D.J., Jacobs T., Shane E., Siris E., et al. (2008) The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab 93: 3462–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran S., Gamble G., Bolland M., Reid I.R., Grey A. (2010) Skeletal effects of interventions in mild primary hyperparathyroidism: a meta-analysis. J Clin Endocrinol. Metab 95: 1653–1662 [DOI] [PubMed] [Google Scholar]
- Schnoke M., Midura S.B., Midura R.J. (2009) Parathyroid hormone suppresses osteoblast apoptosis by augmenting DNA repair. Bone 45: 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B.C., Costa A.G, Cusano N.E., Kousteni S., Bilezikian J.P. (2011) Catabolic and anabolic actions of parathyroid hormone on the skeleton. J Endocrinol Invest 34: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg S.J. (2007) Vitamin D deficiency and primary hyperparathyroidism. J Bone Miner Res 22(Suppl. 2): V100–V104 [DOI] [PubMed] [Google Scholar]
- Silverberg S.J., Lewiecki E.M., Mosekilde L., Peacock M., Rubin M.R. (2009) Presentation of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 94: 351–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg S.J., Locker F.G., Bilezikian J.P. (1996) Vertebral osteopenia: a new indication for surgery in primary hyperparathyroidism. J Clin Endocrinol Metab 81: 4007–4012 [DOI] [PubMed] [Google Scholar]
- Silverberg S.J., Shane E., de la, Cruz L., Dempster D.W., Feldman F., Seldin D., et al. (1989) Skeletal disease in primary hyperparathyroidism. J Bone Mineral Res 14: 283–291 [DOI] [PubMed] [Google Scholar]
- Silverberg S.J., Shane E., Jacobs T.P., Siris E., Bilezikian J.P. (1999) A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med 341: 1249–1255 [DOI] [PubMed] [Google Scholar]
- Vestergaard P., Mollerup C.L., Frokjaer V.G., Christiansen P., Blichert-Toft M., Mosekilde L. (2000) Cohort study of risk of fracture before and after surgery for primary hyperparathyroidism. BMJ 321: 598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard P., Mosekilde L. (2003) Fractures in patients with primary hyperparathyroidism: nationwide follow-up study of 1201 patients. World J Surg 27: 343–349 [DOI] [PubMed] [Google Scholar]
- Vignali E., Viccica G., Diacinti D., Cetani F., Cianferotti L., Ambrogini E., et al. (2009) Morphometric vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 94: 2306–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoehrer R., Dempster D.W., Bilezikian J.P., Zhou H., Silverberg S.J., Shane E., et al. (2008) Bone quality determined by Fourier transform infrared imaging analysis in mild primary hyperparathyroidism. J Clin Endocrinol Metab 93: 3484–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]