

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by the loss of cognitive functions, reflecting pathological damage to the medial prefrontal cortex (mPFC) as well as to the hippocampus and the entorhinal cortex. Astrocytes maintain the internal homeostasis of the CNS and are fundamentally involved in neuropathological processes, including AD. Here, we analysed the astrocytic cytoskeletal changes within the mPFC of a triple transgenic mouse model of AD (3 × Tg-AD) by measuring the surface area and volume of glial fibrillary acidic protein (GFAP)-positive profiles in relation to the build-up and presence of amyloid-β (Aβ), and compared the results with those found in non-transgenic control animals at different ages. 3 × Tg-AD animals showed clear astroglial cytoskeletal atrophy, which appeared at an early age (3 months; 33% and 47% decrease in GFAP-positive surface area and volume, respectively) and remained throughout the disease progression at 9, 12 and 18 months old (29% and 36%; 37% and 35%; 43% and 37%, respectively). This atrophy was independent of Aβ accumulation, as only a few GFAP-positive cells were localized around Aβ aggregates, which suggests no direct relationship with Aβ toxicity. Thus, our results indicate that the progressive reduction in astrocytic branching and domain in the mPFC can account for the integrative dysfunction leading to the cognitive deficits and memory disturbances observed in AD.
Keywords: Alzheimer's disease, astroglia, cognition, medial prefrontal cortex, plasticity
Introduction
Alzheimer's disease (AD) is a chronic, progressive and fatal neurodegenerative disorder manifested by a deterioration of memory and attention as well as an impairment of daily activities (Alzheimer, 1907; Cummings, 2004). These mnesic alterations are accompanied by indifference and apathy (Knopman & Selnes, 2003); behavioural deficits also include aggression, irritation and anxiety (Knopman & Selnes, 2003; Mendez & Cummings, 2003; Harciarek & Jodzio, 2005). These changes affect human identity and alter attentional processes, decision-making, goal-directed behaviour and working memory, which are all controlled by the medial prefrontal cortex (mPFC) (Goldman-Rakic et al. 1984; Goldman-Rakic, 1987; Vertes, 2004).
The mPFC is divided into dorsal (agranular and anterior cingulated cortices) and ventral divisions (infralimbic and prelimbic cortices: IL and PL, respectively), which are anatomically and functionally linked with the limbic system, being directly associated with cognitive, mnesic and emotional processes (Heidbreder & Groenewegen, 2003; Hoover & Vertes, 2007). The mPFC together with the hippocampus are fundamentally responsible for learning and memory, including information consolidation and working memory (Wickelgren, 1979; Otani, 2003; Jay et al. 2004; Funahashi, 2006). The relationship between the hippocampus and the PFC consists of two types of connective pathways, a direct hippocampal projection to the mPFC and an indirect return functional loop, crucial for long-term memory formation, which also involves the active regulatory and relay role of the nucleus reuniens of the thalamus (RE; Jay et al. 1989, 1996; Carr & Sesack, 1996; Buckner et al. 1999, 2000; Vertes, 2004; Vertes et al. 2007). Backward projections of CA1 neurons to the deep layers of the entorhinal as well as the perirhinal and postrhinal cortices strengthen memory-related plasticity due to the fact that the rhinal cortical region is a key intermediary between the hippocampus and the neocortex, being indispensible for the short-term storage and consolidation of specific memory forms (Cousens & Otto, 1998).
It is well known that astroglia significantly contribute to active mnesic-associated processes, including synaptic plasticity (Verkhratsky et al. 2011). Astroglial modulation of synaptic transmission occurs by several mechanisms: first, the control of local ion, neurotransmitter and metabolic homeostasis (Nedergaard & Verkhratsky, 2012); second, astroglia-derived ATP/adenosine, which control transmission in synaptic fields encompassed by astroglial domains (Pascual et al. 2005); and third, astroglial release of d-serine, which controls the activation of N-methyl-d-aspartate receptors (Henneberger et al. 2010). Furthermore, astrocytes are also indispensible for synaptogenesis, synaptic maturation and maintenance (Pfrieger, 2009; Heneka et al. 2010).
Glial cells also have an important pathological potential (Giaume et al. 2007; Rodriguez et al. 2009; Heneka et al. 2010), and their involvement in AD was originally suggested by Alois Alzheimer (Alzheimer, 1910). The main histological hallmarks of AD are amyloid-β-peptide aggregates (Aβ), subsequently developing into senile plaques, and neurofibrillary tangles, filamentous intracellular aggregates of hyperphosphorylated tau protein (Selkoe, 2001). AD progression in the hippocampus and entorhinal cortex is associated with the early atrophy of astrocytes that, in the case of the hippocampus, coexist at later stages of the disease with a population of reactive astroglial cells surrounding the plaques (Olabarria et al. 2010; Yeh et al. 2011).
Here, we report the changes in astroglial morphology that appear in the mPFC of a triple transgenic AD animal model, harbouring the human APP Swedish (KM670/671NL) mutation, the four repeat Tau P301L mutation and the PS1M146V mutation transgenes (Oddo et al. 2003a, b), at different ages that represent and spatio-temporally mimic the different stages of the disease in humans: first, early stages, with the absence of any pathological hallmarks; second, middle stages, with the presence of either Aβ aggregates or early plaques; and third, the late phase, with the presence of both consolidated plaques and neurofibrillary tangles. Furthermore, we try to establish a rationale for how these changes are associated with AD neuropathology and how they can contribute to the behavioural impairments observed at the different disease stages.
Materials and methods
This study was performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) regarding the use of animals in research, and was approved by the Ethical Committee of the Institute of Experimental Medicine of the Academy of Sciences of the Czech Republic, Prague, Czech Republic. All efforts were made to reduce the number of animals.
Mice
Experiments were performed on male 3 × Tg-AD mice and their background-matching controls as described in detail previously (Oddo et al. 2003a, b; Rodriguez et al. 2008, 2009).
Fixation and tissue processing
3 × Tg-AD animals of different ages (3, 9, 12 and 18 months; n = 4 for all ages) and their equivalent non-Tg controls (3, 9, 12 and 18 months; n = 4, 3, 4, 4, respectively) were anaesthetized by an intraperitoneal injection of sodium pentobarbital (50 mg kg−1). The mice were perfused through the aortic arch with 3.75% acrolein (25 mL; Fluka Sigma-Aldrich, Germany) in a solution of 2% paraformaldehyde (Sigma, Germany) and 0.1 m phosphate buffer (PB) pH 7.4, followed by 2% paraformaldehyde (75 mL). The brains were then removed and cut into 4-mm coronal slabs of tissue consisting of the entire rostrocaudal extent of the mPFC. The brain sections were postfixed in 2% paraformaldehyde for 24 h and kept in 0.1 m PB, pH 7.4. Then, they were cut into 40–50-μm-thick coronal sections using a vibrating microtome (MICROM HM 650 V, Thermo Scientific, USA). Free-floating brain sections in 0.1 m PB, pH 7.4 were collected and stored in a cryoprotectant solution containing 25% sucrose and 3.5% glycerol in 0.05 m PB at pH 7.4. For immunohistochemistry, coronal vibratome sections at levels 1.98/1.54 mm anterior to Bregma were selected, according to the mouse brain atlas of Paxinos & Franklin (2004).
Antibodies
A monoclonal mouse antiserum generated against glial fibrillary acidic protein (GFAP) from porcine spinal cord (anti-GFAP; Sigma-Aldrich; #G3893) was used for the determination of glial cytoskeletal properties and changes, while an IgG fraction of rabbit anti-GFAP antiserum (Sigma-Aldrich; #G9269) was used for the analysis of the glial cytoskeleton in relation to Aβ aggregates. For the identification of Aβ deposits, we used a monoclonal mouse antiserum that reacts with abnormally processed isoforms of Aβ, as well as precursor forms, recognizing an epitope within amino acids 3–8 [EFRHDS; anti-Aβ 6E10 (SIG-39300); Signet Laboratories, Dedham, MA, USA]. The immunolabelling pattern we obtained with this antibody is equivalent to that obtained previously in different brain regions (Oddo et al. 2003a). The specificity of the antibodies has been previously verified using immunohistochemistry and Western blotting (Halliday et al. 1996; Eng et al. 2000; Rodriguez et al. 2008). To assess possible non-specific background labelling or cross-reactivity between antibodies derived from different host species, a series of control experiments was performed. The omission of the primary and secondary antibodies from the incubation solutions resulted in the total absence of target labelling (data not shown).
Immunohistochemistry
For the detection and determination of GFAP-positive cells and their relation to Aβ aggregates, we used both single and dual indirect immunofluorescence labelling. To minimize methodological variability, sections through the ventral mPFC of both hemispheres from all animals were processed at the same time under exactly the same experimental conditions. For this procedure, the vibratome sections were first incubated in 1% sodium borohydride (Sigma-Aldrich) for 30 min. The sections were then washed with PB profusely before rinsing in 0.1 m Tris-Saline (TS) for 10 min. Brain sections were then incubated with 0.5% bovine serum albumin (Sigma-Aldrich) in 0.1 m TS and 0.25% Triton X-100 (Sigma-Aldrich) for 30 min. For single-labelling, the sections were incubated for 48 h at room temperature in the primary antibody (mouse anti-GFAP, 1 : 5000; SIGMA, St Louis, MO, USA). The sections were rinsed in 0.1 m TS for 30 min, and incubated in a 1 : 200 dilution of fluorescein (FITC)-conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA, USA) for 1 h at room temperature, then finally rinsed in 0.1 m TS for 30 min. For dual-labelling, the sections were incubated for 48 h at room temperature in a primary antibody cocktail containing: mouse anti-Aβ monoclonal antibody (Aβ; 1 : 2000; Covance, Emeryville, CA, USA); and rabbit anti-GFAP (1 : 5000; SIGMA). Subsequently, Aβ and GFAP immunoreactivities were detected in a sequential manner on the same sections by incubation with rhodamine (TRITC)-conjugated goat anti-mouse and (FITC)-conjugated goat anti-rabbit IgG (Jackson Immunoresearch, West Grove, PA, USA), respectively. Finally, the sections were rinsed with 0.1 m TS for 30 min and permanently mounted in an aqueous medium (Vectashield; Vector Laboratories, Peterborough, UK).
Morphological analysis of the astrocyte cytoskeleton
Astrocytes (n = 35 per animal, with a minimum of five per layer) were imaged using confocal scanning microscopy (Leica TCS SP), recording layers every 0.2 μm. Parallel confocal planes were superimposed, and morphological analysis was carried out by Cell Analyst (Chvatal et al. 2007) using digital filters (average 3 × 3, convolution, gauss 5 × 5, despeckle, simple objects removal) to determine the surface area (S) and volume (V) of the GFAP-stained cytoskeleton of astrocytes.
GFAP-positive cell count in the ventral mPFC
To determine whether the changes in the mPFC GFAP-positive cytoskeletal surface area and volume are linked with changes in the number of astrocytes expressing GFAP, we determined the numerical density (Nv, No. cells/mm3) of GFAP-positive astrocytes in at least three representative non-consecutive sections, analysing an area of 600 000 μm2 in coronal sections of 40 μm thickness, thus representing a total volume of 24 000 000 μm3 per section (for further details, see Olabarria et al. 2010; Yeh et al. 2011). GFAP-positive astrocytes were intensively labelled against a dark background that made them easy to identify with an equal chance of being counted. The number of GFAP-positive astrocytes was determined and quantified blindly on fluorescent microscope images by a single observer to reduce counting bias to a minimum.
Statistical analysis
Unpaired t-tests and one-way anova were used to examine differences in the number, surface area and volume of GFAP-labelled cells between the 3 × Tg-AD and non-Tg animals. Data are expressed as mean ± SEM.
Results
In both non-Tg and 3 × Tg-AD mice, GFAP-immunolabelled astrocytes showed typical characteristics of protoplasmic astrocytes, with numerous, elongated and extended processes arising from the astrocyte somata in a star-shaped radial pattern (Fig. 1A–D). These GFAP-immunoreactive (GFAP-IR) processes differed in thickness and length, from clearly visible solid branches to thin, subtle and elaborated ones, which corresponded to proximal and distal processes, respectively. GFAP-IR astrocytes were much more abundant in layer I, and appeared in lower numbers in layers II–VI. However, independently of the gradient of GFAP-positive astrocytes, the Nv (number of cell/mm3) of GFAP-IR astrocytes was constant and equal at different ages in both non-Tg and 3 × Tg-AD animals, with no significant differences between them (Fig. 2).
Fig. 1.
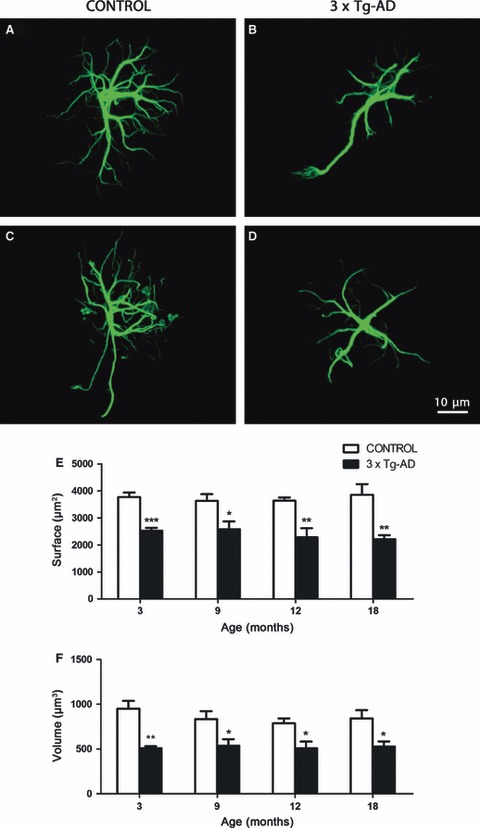
Confocal images showing the classical morphology of GFAP-positive astrocytes in control non-Tg animals and astrocytic atrophy in the 3 × Tg-AD animals at 3 months (A and B, respectively) and 18 months (C and D, respectively) in the mPFC. Bar graphs showing the decreases in the GFAP-positive surface area and volume throughout the whole extent of the mPFC (E, F) in 3 × Tg-AD mice when compared with control animals. Bars represent mean ± SEM.
Fig. 2.
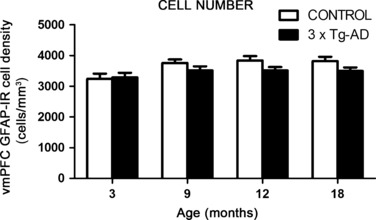
Bar graphs showing the numerical density (number cells/mm3) of glial fibrillary acidic protein-immunoreactive (GFAP-IR) cells in 3 × Tg-AD mice and non-Tg controls. Bars represent mean ± SEM. vmPFC, ventral medial prefrontal cortex.
Early and sustained astroglial cytoskeletal atrophy in 3 × Tg-AD mice
GFAP-positive astrocytes in the 3 × Tg-AD mice showed a significant reduction in their cytoskeletal surface area and volume as early as 3 months old; both parameters decreased by 33% and 47%, respectively, when compared with non-Tg controls (2526.58 ± 103.44 μm2 vs. 3770.23 ± 166.88 μm2, P = 0.0007; 507.78 ± 22.28 μm3 vs. 950.13 ± 86.99 μm3, P = 0.0026). This cytoskeletal atrophy remained at 9 months (a decrease of 29% in surface area, 2580.81 ± 288.53 μm2 vs. 3633.10 ± 244.97 μm2, P = 0.0458; and a decrease of 36% in volume, 536.51 ± 73.30 μm3 vs. 834.16 ± 88.32 μm3, P = 0.0475), 12 months (a decrease of 37% in surface area, 2279.37 ± 340.39 μm2 vs. 3633.35 ± 121.88 μm2, P = 0.0096; and a decrease of 35% in volume, 507.91 ± 74.82 μm3 vs. 787.37 ± 54.76 μm3, P = 0.0236) and 18 months, when the surface area was reduced by 43% (2210.96 ± 143.36 μm2 vs. 3857.58 ± 391.03 μm2, P = 0.0075) and the volume by 37% (526.55 ± 58.08 μm3 vs. 841.94 ± 91.41 μm3, P = 0.0269; Fig. 1E,F).
Layer-specific astrocytic atrophy
Our results show that astrocytic atrophy is not only a generalized phenomenon, but also layer specific. The affected layers at both young (3 months) and old ages (18 months) were the superficial layers 1 and 2 together with the deep layers 4 and 5 (Fig. 3). However, layer 3 only appeared to be affected at advanced ages, starting at 12 months old (Fig. 3c,h).
Fig. 3.
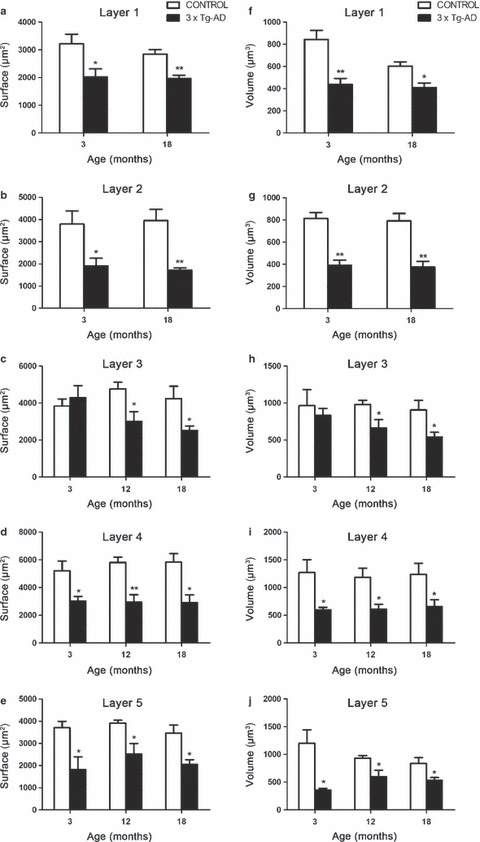
Bar graphs illustrating the decreased GFAP-positive surface area (a–e) and volume (f–j) in the different cortical layers of the mPFC. Bars represent mean ± SEM.
Superficial layers 1–3
In general, astrocytes from the superficial layers in 3 × Tg-AD animals were characterized by shorter, less numerous and rather horizontally oriented processes. At the age of 3 months, astrocytes in superficial layer 1 were reduced in surface area by 37% (2017.35 ± 293.54 μm2 vs. 3219.53 ± 346.18 μm2, P = 0.0381) and in volume by 48% (438.24 ± 55.06 μm3 vs. 844.05 ± 81.32 μm3, P = 0.0061) when compared with those in non-Tg control animals. At very late ages this atrophy, which was absent at middle and advanced ages (9–12 months), reappeared, and the astrocytic cytoskeletal surface area underwent a reduction of 31% (1972.83 ± 111.31 μm2 vs. 2843.57 ± 166.21 μm2, P = 0.0048) along with a 32% decrease in volume (410.70 ± 39.60 μm3 vs. 603.36 ± 35.91 μm3, P = 0.0113; Fig. 3a,f).
Astrocytic atrophy in layer 2 at 3 months was manifested by a 50% reduction in surface area (1900.20 ± 355.75 μm2 vs. 3787.24 ± 595.47 μm2, P = 0.0346) and a 52% reduction in volume (390.97 ± 46.66 μm3 vs. 813.33 ± 52.94 μm3, P = 0.001). Similarly to layer 1 at 18 months, equivalent decreases were observed: 56% and 53% in surface area and volume, respectively (1715.77 ± 92.83 μm2 vs. 3945.10 ± 507.82 μm2, P = 0.005; and 374.96 ± 51.09 μm2 vs. 791.10 ± 66.44 μm2, P = 0.0025; Fig. 3b,g).
The surface area and volume of astrocytes in layer 3 were affected only at advanced and late ages, the changes being significant at 12 months (a decrease of 37%, 3004.46 ± 527.54 μm2 vs. 4763.42 ± 363.08 μm2, P = 0.0334; and a decrease of 32%, 663.33 ± 113.59 μm3 vs. 981.10 ± 55.21 μm3, P = 0.0455, respectively) and at 18 months old (a decline in surface area of 41%, 2521.97 ± 237.91 μm2 vs. 4244.60 ± 661.12 μm2, P = 0.0497; and a decline in volume of 40%, 540.11 ± 65.38 μm3 vs. 906.31 ± 129.89 μm3, P = 0.0454; Fig. 3c,h).
A slight difference was noticeable between astrocytes in the different superficial layers. Astrocytes from layer 2 in comparison to those from layer 1 had slightly more expanded processes, but less numerous than the more bushy astrocytes from the deeper superficial layer 3. At 3 months old in 3 × Tg-AD animals, there were some significant internal differences between the GFAP-positive profiles of layer 3 and those of other layers, despite the fact that this layer was unchanged compared with the same layer in the control group. Layer 3 astrocytes appeared to be larger compared with those in layer 1 (53.08% larger in surface area, 4299.96 ± 644.34 μm2 vs. 2017.35 ± 293.54 μm2, P = 0.0181; and 47.42% larger in volume, 833.54 ± 93.17 μm3 vs. 438.24 ± 55.06 μm3, P = 0.0107) and also those in layer 2 (55.81% in surface area, 4299.96 ± 644.34 μm2 vs. 1900.2 ± 355.75 μm2, P = 0.0178; 53.1% in volume, 833.54 ± 93.17 μm3 vs. 390.97 ± 46.66 μm3, P = 0.0054; Fig. 3); this phenomenon was observed only at early ages and exclusively in 3 × Tg-AD animals, not in control mice.
Deep layers 4 and 5
Astrocytes in the deep layers were star-like and displayed a more typical protoplasmic anatomy (Fig. 4), but with rather thin, numerous, but limited in length processes, sent in all directions. The reduction in astrocytic cytoskeletal branching in layers 4 and 5 was significant not only in the early and late stages of AD, but also at mid-advanced ages (3, 12 and 18 months old).
Fig. 4.
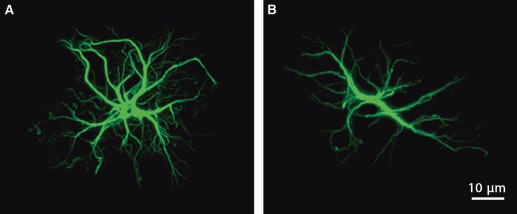
Confocal images showing a typical protoplasmic astrocyte (A) compared with a less classical cortical astrocyte (B) from the deep layers of 3 × Tg-AD mice.
The surface area and volume of astrocytes in layer 4 were decreased at 3 months by 42% (3024.84 ± 329.86 μm2 vs. 5210.35 ± 697.60 μm2, P = 0.0299) and by 53% (597.18 ± 45.41 μm3 vs. 1269.32 ± 232.94 μm3, P = 0.0299), respectively. At 12 months the surface area was decreased by 49% (2944.96 ± 539.49 μm2 vs. 5810.24 ± 383.12 μm2, P = 0.0049) and the volume by 48% (609.86 ± 87.45 μm3 vs. 1183.26 ± 165.95 μm3, P = 0.0223). In the oldest group of animals at 18 months old, the atrophy of mPFC astrocytes in layer 4 remained evident, showing a decrease in both surface area and volume (50% in surface area, 2910.83 ± 556.42 μm2 vs. 5832.46 ± 613.04 μm2, P = 0.0124; and 47% in volume, 654.01 ± 126.84 μm3 vs. 1235.76 ± 200.46 μm3, P = 0.0496; Fig. 3d,i).
The reduction in astrocytic cytoskeletal surface area and volume in layer 5 at 3 months old was manifested to the same extent by a 51% decrease in surface area (1805.06 ± 584.22 μm2 vs. 3702.41 ± 291.62 μm2, P = 0.0271) and a 70% decrease in volume (356.60 ± 30.53 μm3 vs. 1200.08 ± 241.64 μm3, P = 0.0134). At 12 and 18 months old, these reductions persisted even though the differences were less pronounced: 36% and 41% in surface area (2509.24 ± 484.45 μm2 vs. 3907.91 ± 131.85 μm2, P = 0.0318; and 2053.31 ± 205.48 μm2 vs. 3460.72 ± 359.09 μm2, P = 0.0145, respectively) and 36% and 37% in volume (596.90 ± 118.90 μm3 vs. 930.23 ± 46.78 μm3, P = 0.0402; and 528.75 ± 53.96 μm3 vs. 837.37 ± 101.99 μm3, P = 0.0368) at 12 and 18 months, respectively (Fig. 3e,j).
As observed in superficial layers 1 and 2, astrocytes in layer 5 were significantly smaller than those in layer 3. Indeed, layer 5 astrocytes were smaller by 41.98% in surface area (1805.06 ± 584.22 μm2 vs. 4299.96 ± 644.34 μm2, P = 0.0285) and by 43.02% in volume (356.6 ± 30.53 μm3 vs. 833.54 ± 93.17 μm3, P = 0.0028), while no clear differences were evident compared with astrocytes from layer 4.
Intracellular Aβ accumulation but rare astrocytic association with neuropil Aβ aggregates
Up to 18 months old, no plaques were formed in the mPFC of 3 × Tg-AD animals. However, vascular Aβ accumulation and a vast intracellular accumulation of Aβ were observed (Fig. 5A and inset,C), with a minimal tendency to progress to extracellular plaque deposition, except for aggregates (Fig. 5B,C). The intracellular accumulation of Aβ started to appear at about 6 months old, and became more robust with disease progression. Neurons filled with Aβ were located throughout the cortex, with a tendency towards a slightly more prominent presence in the deeper layers (4–5). These neuropathological signs were not generally associated with the presence of neighbouring astrocytes positive for GFAP (Fig. 5B,C) and, if so, only with astrocytes displaying evident atrophic characteristics (Fig. 5B and asterisk).
Fig. 5.
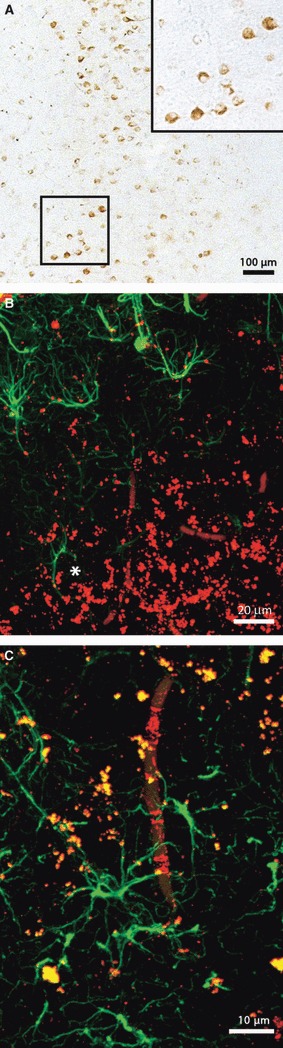
Brightfield micrograph showing the intracellular accumulation of Aβ in the mPFC of an 18-month-old transgenic animal (A). Confocal images illustrating Aβ aggregates in the mPFC, but few GFAP-positive neighbouring astrocytes (B and asterisk), and an Aβ loaded blood vessel surrounded by some reactive astrocytes in an 18-month-old 3 × Tg-AD animal (C).
Discussion
In this study we analysed the GFAP-positive astroglial profiles in the mPFC of 3 × Tg-AD animals at different ages. First, we found that the overall density of GFAP-positive astrocytes does not change significantly between non-Tg and 3 × Tg-AD animals with age; similarly, no evident age-dependent cell loss was observed. Our results are in agreement with our previous findings in the hippocampus and entorhinal cortex in the same animal model where no signs of astrocytic death were found (Olabarria et al. 2010; Yeh et al. 2011). This also relates to other neurological diseases that involve neuroglia, such as schizophrenia, where no significant changes in glial density were found (Selemon et al. 1995, 1998).
The second important and probably more fundamental observation was the identification of a general atrophy of GFAP-positive astrocytes, which was already significant at early pre-symptomatic and pre-pathological ages (3 months), and was sustained during disease progression in all age groups up to 18 months. This atrophy was evidenced by a decrease in the surface area and volume of GFAP-positive profiles; it also showed a clear layer-specific pattern, with layers 1–2 being strongly affected and similar changes being found in the deep layers 4 and 5, while layer 3 was only affected from middle age onwards.
mPFC general atrophy in AD
It is well documented that the PFC is one of the brain regions most sensitive to the detrimental effects of aging (Liu et al. 1996; West, 1996, 2000; Lamar & Resnick, 2004). Age-related degeneration in the PFC is more significant than in other brain areas (Raz et al. 1997). Even more extensive PFC atrophy has been observed in subjects with AD (Salat et al. 1999, 2001). In this regard, Burgmans and colleagues in their recent study (Burgmans et al. 2009) suggested that PFC atrophy is specifically associated with dementia and can be used as a predictor/biomarker of AD. In general, the observed cortical decrease is assumed to be the result of cell shrinkage, rather than a reduction in cell numbers (Kemper, 1994; Uylings et al. 2000).
These results, even if described mainly in neurons, are in agreement with our data, which expand the concept of atrophic cortical astroglia. Changes in the appearance of astrocytes from control mice compared with those from transgenic animals were manifested mainly in thinner and shorter astrocytic processes, with a tendency to be more horizontally oriented (in the superficial layers) or with a preserved star-like protoplasmic look (in the deep layers). Such layer-specific GFAP reductions and alterations also appear in other neurological disorders, such as schizophrenia and depression, for which the mPFC (and particularly layer 5) is highly pathologically relevant. Alterations in the mPFC are even more important for frontotemporal dementia (Miguel-Hidalgo et al. 2000; Martin et al. 2001; Rajkowska et al. 2002).
Regarding the important role of astrocytes in maintaining brain homeostasis, the observed mPFC astroglial atrophy during the progression of AD can result in reduced synaptic coverage, decreased metabolic support to neurons and synapses, and neurotransmitter imbalance, all leading to alterations in the flow and processing of information within the brain (Heneka et al. 2010; Verkhratsky et al. 2010). Considering the arrangement of the afferent and efferent projections of the rodent PL, this brain region is responsible for cognitive functions and is homologous to the dorsolateral PFC in primates and humans. Thus, it is directly involved in the integration and management of sensory and mnemonic information, intellectual functions and actions, all of which are strongly affected in AD. On the other hand, the IL represents a visceromotor centre homologous to the orbitomedial PFC of primates, and appears to be crucial when it comes to reward-related behaviour and initiating the state of the mood (Ongur & Price, 2000; Hoover & Vertes, 2007). Although they perform different functions, the IL and PL are generally treated as a single region of the ventral mPFC (Vertes, 2004), and were treated in the same way in the present study.
The mPFC receives a wide range of afferent projections from such structures as the hippocampus (layers 1–3, 5–6), entorhinal (layers 1–2) as well as the perirhinal (layers 1–3) cortices, and the medial basal forebrain (layers 1, 3, 5–6; Jay & Witter, 1991; Delatour & Witter, 2002; Vertes, 2004; Hoover & Vertes, 2007). Also, the midline RE, the major source of thalamic afferents to the hippocampus, densely innervates the IL and PL, terminating mainly within layers 1 and 5/6 (Wouterlood et al. 1990; Bokor et al. 2002; Vertes, 2006). Even though there are no direct return projections from the mPFC to the hippocampus, the hippocampal terminals form mainly asymmetric synapses on prefrontal pyramidal neurons and mPFC fibres shape the same contacts on the dendritic shafts of those RE cells, which in return project to the hippocampal formation (Hurley et al. 1991; Carr & Sesack, 1996; Vertes, 2004; Fig. 6). Thus, the RE is ideally positioned to strongly influence the activity of the hippocampus and the mPFC as an integral part of the limbic loop between these structures (Di Prisco & Vertes, 2006; Vertes et al. 2007). Also, considering the salient function of the hippocampus/PFC connections in the pathophysiology of major depression and schizophrenia, specific notable alterations could appear due to an altered connectivity of these areas (Szeszko et al. 2003; Goldapple et al. 2004; Jay et al. 2004), associated with a potential homeostatic deficiency due to the observed generalized astrocytic atrophy. In line with the rewiring hypothesis, Dumitriu and colleagues (Dumitriu et al. 2010) found a selective spine loss in aged monkeys in the neurons of layer III in the homologous PFC, suggesting a decreased ability to reconnect PFC circuits with advancing age. Layer III, as an origin and ‘end station’ for cortico-cortical connections, is highly significant for the formation of memory by association, and is assumed to be very sensitive to neurodegenerative changes (Fuster, 2008).
Fig. 6.
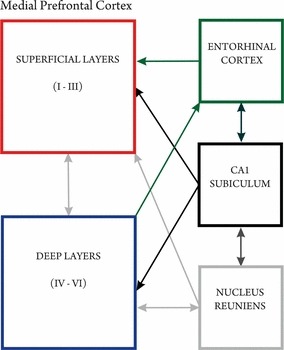
Afferent and efferent projections of the mPFC. Afferences from the limbic system are mainly received from the hippocampal formation, the entorhinal cortex and the thalamic nucleus reuniens, while the mPFC itself projects back indirectly to the hippocampus via the nucleus reuniens of the midline thalamus – an integrating part within this network.
Astrocytic atrophy and its relationship with Aβ pathology
In contrast to other brain regions affected in AD, such as the hippocampus and entorhinal cortex (Olabarria et al. 2010; Yeh et al. 2011), no plaque formation was observed in the mPFC up to very old ages. However, Aβ aggregates were clearly visible and robust, especially in the deep layers. Astrocytes, even if not numerous, were present and had atrophic features. Taking into consideration that astrocytic atrophy in 3 × Tg-AD mice was significant at very early stages, far before any Aβ presence in the PFC, it is clear that the toxicity of Aβ and the observed decrease in the GFAP-positive astroglial surface area and volume are two important, but independent, features of AD pathology in the mPFC.
Conclusion
Once triggered, the process of neurodegeneration resembles a domino effect. The fundamental principle of neurodegeneration is a failure in connectivity within the neural networks, which results in altered synaptic transmission, impaired memory and cognition, and ultimately in neuronal death and atrophy of the brain (Heneka et al. 2010; Verkhratsky & Parpura, 2010).
The global and layer-specific reductions in the GFAP profiles of the astrocytes observed in our study are likely to represent pathological remodelling of a wide range of neuronal–glial and interglial interactions. Astroglial atrophy can impact upon the number and functional status of synapses, leading to decreased connectivity and a neurotransmission imbalance that underlie the progression of AD (Allaman et al. 2011). In this report we also emphasized very early changes in astrocytic GFAP profiles that could signal pathological processes developing well before the appearance of the classical and well-known histological hallmarks of AD.
Acknowledgments
This study was supported by an Alzheimer's Research Trust Programme Grant (ART/PG2004A/1) to J.J.R. and A.V., grants from the Grant Agency of the Czech Republic to J.J.R. (GACR 309/09/1696), to E.S. and J.J.R. (GACR 304/11/0184), and to A.V. (GACR 305/08/1381, GACR 305/08/1384). Support from the Spanish Government, Plan Nacional de I+D+I 2008-2011 and ISCIII- Subdirección General de Evaluación y Fomento de la Investigación (PI10/02738) to J.J.R. and A.V., and the Government of the Basque Country (AE-2010-1-28, AEGV10/16) to J.J.R. is gratefully acknowledged. M.K.-N. is supported by the EC FP7 project AXREGEN (PITN-GA-2008-214003). The authors would also like to thank Miss Chia-Yu Yeh for all of her help in the verification and correction of the fine details of the manuscript, and her support during the submission progress. Finally, the authors would like to express their gratitude to Mr James Dutt for critical reading of the manuscript.
Author contributions
M.K.-N. carried out the immunohistochemical study and contributed to the writing of the manuscript. A.V. participated in the conception of the study and the writing of the manuscript. E.S. contributed to the writing of the manuscript. A.C. contributed to the measurements and data analysis. J.J.R. participated in the conception and design of the study and the writing of the manuscript, as well as coordinating the study. All authors read and approved the final manuscript.
References
- Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiat Phych-Gericht Med. 1907;64:146–148. [Google Scholar]
- Alzheimer A. Beiträge zur Kenntnis der pathologischen Neuroglia und ihrer Beziehungen zu den Abbauvorgängen im Nervengewebe. In: Nissl F, Alzheimer A, editors. Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. , Jena: Gustav Fischer; 1910. pp. 401–562. [Google Scholar]
- Bokor H, Csaki A, Kocsis K, et al. Cellular architecture of the nucleus reuniens thalami and its putative aspartatergic/glutamatergic projection to the hippocampus and medial septum in the rat. Eur J Neurosci. 2002;16:1227–1239. doi: 10.1046/j.1460-9568.2002.02189.x. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Kelley WM, Petersen SE. Frontal cortex contributes to human memory formation. Nat Neurosci. 1999;2:311–314. doi: 10.1038/7221. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Koutstaal W, Schacter DL, et al. Functional MRI evidence for a role of frontal and inferior temporal cortex in amodal components of priming. Brain. 2000;123(Pt 3):620–640. doi: 10.1093/brain/123.3.620. [DOI] [PubMed] [Google Scholar]
- Burgmans S, van Boxtel MP, Smeets F, et al. Prefrontal cortex atrophy predicts dementia over a six-year period. Neurobiol Aging. 2009;30:1413–1419. doi: 10.1016/j.neurobiolaging.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Chvatal A, Anderova M, Kirchhoff F. Three-dimensional confocal morphometry – a new approach for studying dynamic changes in cell morphology in brain slices. J Anat. 2007;210:671–683. doi: 10.1111/j.1469-7580.2007.00724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens G, Otto TA. Induction and transient suppression of long-term potentiation in the peri- and postrhinal cortices following theta-related stimulation of hippocampal field CA1. Brain Res. 1998;780:95–101. doi: 10.1016/s0006-8993(97)01151-7. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Alzheimer's disease. N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- Delatour B, Witter MP. Projections from the parahippocampal region to the prefrontal cortex in the rat: evidence of multiple pathways. Eur J Neurosci. 2002;15:1400–1407. doi: 10.1046/j.1460-9568.2002.01973.x. [DOI] [PubMed] [Google Scholar]
- Di Prisco GV, Vertes RP. Excitatory actions of the ventral midline thalamus (rhomboid/reuniens) on the medial prefrontal cortex in the rat. Synapse. 2006;60:45–55. doi: 10.1002/syn.20271. [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Hao J, Hara Y, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30:7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Funahashi S. Prefrontal cortex and working memory processes. Neuroscience. 2006;139:251–261. doi: 10.1016/j.neuroscience.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Fuster JM. The Prefrontal Cortex. Oxford, UK: Academic Press; 2008. [Google Scholar]
- Giaume C, Kirchhoff F, Matute C, et al. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007;14:1324–1335. doi: 10.1038/sj.cdd.4402144. [DOI] [PubMed] [Google Scholar]
- Goldapple K, Segal Z, Garson C, et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Development of cortical circuitry and cognitive function. Child Dev. 1987;58:601–622. [PubMed] [Google Scholar]
- Goldman-Rakic PS, Selemon LD, Schwartz ML. Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience. 1984;12:719–743. doi: 10.1016/0306-4522(84)90166-0. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Cullen KM, Kril JJ, et al. Glial fibrillary acidic protein (GFAP) immunohistochemistry in human cortex: a quantitative study using different antisera. Neurosci Lett. 1996;209:29–32. doi: 10.1016/0304-3940(96)12592-1. [DOI] [PubMed] [Google Scholar]
- Harciarek M, Jodzio K. Neuropsychological differences between frontotemporal dementia and Alzheimer's disease: a review. Neuropsychol Rev. 2005;15:131–145. doi: 10.1007/s11065-005-7093-4. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. 2003;27:555–579. doi: 10.1016/j.neubiorev.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Rodriguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Res Rev. 2010;63:189–211. doi: 10.1016/j.brainresrev.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, et al. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Hurley KM, Herbert H, Moga MM, et al. Efferent projections of the infralimbic cortex of the rat. J Comp Neurol. 1991;308:249–276. doi: 10.1002/cne.903080210. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Jay TM, Glowinski J, Thierry AM. Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain Res. 1989;505:337–340. doi: 10.1016/0006-8993(89)91464-9. [DOI] [PubMed] [Google Scholar]
- Jay TM, Burette F, Laroche S. Plasticity of the hippocampal-prefrontal cortex synapses. J Physiol Paris. 1996;90:361–366. doi: 10.1016/s0928-4257(97)87920-x. [DOI] [PubMed] [Google Scholar]
- Jay TM, Rocher C, Hotte M, et al. Plasticity at hippocampal to prefrontal cortex synapses is impaired by loss of dopamine and stress: importance for psychiatric diseases. Neurotox Res. 2004;6:233–244. doi: 10.1007/BF03033225. [DOI] [PubMed] [Google Scholar]
- Kemper TL. Neuroanatomical and neuropathological changes during aging and in dementia. In: Albert ML, Knoepfel EJE, editors. Clinical Neurology of Aging. , New York: Oxford University Press; 1994. pp. 3–67. [Google Scholar]
- Knopman DS, Selnes OA. Neuropsychology of dementia. In: Heilman KM, Valenstein E, editors. Clinical Neuropsychology. , New York: Oxford University Press; 2003. pp. 574–616. [Google Scholar]
- Lamar M, Resnick SM. Aging and prefrontal functions: dissociating orbitofrontal and dorsolateral abilities. Neurobiol Aging. 2004;25:553–558. doi: 10.1016/j.neurobiolaging.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Liu X, Erikson C, Brun A. Cortical synaptic changes and gliosis in normal aging, Alzheimer's disease and frontal lobe degeneration. Dementia. 1996;7:128–134. doi: 10.1159/000106867. [DOI] [PubMed] [Google Scholar]
- Martin JA, Craft DK, Su JH, et al. Astrocytes degenerate in frontotemporal dementia: possible relation to hypoperfusion. Neurobiol Aging. 2001;22:195–207. doi: 10.1016/s0197-4580(00)00231-1. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Cummings JL. Dementia – a Clinical Approach. Philadelphia, PA: Butterworth-Heinemann (Elsevier); 2003. [Google Scholar]
- Miguel-Hidalgo JJ, Baucom C, Dilley G, et al. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol Psychiatry. 2000;48:861–873. doi: 10.1016/s0006-3223(00)00999-9. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Verkhratsky A. Artifact versus reality – How astrocytes contribute to synaptic events? Glia. 2012;60:1013–1023. doi: 10.1002/glia.22288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, et al. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Olabarria M, Noristani HN, Verkhratsky A, et al. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia. 2010;58:831–838. doi: 10.1002/glia.20967. [DOI] [PubMed] [Google Scholar]
- Ongur D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10:206–219. doi: 10.1093/cercor/10.3.206. [DOI] [PubMed] [Google Scholar]
- Otani S. Prefrontal cortex function, quasi-physiological stimuli, and synaptic plasticity. J Physiol Paris. 2003;97:423–430. doi: 10.1016/j.jphysparis.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 2004. [Google Scholar]
- Pfrieger FW. Roles of glial cells in synapse development. Cell Mol Life Sci. 2009;66:2037–2047. doi: 10.1007/s00018-009-0005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, et al. Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res. 2002;57:127–138. doi: 10.1016/s0920-9964(02)00339-0. [DOI] [PubMed] [Google Scholar]
- Raz N, Gunning FM, Head D, et al. Selective aging of the human cerebral cortex observed in vivo: differential vulnerability of the prefrontal gray matter. Cereb Cortex. 1997;7:268–282. doi: 10.1093/cercor/7.3.268. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Jones VC, Tabuchi M, et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One. 2008;3:e2935. doi: 10.1371/journal.pone.0002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Jones VC, Verkhratsky A. Impaired cell proliferation in the subventricular zone in an Alzheimer's disease model. NeuroReport. 2009;20:907–912. doi: 10.1097/WNR.0b013e32832be77d. [DOI] [PubMed] [Google Scholar]
- Salat DH, Kaye JA, Janowsky JS. Prefrontal gray and white matter volumes in healthy aging and Alzheimer disease. Arch Neurol. 1999;56:338–344. doi: 10.1001/archneur.56.3.338. [DOI] [PubMed] [Google Scholar]
- Salat DH, Kaye JA, Janowsky JS. Selective preservation and degeneration within the prefrontal cortex in aging and Alzheimer disease. Arch Neurol. 2001;58:1403–1408. doi: 10.1001/archneur.58.9.1403. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Abnormally high neuronal density in the schizophrenic cortex. A morphometric analysis of prefrontal area 9 and occipital area 17. Arch Gen Psychiatry. 1995;52:805–818. doi: 10.1001/archpsyc.1995.03950220015005. discussion 819–820. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Comp Neurol. 1998;392:402–412. [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Szeszko PR, Goldberg E, Gunduz-Bruce H, et al. Smaller anterior hippocampal formation volume in antipsychotic-naive patients with first-episode schizophrenia. Am J Psychiatry. 2003;160:2190–2197. doi: 10.1176/appi.ajp.160.12.2190. [DOI] [PubMed] [Google Scholar]
- Uylings HBM, West MJ, Coleman PD. Neuronal and cellular changes in the aging brain. In: Clark CM, Trojanowski JQ, et al., editors. Neurodegenerative Dementias. , New York: McGraw-Hill; 2000. pp. 61–76. [Google Scholar]
- Verkhratsky A, Parpura V. Recent advances in (patho)physiology of astroglia. Acta Pharmacol Sin. 2010;31:1044–1054. doi: 10.1038/aps.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Olabarria M, Noristani HN, et al. Astrocytes in Alzheimer's disease. Neurotherapeutics. 2010;7:399–412. doi: 10.1016/j.nurt.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Parpura V, Rodriguez JJ. Where the thoughts dwell: the physiology of neuronal-glial “diffuse neural net”. Brain Res Rev. 2011;66:133–151. doi: 10.1016/j.brainresrev.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142:1–20. doi: 10.1016/j.neuroscience.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Vertes RP, Hoover WB, Szigeti-Buck K, et al. Nucleus reuniens of the midline thalamus: link between the medial prefrontal cortex and the hippocampus. Brain Res Bull. 2007;71:601–609. doi: 10.1016/j.brainresbull.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RL. An application of prefrontal cortex function theory to cognitive aging. Psychol Bull. 1996;120:272–292. doi: 10.1037/0033-2909.120.2.272. [DOI] [PubMed] [Google Scholar]
- West R. In defense of the frontal lobe hypothesis of cognitive aging. J Int Neuropsychol Soc. 2000;6:727–729. doi: 10.1017/s1355617700666109. discussion 730. [DOI] [PubMed] [Google Scholar]
- Wickelgren WA. Chunking and consolidation: a theoretical synthesis of semantic networks, configuring in conditioning, S–R versus congenitive learning, normal forgetting, the amnesic syndrome, and the hippocampal arousal system. Psychol Rev. 1979;86:44–60. [PubMed] [Google Scholar]
- Wouterlood FG, Saldana E, Witter MP. Projection from the nucleus reuniens thalami to the hippocampal region: light and electron microscopic tracing study in the rat with the anterograde tracer Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1990;296:179–203. doi: 10.1002/cne.902960202. [DOI] [PubMed] [Google Scholar]
- Yeh CY, Vadhwana B, Verkhratsky A, et al. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer's disease. ASN Neuro. 2011;3:271–279. doi: 10.1042/AN20110025. [DOI] [PMC free article] [PubMed] [Google Scholar]