

Abstract
Dietary copper deficiency causes cardiac hypertrophy and its transition to heart failure in a mouse model. Copper repletion results in a rapid regression of cardiac hypertrophy and prevention of heart failure. The present study was undertaken to understand dynamic changes of cardiomyocytes in the hypertrophic heart during the regression. Dams of FVB mice were fed copper deficient diet (0.3 mg Cu/kg) starting from day 3 post delivery and weanling pups were fed the same diet until copper repletion (6.0 mg Cu/kg) in the diet at 31 days of age. Heart samples were obtained at the end of copper deficient feeding or at 3, 7, 14 or 28 days after copper repletion. Copper deficiency resulted in increases in the size and reduction in the number of cardiomyocytes in the heart. Copper repletion led to regression in the size of hypertrophic cardiomyocytes and normalization of the total number of cardiomyocytes. Although a direct reduction in the cell size would be significantly responsible for the regression of heart hypertrophy, some hypertrophied cardiomyocytes upon copper repletion reentered the cell cycle as determined by Ki-67 staining in the cardiomyocyte-specific α-sarcomeric actin stained cells and underwent division as determined by a mitosis-specific marker phosphor-histone H3. Quantitative analysis indicated that the replication of hypertrophic cardiomyocytes made a contribution about 1/3 to the total mitosis of the regenerated myocardium. This study suggests that a direct reduction in the size of some hypertrophied cardiomyocytes and a replication of other hypertrophied cardiomyocytes with reduced size make a significant contribution to the regression of copper deficient heart hypertrophy, leading to normalization of the size and the number of cardiomyocytes in the heart.
Keywords: Cell cycle, copper, hypertrophy, mitosis, myocardial regeneration, regression
Introduction
Copper (Cu) is an essential trace element and dietary Cu restriction leads to cardiac hypertrophy (1). Cu deficient heart hypertrophy in mouse and rat models has characteristic structural, biochemical, and signaling alterations resembling those induced by pressure overload (1), and transits to heart failure (2). We have recently observed that dietary Cu repletion results in a rapid regression of the hypertrophy and a complete functional recovery (3). The regression of heart hypertrophy and functional recovery indicate a cytokinetic change in the hypertrophic cardiomyocytes. Possible mechanisms underlying the change include apoptosis leading to a clean up of the hypertrophic cardiomyocytes, a reduction in cellular volume making the hypertrophic myocytes smaller by an unknown mechanism, or mitosis of the hypertrophic cardiomyocytes with reduced cell size leading to myocardial regeneration.
Apoptosis of cardiomyocytes in the failing heart is a major cellular event involved in the pathogenesis (4). Our early studies have shown that dietary Cu deficiency causes myocardial apoptosis (5), which would be highly responsible for the transition from cardiac hypertrophy to heart failure. Defective myocardial function is associated with the loss of cardiomyocytes due to apoptosis. In the regression induced by Cu repletion in Cu deficiency-induced hypertrophic hearts, the depressed function was completely recovered (3) and myocardial structures were normalized as examined by both light and electron microscopes (3). Therefore, it is unlikely that apoptosis can be responsible for the regression of heart hypertrophy induced by Cu repletion.
Heart hypertrophy developed under physiological stimulations such as exercise-induced myocardial growth is reversible (6), although molecular mechanisms involved in the regression of physiological heart hypertrophy have not been fully understood. At the cellular level, the size of hypertrophied cardiomyocytes has to become smaller or normalized in order for the regression of heart hypertrophy to occur. This process would be accomplished by a direct reduction in the size of the hypertrophied cardiomyocytes. Under pathological condition, heart hypertrophy can also be reversed under different conditions in mice (7,8). This regression, however, would be more complicated than a simple reduction in the size of hypertrophied cardiomyocytes under physiological conditions. The functional recovery of the failing heart would demand myocardial regeneration, which requires repletion of the lost cardiomyocytes during the transition from cardiac hypertrophy to heart failure.
Myocardial regeneration through mitosis of the hypertrophic cardiomyocytes with reduced cell size potentially is an attractive mechanism for regression of heart hypertrophy and recovery of myocardial function observed in the Cu deficient hearts. Although the view of the adult mammalian heart as a terminally differentiated organ without regenerative capacity remains dominant (9), recently documented evidence challenges this belief. Several studies have shown the presence of cycling ventricular myocytes in the adult mammalian heart under both physiological and pathological conditions (10-12). It is thus important to know whether or not the Cu repletion-induced regression of heart hypertrophy and recovery of cardiac function is associated with cell cycle re-entry and mitosis of the cardiomyocytes.
The present study was undertaken to determine (1) cytokinetic changes in Cu-induced regression of heart hypertrophy in a mouse model and (2) potential contributions of mitosis of hypertrophic cardiomyocytes to myocardial regeneration. We found that besides a direct reduction in the size of hypertrophied cardiomyocytes, a replication of hypertrophic cardiomyocytes would make a significant contribution to the regression of heart hypertrophy, although differentiation of other types of cells to cardiomyocytes may also play an important role in the regeneration process.
Materials and Methods
Experimental animals and procedure
FVB mice obtained from Jackson Laboratory (Bar Harbor, ME) were maintained at the University of Louisville animal facilities and housed in plastic cages at 22°C on a 12-hr light/dark cycle. Dams of the pups were fed copper-deficient (CuD) AIN-93 diet containing 0.3 mg Cu/kg or copper-adequate (CuA) diet containing 6.0 mg Cu/kg starting on the third day after delivery. The AIN-93 powder diet was prepared according to a report published previously (13). After the pups were weaned at 21 days of age, they were fed the same diet as their respective dam for seven additional days (31 days of age), at which point some of the CuD mice were sacrificed and others were fed the same diet or switched to CuA diet for additional one or two weeks (6-12 mice in each group). Mice had free access to double-distilled water. Cages, feeding jars, and water bottles were rinsed regularly with water containing EDTA first and then with distilled water. The parallel CuA mice were sacrificed at the same time as feeding and age controls. At the conclusion of each feeding experiment, mice were anesthetized with avertin (0.4 mg/g), and blood was drawn from dorsal vena cava. Plasma was collected by centrifugation at 10,000 rpm for 10 min and stored at −80°C until used. Hearts were removed, perfused with PBS, and processed for the analyses below. All procedures were approved by the University of Louisville IACUC, which is certified by AAALAC.
Cardiac hypertrophy
At the organ level, whole hearts were fixed with 10% neutral formalin and embedded in paraplast. Longitudinal sections were cut at 5 μm and stained with hematoxylin and eosin. Whole heart images were captured using a Nikon steromicroscope. At the cellular level, cardiomyocytes on histological sections were stained by immunofluorescence with cytoplasmic marker α-sarcomeric actin. Cross sections of myocardium were rehydrated and incubated with 5% normal donkey serum for 30 min to block non-specific binding sites. Sections were stained with a monoclonal mouse anti-α-sarcomeric actin antibody and Cy3-labeled donkey anti-mouse antibody to detect cardiomyocytes. Digital images with cross-sectioned cardiomyocytes were captured with 40× objective, and for each histological section of the myocardium from 6-8 mice, the diameters of 300 cardiomyocytes were measured and the total number of cardiomyocytes in a 25 mm2 were counted.
Immunofluorescence detection of Ki-67, phospho-histone 3 and myocte enhancing factor-2 (MEF-2)
Double staining of Ki-67/α-sarcomeric actin and phospho-histone 3/α-sarcomeric actin were performed for detection of proliferating and mitotic cardiomyocytes. For staining of Ki-67/α-sarcomeric actin, Ki-67 was first stained by incubation with rabbit anti-Ki-67 antibody (Vector Laboratories, Burlingame, CA) and FITC conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), and then α-sarcomeric actin was stained by incubation with a monoclonal mouse anti-a-sarcomeric actin antibody and FITC-labeled donkey anti mouse antibody. For staining of phospho-histone 3/α-sarcomeric actin, phospho-histone 3 was first stained with rabbit anti-phospho-histone 3 antibody (Vector Laboratories) and FITC conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories), and then α-sarcomeric actin was stained by incubation with a monoclonal mouse anti-α-sarcomeric actin antibody and FITC-labeled donkey anti mouse antibody. For triple staining of phospho-histone 3, MEF-2 and α-sarcomeric actin, the histological sections were first stained by incubation with rabbit anti-phospho-histone 3 antibody (Vector Laboratories) and FITC conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories), and then stained by incubation of goat anti-MEF-2 antibody and AMC-labeled donkey anti-goat antibody, followed by staining with monoclonal mouse anti-α-sarcomeric actin antibody and Cy3-labeled donkey anti mouse antibody.
Electron microscopy (EM)
Heart tissues used for EM study were obtained through an in situ perfusion procedure described previously (14). The free wall of left ventricle was cut into about 1-mm3. For conventional EM, samples were fixed in 3% glutaraldehyde in 0.1 mM sodium cacodylate buffer, pH 7.4, for 2 h at 4 °C and post-fixed in 1% osmium tetroxide. The samples were embedded in resin, and ultrathin sections were cut and stained with uranyl acetate and lead citrate. For immunogold EM, samples were fixed in 2% freshly depolymerized paraformaldehyde and 0.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, at 4 °C for 2 hr. After rinsing in sodium cacodylate buffer, the samples were partially dehydrated with 50% and 70% ethanol and embedded in LR White resin. Ultrathin sections were incubated with the rabbit anti-ki-67 antibody overnight at 4 °C. After rinsing in 0.01 M PBS (pH 8.2), the ultrathin sections were incubated in 10-nm gold conjugated goat anti-rabbit antibody (British BioCell, Cardiff, UK) diluted in 0.01 M PBS (pH 8.2) for 2 hr. The ultrathin sections were then rinsed in distilled water and counterstained with uranyl acetate and lead citrate.
TUNEL assay
An ApopTag in situ detection kit was used according to the manufacture’s instruction and following the procedure described previously (5). Briefly, the histological sections were pretreated with H2O2 and incubated with the reaction mixture containing TdT and digoxigenin-conjugated dUTP for 1 hr at 37 °C. Labeled DNA was visualized with peroxidase-conjugated anti-digoxigenin antibody with DAB as the chromagen. Rat mammary gland tissue provided in the kit was used as positive control. For negative control, TdT was routinely omitted from the reaction mixture.
Statistical analysis
Data were analyzed by one-way ANOVA followed by a Duncan’s multiple-range test for further determination of the significance of differences. Differences among groups were considered significant at p < 0.05.
Results
Reduction in the size of hypertrophied cardiomyocytes by Cu repletion in hypertrophic hearts
After mice were fed Cu deficient diet for 4 weeks, the heart size was increased as measured by heart weight to body weight ratio (9.7 ± 0.5 vs 5.2 ± 0.2 mg/g). Histological sections of the whole heart showed increased wall thickness (Fig 1). Cu repletion resulted in regression of the heart size (6.0 ± 0.2 mg/g) and normalized wall thickness (Fig 1). To explore the cellular events of Cu repletion-induced regression of heart hypertrophy, we measured the size of cardiomyocytes at different stages of the regression. Histological sections of the myocardium were stained with α-sarcomeric actin antibody to label cardiomyocytes specifically. In the hypertrophic myocardium, the average size of cardiomyocytes was significantly increased (Fig 2). During the regression, a progressive decrease in the number of hypertrophic myocytes and an increase in the number of normal-sized cardiomyocytes were observed 3 days and 7 days after dietary Cu repletion. Normalization in the size of the cardiomyocytes within 2 weeks and in the number within 4 weeks was observed (Fig 2). Examination through EM we found mitochondrial swelling and disorganization, myofibrillar damage in cardiomyocytes from the hypertrophic myocardium, and transitional improvement in the morphological changes after 3 and 7 days of Cu repletion and a normalization within 2 weeks of Cu repletion (Fig 3).
Fig 1.
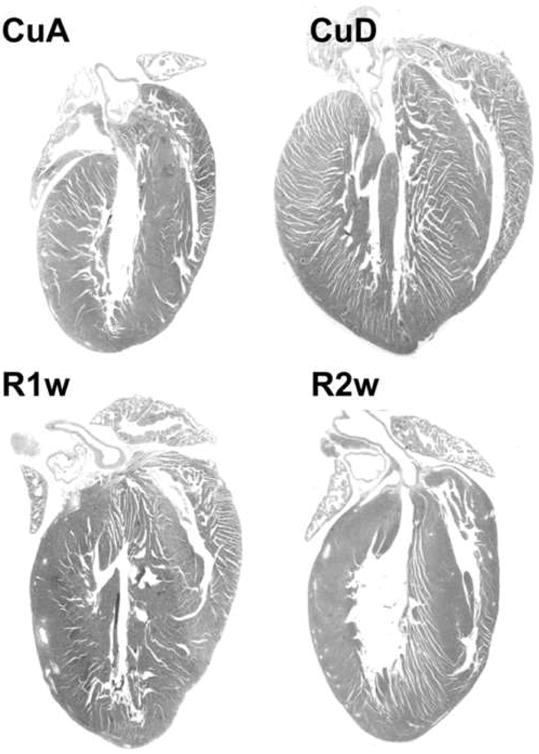
Regression of Cu deficient heart hypertrophy by Cu repletion. Mice were fed Cu-deficient diet (CuD) along with Cu-adequate controls (CuA) for 4 w, and then switched to Cu-adequate diet for 2 w. The regression of heart hypertrophy was examined at 1 w (R1w) and 2 w (R2w) after Cu repletion. Hypertrophy was evidenced by increased wall thickness and decreased chamber lumen and regression evidenced by reversal of the changes.
Fig 2.
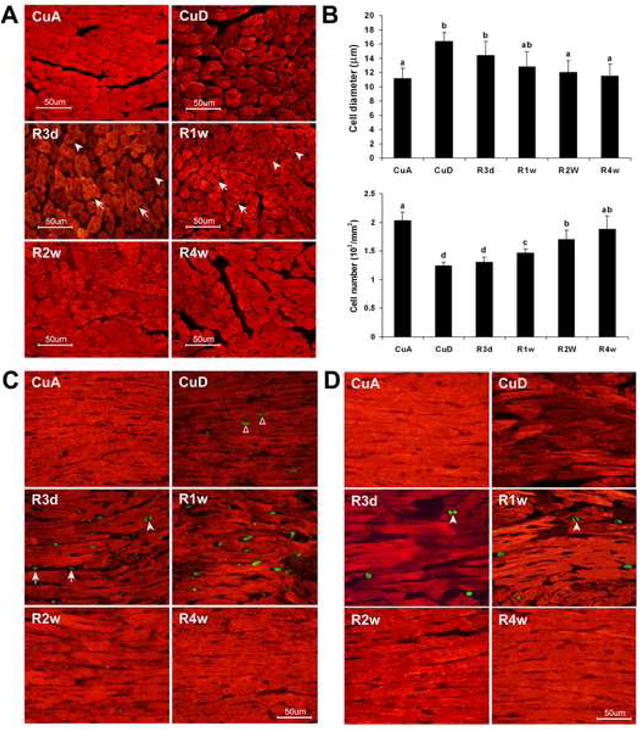
Regression of Cu deficiency-induced cardiomyocyte hypertrophy by Cu repletion. A. α-Sarcomeric actin staining of cardiomyocytes (red) shows Cu deficiency (CuD) increases the cell size compared to Cu adequate (CuA). Cu repletion for 3 d (R3d), 1 w (R1w), 2 w (R2w), or 4 w (R4w) gradually decreases the cell size and increases the number of cells. B. Quantitative analysis of the diameter and number of cardiac myocytes. Digital images were captured with 40× objective and for each histological section, the diameter of 300 myocytes was measured and the total number in a 25 mm2 was counted. The values (mean ± SEM, n=6-8) do not share the same letter are significantly different (p<0.05). C. Double staining of α-sarcomeric actin (red) and Ki-67 (green) to visualize co-localization of Ki-67 positive nuclei and cardiomyocyte cytoplasm. All of the Ki-67-positive nuclei localized in the cardiomyocytes in Cu deficient heart (CuD). The Ki-67 positive nuclei were also found in cells that were not stained by α-sarcomeric actin obtained from Cu replete mice (R3d and R1w). D. Double staining of α-sarcomeric actin (red) and phospho-histone H3 (green) to identify mitotic nuclei and their co-localization with myocyte cytoplasm. No mitotic nuclei found in the CuD, but extensively in the R3d and R1w hearts. Reduced intensity of α-sarcomeric actin staining in the CuD myocardium and gradually regaining the intensity of α-sarcomeric actin staining in the R3d and R1w hearts and normalizing 2 w after copper repletion (R2w) were observed in A, C, and D.
Fig 3.

Electron micrograph of cardiac myocytes obtained from Cu deficient (CuD) mice in comparison with that obtained from Cu adequate (CuA). Cu repletion for 3 d (R3d), 1 w (R1w), 2 w (R2w), or 4 w (R4w) caused a progressive recovery of the degenerated changes in the myocytes. Mitochondrial swelling with vacuolar disorganization and disarray of cristae were observed in the CuD myocardium and were completely recovered 2 w after Cu repletion. Myofibrillar damage was also observed. M: mitochondria, Bar: 3 μm.
Cu repletion-induced cell cycle re-entry of hypertrophic cardiomyocyte
We performed a TUNEL assay to detect apoptotic cardiomyocytes and found that, although there were a few TUNEL-positive cardiomyocytes in the Cu deficient hearts, no-TUNEL positive cardiomyocytes were detected in any myocardial tissue sections obtained from the Cu-replete mice (data not shown), excluding the possibility that apoptosis may be responsible for the clearance of the hypertrophic cardiomyocytes. Therefore, a direct reduction in the size would be ascribed to the normalization of the hypertrophied cardiomyocytes. The normalization of the number of cardiomyocytes in the heart, which had a reduction in the hypertrophied myocardium, would require repletion of the lost cardiomyocytes.
Hypertrophic cardiomyocytes may go through mitosis with reduced cell size leading to normalization in both cellular volume and the total number. To test this, we first used Ki-67, a nuclear protein present only in proliferating cells (15), to identify the cells in the cell cycle. This protein is expressed during G1, S, G2 and metaphase, but is neither present in quiescent cells nor involved in DNA repair (15). We found a few Ki-67-positive cells in the Cu deficient hypertrophic hearts, but the number of the Ki-67-positive cells was significantly increased in the myocardium obtained from mice with Cu repletion for 3 days or 7 days (Fig 2C and 4C). There were very few Ki-67-positive cells after 2 weeks of Cu repletion (Fig 2C and 4C). We also found that the Ki-67-positive cells in Cu-deficient myocardium were cardiomyocytes, as they were dually labeled with α-sarcomeric actin, whereas Ki-67-positive cells in Cu-replete hearts likely included other cell types (Fig 2C).
Fig 4.
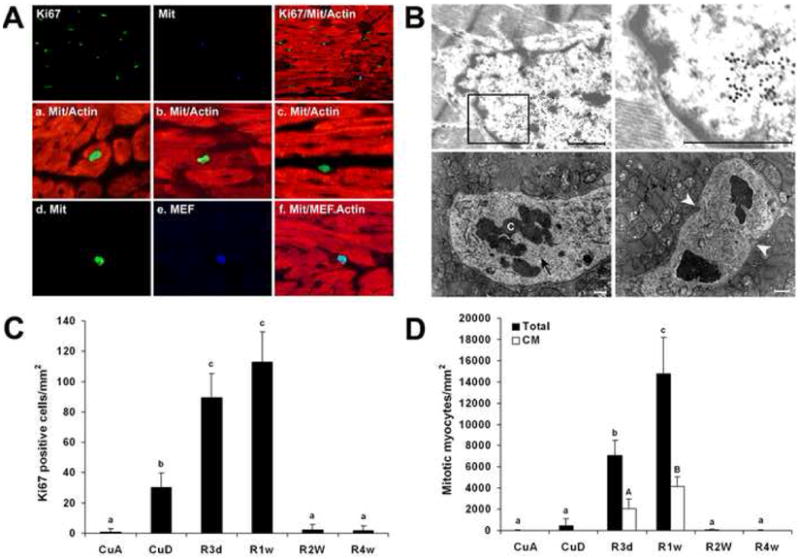
Identification of the origin of mitotic cells in hypertrophic myocardium undergoing regeneration (cu repletion for 1 w). A. Dual staining of Ki-67 (blue) and phospho-histone H3 (green) for visualizing co-localization of the mitotic nuclei with the Ki-67 cell cycle-positive marker (top row). All of the mitotic positive nuclei are Ki-67-positive (arrowheads), but not vice versa. a, phospho-histone H3 positive nucleus localized in hypertrophic cardiomyocyte; b, phospho-histone H3 nucleus localized in hypertrophic myocyte with degenerated cytoplasm (indicated by decreased intensity of α-sarcomeric actin staining); c, the nucleus stained by phospho-histone H3 but not localized in myocyte. The micrographs of d, e, and f show a mitotic positive nucleus (d) is also MEF-2 positive, a cardiomyocyte-specific marker (e), although the nucleus is not localized in myocyte (f). B. Identification of cell cycle-active nuclei in the myocardium by electron microscope. a, immunogold (20 nm gold) electron micrograph of Ki-67 in cardiomyocyte, demonstrating the cell cycle active; b, higher magnification of the framed area of the image a; c, electron micrograph of anaphase nucleus; and d, early telophase nucleus. The disorganized mitochondria are also shown in c and d, demonstrating the mitotic nuclei localized in the degenerated cardiomyocyte. C. Quantitative analysis of Ki-67-positive cells under different conditions as shown in Fig 2C. D. Mitotic nuclei in hypertrophic cardiomyocytes (CM) were differentiated as described in the text and the total mitotic nuclei include all phospho-histone H3-positive cells. The number of the Ki-67- or phospho-histone H3-positive nuclei among 10,000 nuclei in each histological section was counted. Each data point was obtained from 6 to 8 heart samples and expressed as means ± SEM, and the mean values do not share the same letter are significantly different (p<0.05).
Mitosis of hypertrophic cardiomyocytes induced by Cu repletion
To determine if all or any of these Ki-67-positive cells undergo mitosis, a mitosis-specific phospho-histone H3 antibody (16) was used. We found non mitotic cells in most cases or only one mitotic cell for the most in any histological section from the Cu deficient hypertrophic myocardium, but a large number of mitotic cells from the myocardium of mice fed Cu-replete diet for 3 days or 7 days (Fig 2D and 4C). Considerably fewer mitotic cells than Ki-67-positive cells were present at any given time under all the conditions (Fig 2 and 4), but all mitotic cells were Ki-67-positive (Fig 4A). Mitosis of hypertrophic cardiomyocytes was identified by dual immunohistochemical staining with a-sarcomeric actin and phospho-histone H3 in hypertrophic cardiomyocytes (Fig 4A). To further define that duplication of the existing cardiomyocytes plays a role in myocardial regeneration, we used EM and observed that Ki-67 immunogold staining was localized in the nuclei of cardiomyocytes (Fig 4B). We also found cardiomyocytes with disorganized organelles in the process of mitosis (Fig 4B).
We determined quantitatively the contribution of the duplication of hypertrophic cardiomyocytes to the regenerated myocardium. Only those mitotic myocytes clearly showing hypertrophy were counted as duplicating cardiomyocytes. Other mitotic nuclei that were not in the hypertrophied cardiomyocytes were counted only for the total number of the mitotic nuclei. However, these latter nuclei were found to contain the myocyte-specific transcription factor, MEF-2, demonstrating that these cells were either of myocyte origin or readily differentiated to myocyte (Fig 4A). With the exclusion of this type of cells, we found that about 1/3 of the mitotic cells were hypertrophic cardiomyocytes in the regenerating myocardium, but none of the mitotic myocytes were present in Cu-deficient hypertrophic myocardium (Fig 4C).
Discussion
The results here demonstrate that Cu repletion-induced regression of heart hypertrophy is associated with normalization of the size of the hypertrophied cardiomyocytes as well as the total number of cardiomyocytes in the heart. This process is likely accomplished by a combination of several cellular dynamic changes. It appeared that a direct reduction in the size of the hypertrophied cardiomyocytes was involved in this process. The division of the hypertrophic cardiomyocytes with reduced cell size was also detected. In addition, it was possible that cardiac progenitor cells were involved in the regeneration process.
A direct reduction in the size by Cu supplementation of hypertrophied cardiomyocytes has been observed in vitro in our recent studies (17). Primary cultures of neonatal rat cardiomyocytes were treated with phenylephrine (PE) at a final concentration of 100 μM in cultures and the cells became hypertrophied after the treatment with PE for 48 hrs. In that study, we have found that the purity of cardiomyocytes in the cultures was more than 95%, determined by flow cytometry cell sorting of α-sarcomeric actin stained cardiomyocytes. The hypertrophied cardiomyocytes had increased expression of β-myosin heavy chain protein, α-skeletal actin, and atrial natriuretic peptide, along with increased cell volume and protein content. The hypertrophied cardiomyocytes were exposed to Cu sulfate at a final concentration of 5 μM in cultures. This Cu treatment reduced the size of the hypertrophied cardiomyocytes, as measured by flow cytometry, protein content in cells and cell volume. Cell cycle analysis and cell sorting of phosphor-histone H3 labeled cardiomyocytes indicated that cell division was not involved in the Cu-induced regression of cardiomyocyte hypertrophy. This in vitro study thus demonstrates that Cu reduces the size of hypertrophied cardiomyocytes leading to regression of cardiac hypertrophy in the primary cultures of neonatal rat cardiomyocytes.
Re-entry into cell cycle and mitosis of the hypertrophic cardiomyocytes would make a critical contribution to the regression of heart hypertrophy in vivo. This contribution to the regression of heart hypertrophy was not observed in our in vitro studies using primary cultures of neonatal rat cardiomyocytes. In the hypertrophied heart, the hypertrophic cardiomyocytes were Ki-67-positive. In the regenerating myocardium, the mitotic cardiomyocytes were those having increased volume. The time course of the regression of the hypertrophic cardiomyocytes was coincident with the mitotic active period and closely correlated with the time course of the myocardial regeneration. At the end of the two-week regression, the hypertrophic cardiomyocytes disappeared and mitosis ceased, further indicating the contribution of duplication of hypertrophic cardiomyocytes to myocardial regeneration.
The rapid regression of heart hypertrophy following Cu repletion was initially observed in the Cu deficiency-induced heart hypertrophy in mouse model. Our recent studies have shown that dietary supplementation with physiologically relevant levels of Cu also made a rapid regression of heart hypertrophy induced by pressure overload in mouse model (18). These observations suggest that pathological heart hypertrophy would be like physiological heart hypertrophy in which the regression of the hypertrophy occurs. This notion is also supported by recent human studies that the implantation of left ventricular assist devices in cardiac failure patients can result in an improved geometry of the hypertrophied heart at both organ and cellular levels, thus leading to improved left ventricular function as a result of a reduction in wall stress and improved mechanical performance (19-21). We show here that differentiated cardiomyocytes have the capacity for self-regeneration. The potential for cell replication is likely retained in the cell, but for some reasons is inhibited in the differentiated cardiomyocytes. In the Cu-deficient heart, the hypertrophic cardiomyocytes were stimulated to re-enter the cell cycle, but prevented from undergoing mitosis. On Cu repletion, some critical factor(s) would be available to trigger, or to remove the inhibition of, the transition to mitosis. While Cu repletion initiated this mitotic transition in the present study, mechanical intervention using left ventricular assist devices has apparently caused analogous changes in patients with failing hearts (19-21).
A clinically relevant question is whether or not Cu supplementation to patients with heart disease would be beneficial. Previous studies have documented that the hearts of people who died from ischemic heart disease are low in Cu (22-25), although the mechanism for Cu loss in the ischemic heart has not been explored. Furthermore, Cu deficiency leads to cardiac ischemic injury and hypertrophic cardiomyopathy (1,2,26). Multiple signaling pathways and cross interactions between the pathways are involved in the regulation of cardiac hypertrophy (27). Therefore, pressure overload and dietary Cu deficiency would activate the same cascade and once the cascade of signaling transduction that leads to hypertrophy is activated, cardiac hypertrophy would be a common endpoint. To prevent cardiac hypertrophy an intervention targeting the etiology would be effective; however, to reverse the cardiac hypertrophy that has already developed an etiology-targeted approach would not be effective. Therefore, Cu repletion-induced regression of heart hypertrophy observed in this study would not be simply explained as a counter action of Cu repletion to Cu deficiency. This was further elucidated in the study of Cu supplementation reverses pressure overload-induced heart hypertrophy (20). These results suggest that a shift from the myocardial remodeling to a reverse remodeling program may be activated by Cu repletion.
In summary, Cu repletion results in a rapid regression of heart hypertrophy and prevents heart failure induced by dietary Cu restriction. Although a direct reduction in the size of the hypertrophied cardiomyocytes is importantly involved in the regression of heart hypertrophy, the hypertrophied cardiomyocytes have the potential to replicate, making an important contribution to the repletion of the lost cardiomyocytes. The hypertrophied cardiomyocytes have the potential to re-enter the cell cycle, but undergo mitosis only during Cu repletion-induced regression. In addition, non-myocyte proliferation and differentiation to cardiomyocytes may be involved in the regeneration of myocardium. The combination of these processes in response to Cu repletion contributes to the regression of heart hypertrophy, including the reduction in the size of the hypertrophied cardiomyocytes and normalization of the total number of cardiomyocytes.
Acknowledgments
The authors thank Xinggu Sun for technical assistance. This study was support in part by the National Institutes of Health grant HL63760. YJK is a Distinguished University Scholar of the University of Louisville.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Medeiros DM, Wildman RE. Newer findings on a unified perspective of copper restriction and cardiomyopathy. Proc Soc Exp Biol Med. 1997;215:299–313. doi: 10.3181/00379727-215-44141. [DOI] [PubMed] [Google Scholar]
- 2.Elsherif L, Ortines RV, Saari JT, Kang YJ. Congestive heart failure in copper-deficient mice. Exp Biol Med. 2003;228:811–817. doi: 10.1177/15353702-0322807-06. [DOI] [PubMed] [Google Scholar]
- 3.Elsherif L, Wang L, Saari JT, Kang YJ. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. J Nutr. 2004;134:855–860. doi: 10.1093/jn/134.4.855. [DOI] [PubMed] [Google Scholar]
- 4.Sabbah HN, Sharov VG. Apoptosis in heart failure. Prog Cardiovasc Dis. 1998;40:549–562. doi: 10.1016/s0033-0620(98)80003-0. [DOI] [PubMed] [Google Scholar]
- 5.Kang YJ, Zhou ZX, Wu H, Wang GW, Saari JT, Klein JB. Metallothionein inhibits myocardial apoptosis in copper-deficient mice: role of atrial natriuretic peptide. Lab Invest. 2000;80:745–757. doi: 10.1038/labinvest.3780078. [DOI] [PubMed] [Google Scholar]
- 6.Richey PA, Brown SP. Pathological versus physiological left ventricular hypertrophy: a review. J Sports Sci. 1998;16:129–141. doi: 10.1080/026404198366849. [DOI] [PubMed] [Google Scholar]
- 7.Kishimoto I, Rossi K, Garbers DL. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci U S A. 2001;98:2703–2706. doi: 10.1073/pnas.051625598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem. 2003;278:47694–47699. doi: 10.1074/jbc.M309661200. [DOI] [PubMed] [Google Scholar]
- 9.Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, Crow MT, Lakatta EG. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res. 1994;75:23–32. doi: 10.1161/01.res.75.1.23. [DOI] [PubMed] [Google Scholar]
- 10.Flesch M, Erdmann E, Bohm M. Changes in beta-adrenoceptors and G-proteins during the transition from cardiac hypertrophy to heart failure. J Card Fail. 1996;2:S35–S43. doi: 10.1016/s1071-9164(96)80057-1. [DOI] [PubMed] [Google Scholar]
- 11.Grossman W. Diastolic dysfunction in congestive heart failure. N Engl J Med. 1991;325:1557–1564. doi: 10.1056/NEJM199111283252206. [DOI] [PubMed] [Google Scholar]
- 12.Chemla D, Coirault C, Hebert JL, Lecarpentier Y. Mechanics of Relaxation of the Human Heart. News Physiol Sci. 2000;15:78–83. doi: 10.1152/physiologyonline.2000.15.2.78. [DOI] [PubMed] [Google Scholar]
- 13.Reeves PG, Nielsen FH, Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 14.Kang YJ, Chen Y, Yu A, Voss-McCowan M, Epstein PN. Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J Clin Invest. 1997;100:1501–1506. doi: 10.1172/JCI119672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 16.Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 1999;97:99–109. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Y, Jiang Y, Kang YJ. Copper reverses cardiomyocyte hypertrophy through vascular endothelial growth factor-mediated reduction in the cell size. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang Y, Reynolds C, Xiao C, Feng W, Zhou Z, Rodriguez W, Tyagi SC, Eaton JW, Saari JT, Kang YJ. Dietary copper supplementation reverses hypertrophic cardiomyopathy induced by chronic pressure overload in mice. J Exp Med. 2007;204:657–666. doi: 10.1084/jem.20061943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zafeiridis A, Jeevanandam V, Houser SR, Margulies KB. Regression of cellular hypertrophy after left ventricular assist device support. Circulation. 1998;98:656–662. doi: 10.1161/01.cir.98.7.656. [DOI] [PubMed] [Google Scholar]
- 20.Mancini DM, Beniaminovitz A, Levin H, Catanese K, Flannery M, DiTullio M, Savin S, Cordisco ME, Rose E, Oz M. Low incidence of myocardial recovery after left ventricular assist device implantation in patients with chronic heart failure. Circulation. 1998;98:2383–2389. doi: 10.1161/01.cir.98.22.2383. [DOI] [PubMed] [Google Scholar]
- 21.Muller J, Wallukat G, Weng YG, Dandel M, Spiegelsberger S, Semrau S, Brandes K, Theodoridis V, Loebe M, Meyer R, Hetzer R. Weaning from mechanical cardiac support in patients with idiopathic dilated cardiomyopathy. Circulation. 1997;96:542–549. doi: 10.1161/01.cir.96.2.542. [DOI] [PubMed] [Google Scholar]
- 22.Zama N, Towns RLR. Cardiac copper, magnesium, and zinc in recent and old myocardial infarction. Biol Trace Elem Res. 1986;10:201–208. doi: 10.1007/BF02795618. [DOI] [PubMed] [Google Scholar]
- 23.Wester PO. Trace elements in human myocardial infarction determined by neutron activation analysis. Acta Med Scand. 1965;178:765–788. doi: 10.1111/j.0954-6820.1965.tb04329.x. [DOI] [PubMed] [Google Scholar]
- 24.Chipperfield B, Chipperfield JR. Differences in metal content of the heart muscle in death from ischemic heart disease. Am Heart J. 1978;95:732–737. doi: 10.1016/0002-8703(78)90503-3. [DOI] [PubMed] [Google Scholar]
- 25.Anderson TW, Neri LC, Schreiber GB, Talbot FD, Zdrojewski A. Letter: Ischemic heart disease, water hardness and myocardial magnesium. Can Med Assoc J. 1975;113:199–203. [PMC free article] [PubMed] [Google Scholar]
- 26.Klevay LM, Saari JT. Comparative responses of rats to different copper intakes and modes of supplementation. Proc Soc Exp Biol Med. 1993;203:214–220. doi: 10.3181/00379727-203-43594. [DOI] [PubMed] [Google Scholar]
- 27.Schaub MC, Hefti MA, Harder BA, Eppenberger HM. Various hypertrophic stimuli induce distinct phenotypes in cardiomyocytes. J Mol Med. 1997;75:901–920. doi: 10.1007/s001090050182. [DOI] [PubMed] [Google Scholar]