

Abstract
A series of inhibitors with a squaramide core was synthesized following its discovery in a high-throughput screen for novel inhibitors of a transcription-coupled translation assay using Escherichia coli S30 extracts. The inhibitors were inactive when the plasmid substrate was replaced with mRNA, suggesting they interfered with transcription. This was confirmed by their inhibition of purified E. coli RNA polymerase. The series had antimicrobial activity against efflux-negative strains of E. coli and Haemophilus influenzae. Like rifampin, the squaramides preferentially inhibited synthesis of RNA and protein over fatty acids, peptidoglycan, and DNA. However, squaramide-resistant mutants were not cross-resistant to rifampin. Nine different mutations were found in parts of rpoB or rpoC that together encode the so-called switch region of RNA polymerase. This is the binding site of the natural antibiotics myxopyronin, corallopyronin, and ripostatin and the drug fidaxomicin. Computational modeling using the X-ray crystal structure of the myxopyronin-bound RNA polymerase of Thermus thermophilus suggests a binding mode of these inhibitors that is consistent with the resistance mutations. The squaramides are the first reported non-natural-product-related, rapidly diversifiable antibacterial inhibitors acting via the switch region of RNA polymerase.
INTRODUCTION
Clinical resistance to currently prescribed antibiotics is on the rise, thus increasing the need for new classes of antimicrobials that can circumvent emerging resistance mechanisms (10). There are still only a few enzymes that are essential for bacterial growth and have been clinically validated as antibacterial targets. All clinical antibacterial protein translation inhibitors have so far been identified by cell-based screening efforts with compounds from natural sources (8). New, small inhibitors might be found by screening small-molecule libraries for inhibitors of the translation machinery with an in vitro system, such as transcription-coupled translation in bacterial S30 extracts.
Here, we report the discovery of squaramides as inhibitors of RNA polymerase (RNAP) that resulted from such a screening effort. The antimicrobial activity against an efflux-negative strain of H. influenzae was exploited to show that squaramides mediate their inhibitory activity via the switch region of RNAP. Their mode of action therefore is similar to that of the natural compounds myxopyronin, corallopyronin, ripostatin, and fidaxomicin (26) rather than that of rifamycins, which bind closer to the catalytic site and prevent RNA extension (7). This is the first report of rapidly diversifiable small-molecule inhibitors of RNAP with that mode of inhibition, supporting the use of a transcription-coupled translation assay to find novel inhibitory scaffolds of the RNAP switch region in small-molecule collections.
MATERIALS AND METHODS
Bacterial strains.
E. coli RNAP and S30 extracts were isolated from E. coli MRE600 (ATCC 2941). For susceptibility studies E. coli ATCC 27325 and H. influenzae ATCC 51907 were used, which were also the parental strains of E. coli tolC::Tn10 and H. influenzae acrB::cat, respectively.
Purification of E. coli RNA polymerase.
Purification of E. coli RNAP was based upon the procedure developed by Burgess and Jendrisak (4). The enzyme was purified from E. coli cultures grown in 5 liters Terrific Broth in a Bioflo 3000 fermentor (New Brunswick Scientific, Edison, NJ) at 37°C with constant agitation at 300 rpm and harvested at an optical density at 600 nm (OD600) of up to 17. The resulting 120-g wet weight of frozen cell paste was resuspended in 200 ml of lysis buffer consisting of 25 mM Tris-HCl (pH 8.0), 1 mM EDTA, 10 mM dithiothreitol (DTT), 10 mM MgCl2, 10% (vol/vol) glycerol, 20 mM spermidine, and five protease inhibitor cocktail tablets (Roche Molecular Biochemical, Indianapolis, IN). Cells were disrupted by a French press at 18,000 lb/in2 twice, and the crude extract was centrifuged at 150,000 × g for 30 min at 4°C. Solid ammonium sulfate (0.35 g/ml) was added to the supernatant, which was mixed at 4°C for 1 h and then centrifuged at 100,000 × g for 20 min at 4°C. The pellets were suspended in 100 ml of buffer A, consisting of 25 mM Tris-HCl (pH 8.0), 1 mM EDTA, 10 mM DTT, 10 mM MgCl2, and 10% (vol/vol) glycerol, and dialyzed against 4 liters of buffer A at 4°C overnight. The dialyzed sample was centrifuged at 10,000 × g at 4°C for 30 min to remove insoluble proteins. The supernatant was loaded at a flow rate of 3.0 ml/min onto a 300-ml Q-Sepharose HP (XK 50/30) column (GE Healthcare Life Sciences, Piscataway, NJ) preequilibrated with buffer A. The column was washed with buffer A, and the protein was eluted with 0.35 M NaCl in buffer A. Fractions containing E. coli RNAP were identified by Western blotting with anti-E. coli RNAP α subunit monoclonal antibody (Neoclone, Madison, WI), pooled, and dialyzed against 2 liters of buffer A at 4°C overnight. The dialyzed sample was loaded at a flow rate of 2.0 ml/min onto a 60-ml Q-Sepharose HP (XK 26/20) column (GE Healthcare Life Sciences) preequilibrated with buffer A. The column was then washed with buffer A, and the protein was eluted by a linear gradient from 0 to 1 M NaCl in buffer A. Fractions containing RNAP were pooled and dialyzed against 1 liter of buffer A overnight at 4°C. The dialyzed sample was loaded at a flow rate of 1.5 ml/min onto a 20-ml heparin Sepharose CL-6B (HR16/10) column (GE Healthcare Life Sciences) preequilibrated with buffer A. After the column was washed with 100 ml of buffer A, the protein was eluted by a linear gradient from 0 to 1 M NaCl in buffer A. The fractions containing holoenzyme with subunit α2β′βσ were pooled and dialyzed against 1 liter of 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 0.1 mM EDTA, 1 mM DTT, and 50% (vol/vol) glycerol. The final yield of RNAP of about 30 mg was stored at −20°C.
RNA polymerase inhibition assay.
Assays (30 μl) based on that described by Johnson et al. (16) contained 3 nM purified E. coli RNAP holoenzyme; 2.4 μg/ml of plasmid NTC-7382-41H-HA (Nature Technologies, Lincoln, NE); 180 μM (each) ATP, CTP, GTP, and UTP; and 0.1 U/ml inorganic pyrophosphatase (Roche Applied Science) in buffer consisting of 50 mM HEPES-NaOH (pH 8.0), 0.1 M trehalose, 80 mM NH4Cl, 4 mM MgCl2, 2 mM dithiothreitol, 0.8 mM spermidine, and 0.0045% Tween 20. The reactions also contained various concentrations of test compounds dissolved in dimethylsulfoxide (DMSO) and diluted 50-fold in the assay. Compounds were incubated with DNA, after which the reactions were started with RNAP. For 100% inhibition, the reaction mixtures contained 200 nM actinomycin D (50% inhibitory concentration [IC50], 2.3 nM). Reactions were quenched after 1 h with 30 μl of malachite green/molybdate reagent (15). Absorbance at 650 nm was determined after 5 min to measure the concentration of inorganic phosphate.
Construction of luciferase reporter plasmid.
Plasmid pLH1824, used as the reporter plasmid, was constructed by amplification of lacIq from pMAL-p2X (New England BioLabs, Ipswich, MA) and cloning into the XhoI site of pBest (Promega). Oligonucleotides used for amplification were 5′-ACGCTCGAGCCGACACCATCGAATGGTGCAAAACC-3′ and 5′-ACGTCTCGAGTCACTGCCCGCTTTCCAGTCGGGAAACC-3′ (XhoI sites are underlined). The desired orientation, unidirectional transcription of lacIq with luc, was confirmed by nucleotide sequencing.
Preparation of S30 extract.
The method for preparation of S30 extract was modified from the procedure of Pratt (22). E. coli was grown to an OD650 of 2.5 in growth medium containing 0.6% KH2PO4, 2.9% K2HPO4, 1% yeast extract, and 1% glucose in a 100-liter fermentation volume (37°C, 200 rpm agitation, 30% dissolved oxygen). Cells were harvested by centrifugation for 15 min at 3,500 × g at 4°C and washed twice in cold wash buffer consisting of 10 mM Tris-acetate (pH 7.4), 14 mM magnesium acetate, 60 mM KCl, 6 mM β-mercaptoethanol (BME), and 50 μg/ml phenylmethanesulfonyl fluoride (PMSF). Pellets were resuspended in 4 ml of cold wash buffer without BME per g of cell paste, followed by cell lysis by two passes through a French press at 10,000 lb/in2 and centrifugation at 30,000 × g for 30 min at 4°C. The supernatant was centrifuged a second time, resulting in crude S30 extract. Preincubation mix (1 ml), consisting of 0.75 M Tris-acetate (pH 7.4), 7.5 mM DTT, 21 mM magnesium acetate, 75 μM (each) the 20 amino acids, 6 mM ATP (Sigma-Aldrich, St. Louis MO), 2% phosphoenolpyruvate-trisodium salt (Chem-Impex International, Wood Dale, IL), and 5 U of pyruvate kinase (Sigma-Aldrich), was added per 6 ml of crude extract, followed by shaking at 100 rpm at room temperature for 60 min. The resulting product was dialyzed against 2 volumes of dialysis buffer at 4°C consisting of 10 mM Tris-acetate (pH 7.4), 14 mM magnesium acetate, 60 mM potassium acetate, and 1 mM DTT in dialysis tubing (molecular weight cutoff, 6,000 to 8,000; Spectrum Labs, Rancho Dominguez CA) with 4 buffer changes at 1-h intervals. The extract was aliquoted in 50-ml volumes and stored at −80°C.
S30 protein concentration measurement.
S30 concentration was determined using the microtiter plate procedure of the Bio-Rad protein assay kit II (Bio-Rad Life Sciences, Hercules, CA). Absorbance at 595 nm was measured with a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Transcription-coupled translation assay.
The transcription-coupled translation assay was performed essentially according to Zubay (28) and Pratt (22). Reagent 1 contained S30 extract, which was diluted to 4 mg/ml in S30 buffer consisting of 10 mM Tris-acetate (pH 7.4), 60 mM potassium acetate, and 14 mM magnesium acetate. Reagent 2 consisted of 0.5 mM (each) ATP, CTP, UTP, and GTP (Chem-Impex International, Wood Dale, IL), 20 mM phosphoenolpyruvate (Chem-Impex International), 100 μg/ml E. coli tRNA (Roche Diagnostics Corp., Indianapolis, IN), 20 μg/ml folinic acid, 1 mM cyclic AMP (cAMP), 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG), 0.2 mM DTT, 30 mg/ml polyethylene glycol 8000, 0.5 mM (each) all 20 of the translated amino acids, 2 U/ml pyruvate kinase, and 40 μg/ml pLH1824 reporter plasmid DNA. Both reagents were allowed to preincubate at room temperature for 1 h.
Compounds were dissolved at up to 10 mM in DMSO and 2-fold serially diluted in DMSO. For 100% inhibition, the reaction mixtures contained 10 μM erythromycin (IC50 = 0.4 μM). Diluted compound (0.3 μl) was added to 384-well white polystyrene plates (Corning, Inc., Lowell, MA). Reagent 1 (15 μl) was added to the compound plates, avoiding the formation of air bubbles, followed by 15 μl of reagent 2, after which the plate was briefly shaken in an Eppendorf Mixmate plate shaker. The plates were sealed with foil and incubated at room temperature for 1 h, avoiding temperature fluctuations due to drafts or sunlight. Subsequently, 15 μl of luciferin developer consisting of 0.4 μM lithium salt of coenzyme A (Sigma), 0.7 μM D-luciferin (Gold Biotechnology, St. Louis MO), 0.8 μM ATP, 20 mM Tricine (pH 7.8), 1 mM magnesium carbonate, 0.1 mM EDTA, 2.3 mM magnesium sulfate, and 33 mM DTT was added, and light production was measured immediately using a Pherastar plate reader (BMG Labtech, Ortenberg, Germany).
Artifact correction assay.
Compounds which interfere with luciferase detection were identified as assay artifacts by addition of 1 nM luciferase protein (Promega, Madison, WI) and omission of luciferase reporter plasmid pLH1824 from the transcription-coupled translation assay as described above. Artifact assays were run in parallel with the activity assay and used to correct the latter (25).
Translation assay.
In order to distinguish between transcription and translation inhibitors, luciferase reporter plasmid pLH1824 used in the E. coli coupled T/T assay was replaced with purified luciferase mRNA (20 μg/ml). mRNA template was generated using the RiboMax large-scale RNA production system and linear SP6 luciferase DNA (Promega, Madison, WI).
Susceptibility testing.
MICs were determined using broth microdilutions according to the guidelines of the Clinical and Laboratory Standards Institute (9).
Inhibition of macromolecular synthesis.
The procedure was essentially that according to Hilliard et al. (14), with modifications published previously (5).
Rabbit reticulocyte translation assay.
The rabbit reticulocyte translation assay was conducted according to the instructions of the manufacturer (Promega, Madison WI) at a volume of 10 μl in 384-well white polystyrene plates with a final DMSO concentration of 1% (vol/vol) and up to 100 μM test compound. After 1 h at room temperature, 2 μl was transferred to a white 384-well polystyrene plate to which 43 μl of luciferin developer was added. Luminescence was measured as described above.
Physicochemical and biophysical properties.
Plasma protein binding and logD determination were measured according to Reck et al. (23).
Equilibrium solubility was determined for 50-μl samples with 10 mM compound dissolved in DMSO. The solvent was removed by drying under vacuum at 40°C, after which the samples were dissolved at 25°C in 300 μl 0.1 M sodium phosphate buffer (pH 7.4) with agitation for 24 h. Samples were filtered through GF/B filtration plates (GE Healthcare Life Sciences) to remove undissolved material, after which the filtrates were quantitated using liquid chromatography-mass spectrometry (LC-MS).
Clearance in rat hepatocytes was determined by incubating compounds at 1 μM with rat hepatocytes at 106/ml in 500 μl Leibovitz L-15 medium (Life Technologies) for 2 h. Samples of 50 μl were added to 150 μl stop solution (125 ng/ml carbutamide [Aldrich], 0.1% formic acid in acetonitrile) and analyzed by LC-MS.
Isolation of resistant mutants.
Mutants of H. influenzae acrB were isolated as described previously (6). In short, H. influenzae acrB was grown overnight at 37°C in an atmosphere containing 5% (vol/vol) CO2 on Haemophilus test medium (HTM) agar (9), and the resulting colonies were scraped off and suspended in HTM broth. Volumes of 100 μl containing 10, 100, and 1,000 CFU were spread on chocolate II agar plates (Becton, Dickinson, Franklin Lakes, NJ) to determine the titer. Volumes of 100 μl containing 105, 106, 107, and 108 CFU were spread on HTM agar plates containing 15 μg/ml filter-sterilized hematin (Sigma), 15 μg/ml filter-sterilized NAD (Sigma), and various 2-fold serial dilutions of compound 1, ranging from 3.12 to 25 μM. The lowest titer was used to determine the plate MIC of compound 1 at 12.5 μM. At higher compound concentrations, resistant colonies appeared after 24 to 48 h at 37°C with 5% (vol/vol) CO2 at a frequency of approximately 10−7.
DNA manipulation and sequencing.
Genomic DNA was isolated using a Maxwell 16-cell DNA purification kit (Promega, Madison WI). Custom-made oligonucleotides were obtained from Eurofins MWG Operon (Huntsville, AL). Amplification reactions were performed using high-fidelity PCR master mix (Roche Diagnostics, Indianapolis, IN). The products were isolated using QIAquick PCR purification kits (Qiagen, Valencia, CA). Sequencing reactions were performed by Eurofins MWG Operon.
Preparation of protein structure for modeling.
The X-ray crystal structure of the RNAP-myxopyronin complex from Thermus thermophilus (Protein Data Bank [PDB] code 3DXJ [21]) was used to provide a possible provisional model of the binding of compounds 1 to 9, since there is no available X-ray crystal structure of myxopyronin bound to E. coli RNAP. This crystal structure contains two RNAP-myxopyronin complexes in the unit cell; the first is comprised of chains A to F, and the second is formed by chains K to P. The first RNAP complex (chains A to F) was selected for our docking model because the average B-factor of the bound myxopyronin ligand was significantly lower than the average B-factor of the myxopyronin ligand bound to the second RNAP complex (69 versus 84). The lower B-factor indicates that the ligand coordinates are resolved better in the first complex structure. The conformation of the binding pocket residues in the first complex structure will likely be more relevant to binding as a result. The myxopyronin ligand, ions, and cosolutes were removed using the MOE 2011.11 modeling software (Chemical Computing Group, Montreal, Canada). The RNAP receptor model was saved as a PDB file to be prepared for docking.
The docking model was first validated by docking in many near-native poses of myxopyronin. The top 3 ranked poses fell between a root mean square deviation of 2.2 and 2.3 Å on the X-ray crystal structure coordinates of the ligand. Qualitatively, the top-ranked docking poses reproduced the crystallographic binding mode of myxopyronin well (see Fig. 3). Both the carbamate and dienone side chains were oriented in the appropriate pockets and reproduced most of the important interactions observed in the crystal structure. The pyrone ring was appropriately oriented despite the lack of many direct interactions between the ring and the pocket. In the top-ranked docked poses, the pyrone ring was flipped by 180 degrees relative to the crystal pose which, other than altering the position of the ether oxygen in the ring, was a trivial inversion due to the shape symmetry of the pyrone ring. Most importantly, the carbonyl oxygens on the ring were oriented nearly identically in both the docked pose and the crystal structure, as these two groups form direct or water-mediated hydrogen bonds with the protein.
Fig 3.

Self-docking of myxopyronin into the switch region of RNA polymerase. The RpoB and RpoC proteins are shown as red and blue ribbons, respectively, illustrating that the switch region is formed by the RpoB-RpoC interface. SW1 and SW2, which are part of RpoC, are shown in pink; the dienone tail points to SW1, whereas the pyrone ring mainly interacts with SW2. The partial rpoB helix to the left of myxopyronin is part of the helix-turn-helix that is sandwiched between SW3 and SW4, which are not visible. The predicted binding mode of myxopyronin displays two minor deviations from the crystallographic mode: the dienone tail in the docked pose is rotated by approximately 60 degrees with respect to the crystal pose, and the pyrone ring is flipped by approximately 180 degrees. Although the flip of the pyrone ring modifies the location of the ether oxygen in the ring, the overall binding mode from the docking is very similar to the crystallographic binding mode.
As a control to explore the dependence of these results on the RNAP structure used, we repeated the docking of compounds 1 to 9 to a different structure of RNAP from T. thermophilus (PDB 3EQL [3]). Qualitatively, the binding modes of the squaramides predicted by this second docking were equivalent to those predicted by the first docking. The phenyl tail of the squaramide ligands was predicted to bind specifically to the dienone pocket, and the squaramide core appeared to mimic the pyrone ring. The position of the oxazole tail was even less well determined in the second docking, as it appears to make no highly specific interactions with this conformation of the receptor.
Docking.
The Gold 5.1 software (CCDC, Cambridge, United Kingdom) was used to dock compounds 1 to 9 to the RNAP receptor structure. Using the Hermes molecular editor (CCDC, Cambridge, United Kingdom), hydrogen atoms were added to the receptor atoms, and the binding site was defined by including all residues within 6.0 Å of the myxopyronin ligand coordinates. After this preparation step, the receptor was saved in MOL2 format for docking with Gold. Ten ligands, compounds 1 to 9 and myxopyronin, were prepared for docking by first converting the structures to isomeric smile format using the Babel software (OpenEye Scientific Software, Santa Fe, NM) in order to eliminate any conformational bias from the docking. Next, using the Omega2 program (OpenEye Scientific Software, Santa Fe, NM), one low-energy conformation of each ligand was generated. Gold flexible ligand docking generated 30 poses of each ligand, which were ranked using the Goldscore scoring function. Default values were used for all other docking parameters.
Docked ligand poses were rescored using a first-principle molecular mechanics-based scoring function that incorporated the Poisson-Boltzmann implicit solvation model to account for protein and ligand desolvation energies. Using Zapbind (OpenEye Scientific Software, Santa Fe, NM), the electrostatic component of the binding free energy was calculated. Subsequently, a Lennard-Jones term, with a 6 to 9 softened repulsive potential to account for Van der Waals interactions between the protein and the ligand, was added. The softened repulsive potential was used to compensate for our treatment of the receptor as a rigid body, despite the likelihood of allosteric motions of the switch region.
Homology modeling of resistance mutations.
To understand the mechanisms by which the observed mutations could reduce the target potency of compounds 1 to 9, homology models were constructed to predict the approximate structure of the nine mutations listed in Table 2. The homology modeling module in the Molecular Operating Environment (MOE) 2011.11 (Chemical Computing Group, Montreal, Canada) software was used to introduce each of the nine mutations into the first RNAP assembly (chains A to F) in the 3DXJ crystal structure. The mutated residues were repacked and energy minimized using default values for the homology modeling parameters in the MOE.
Table 2.
Antimicrobial activity of squaramide RNA polymerase inhibitors 1 to 9a
| Compound | Activity against H. influenzae and mutants |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parent | RpoB |
RpoC |
||||||||
| Q1258H | E1280K | D1297E | P1321S | I1327T | A1323V | A1323T | L1333F | K1349Q | ||
| 1 | 12.5 | 100 | 50 | 141 | 141 | 141 | 200 | 200 | 200 | 25 |
| 2 | 71 | 400 | 200 | 400 | 400 | 800 | 400 | 800 | 400 | 141 |
| 3 | 18 | 282 | 50 | 400 | 400 | 50 | >800 | 400 | 400 | 100 |
| 4 | 35 | 100 | 100 | 100 | 141 | 141 | 200 | 200 | 141 | 35 |
| 5 | 71 | 400 | 141 | 400 | 400 | 141 | >800 | 800 | 400 | 282 |
| 6 | 141 | >800 | 200 | >800 | >800 | 50 | >800 | >800 | >800 | 800 |
| 7 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| 8 | 200 | 141 | 200 | 100 | 200 | 200 | 200 | 141 | 100 | 100 |
| 9 | 200 | 200 | 200 | 100 | 200 | 200 | 200 | 200 | 141 | 100 |
| Rifampin | 0.1 | 0.1 | 0.14 | 0.1 | 0.1 | 0.1 | 0.14 | 0.14 | 0.1 | 0.1 |
| Linezolid | 8.9 | 12.5 | 8.9 | 6.3 | 12.5 | 12.5 | 8.9 | 12.5 | 12.5 | 6.3 |
Activity was measured as MICs (in μM; geometric means are shown [n = 2]) against H. influenzae acrB::cat and mutants isolated as being resistant to compound 1. Altered amino acids are indicated using E. coli numbering.
Compound syntheses.
Synthesis of compound 1 and analogs followed standard solution-phase organic synthesis and purification.
3-Amino-4-ethoxycyclobut-3-ene-1,2-dione.
A solution of 3,4-diethoxycyclobut-3-ene-1,2-dione (2.5 g, 14.69 mmol) in methanol (15 ml) was treated with ammonia (7 M in methanol, 5 ml, 35 mmol) and stirred at room temperature for 16 h. The resulting suspension was filtered and the filtrate concentrated to give crude product. This material was suspended in a 1:1 mixture of ethyl acetate and hexanes (70 ml), heated to 60°C for 2 min, cooled to room temperature, and filtered to give the title compound as an off-white solid (1.54 g, 10.91 mmol, 74.3%). 1H nuclear magnetic resonance (NMR) (300 MHz, DMSO-d6) d ppm 1.20 to 1.55 (m, 3H), 4.64 (quartet [q], J = 6.97 Hz, 2H), 8.30 (broad singlet [br. s], 2H).
N-(2-Ethoxy-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide.
A stirred solution of 3-amino-4-ethoxycyclobut-3-ene-1,2-dione (2,050 mg, 14.53 mmol) in tetrahydrofuran (60 ml) was treated with sodium hydride (1,400 mg, 35 mmol) to give a bright yellow suspension. This was stirred at room temperature for 20 min, followed by treatment with 3,5-dimethylisoxazole-4-sulfonyl chloride (3,694 mg, 18.88 mmol) in one portion. This suspension was stirred at room temperature for 24 h, after which solvents were removed under reduced pressure. The resulting red paste was suspended in hot ethyl acetate (50 ml), neutralized with 2 N HCl in ether (10 ml) and filtered hot. The filtrate was concentrated and the resulting residue purified on silica gel eluting with ethyl acetate, followed by a gradient of 0 to 10% methanol in dichloromethane, to give the title compound as a yellow oil (2.07 g, 6.89 mmol, 47.5% yield). 1H NMR (300 MHz, DMSO-d6) d ppm 1.22 to 1.35 (m, 3H), 2.26 (s, 3H), 2.55 (s, 3H), 4.56 (q, J = 6.97 Hz, 2H).
3,5-Dimethyl-N-{2-[4-(4-methylbenzyl)piperidin-1-yl]-3,4-dioxocyclobut-1-enyl}isoxazole-4-sulfonamide (compound 1).
A solution of N-(2-ethoxy-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (75 mg, 0.25 mmol), 4-(4-methylbenzyl)piperidine hydrochloride (1.25 mmol), and diisopropylethylamine (0.75 ml) in 1,4-dioxane (8 ml) was heated to 60°C for 18 h. Solvents were removed under reduced pressure, and the resulting oil was purified by silica gel chromatography, eluting a linear gradient of 50 to 100% acetone in ethyl acetate. The desired product was then precipitated from a hot ethyl acetate hexane mixture to give the title compound as an off-white powder (58 mg, 53% yield). 1H NMR (400 MHz, DMSO-d6) d ppm 1.19 (qd, J = 12.17, 3.66 Hz, 2H), 1.62 (d, J = 12.88 Hz, 2H), 1.73 (td, J = 6.95, 4.04 Hz, 1H), 2.27 (s, 4H), 2.29 (s, 3H), 2.48 (d, J = 6.90 Hz, 1H), 2.56 (s, 3H), 2.89 to 3.04 (m, 2H), 4.48 (d, J = 11.87 Hz, 2H), 5.50 to 5.95 (m, 1H), 7.01 to 7.13 (m, 4H).
Compounds 2, 4, 8, and 9 were synthesized in an analogous manner, substituting the appropriate sulfonyl chlorides and amines. For compounds 3 and 5, Boc-protected amino piperidine was used. The resulting Boc-intermediate was deprotected with anhydrous HCl, which was then subjected to reductive amination with benzaldehydes to give the desired compounds. Compound 6 was synthesized in a similar manner by reacting with 2-methyl-4-piperidone, followed by reductive amination with a benzylamine. The synthesis of compound 7 was achieved by reacting dimethyl squarate with benzyl piperidine in methanol at room temperature. This intermediate was treated with bromophenylsulfonamide and sodium hydride in dioxane at reflux to give the desired sulfonamide 7. Compound purity was determined by LC-MS shortly before biological testing and was >90%.
RESULTS
A high-throughput screen was performed on the AstraZeneca corporate compound collection using a transcription-coupled translation assay with S30 extracts of E. coli. The reporter gene was luc, which, when transcribed, translated, and incubated with suitable reagents, including luciferin, resulted in luminescence. Compounds were tested at 10 μM (adding 1% [vol/vol] DMSO) using 1% DMSO as the maximum signal and 10 μM erythromycin as the minimum signal. Inhibitory samples were further tested with serial dilutions to determine their IC50s. Compounds were abandoned if their highest soluble concentrations were lower than their IC50s. Multiple inhibitory scaffolds were identified and were separated into transcription and translation inhibitors by measuring their activity when the plasmid was replaced with luc mRNA, thus bypassing the requirement of transcription. One compound was found to inhibit the transcription process. Testing the compound in an E. coli RNA polymerase assay showed it to be an inhibitor of this enzyme, although its IC50 was higher than that in the transcription-coupled translation assay (Table 1).
Table 1.
Biochemical and antimicrobial characterization of squaramide RNA polymerase inhibitorsa
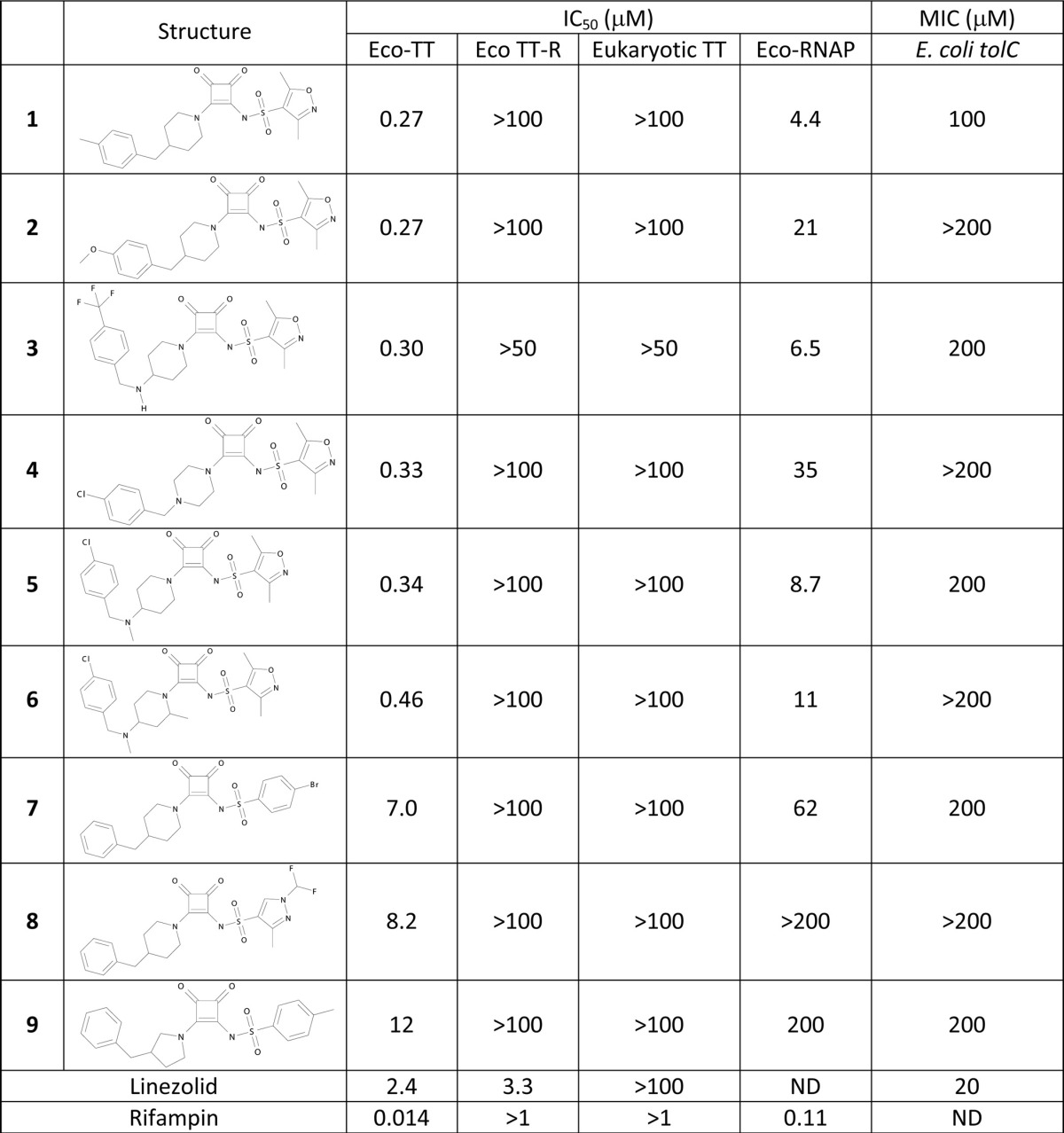
IC50s (in μM) are the averages from at least two measurements, and Hill slopes were between 0.7 and 1.7. MICs are in μM. TT, transcription-coupled translation; TT-R, TT with mRNA; eukaryotic TT, rabbit reticulocytes; RNAP, RNA polymerase; ND, not determined.
A synthetic chemistry program based on this scaffold was initiated, resulting in compounds with IC50s as low as 0.3 μM in the transcription-coupled translation assay. A common chemical feature of all active compounds was the squaramide core that has been described as a urea isostere with reasonable physicochemical and biophysical properties for drug discovery purposes (19, 27). For instance, compounds 1 to 9 showed high aqueous solubility (>1 mM), reasonable logD values (−0.1 to 0.8), human serum protein binding up to 7% free, and, when measured, low clearance in rat hepatocytes (2 to 7 μl/min/106 cells). No antimicrobial activity (MICs of >200 μM) against wild-type strains of Staphylococcus aureus, Streptococcus pneumoniae, Pseudomonas aeruginosa, Klebsiella pneumoniae, and E. coli was obtained (data not shown), although weak activity against efflux-negative E. coli tolC (11) was observed (Table 1). Weak antimicrobial activity against H. influenzae, with MICs of 100 to 200 μM, was observed for compounds 1, 3, 4, 6, and 7, whereas compounds 2, 5, 8, and 9 were inactive. However, activity against efflux-negative H. influenzae acrB (24) was sufficient to determine the mode of antibacterial action of these compounds.
Inhibition of incorporation of the biosynthetic precursors thymidine, uridine, leucine, acetic acid, and N-acetylglucosamine into DNA, RNA, protein, fatty acid, and peptidoglycan, respectively, was determined in H. influenzae acrB. Rifampin inhibited uridine incorporation by 50% at 0.2 μM, a value close to its MIC against this strain (Fig. 1 and Table 2). Leucine incorporation was inhibited similarly, consistent with the very close coupling between transcription and translation. Precursor incorporation into other macromolecules was inhibited only at rifampin concentrations that were at least 10-fold higher. Compound 1 inhibited leucine and uridine incorporation by 50% at 5 and 10 μM, respectively, matching its MIC. Inhibition of incorporation of the other precursors, however, required 25 to 50 μM (Fig. 1). Similar results were obtained with compounds 2 to 6 (data not shown). These results support inhibition of transcription as the mode of action for compounds 1 to 6. Compound 7, however, did not display this pattern. Leucine and uridine incorporation were inhibited by 50% at 75 and 50 μM, respectively, at which concentrations N-acetylglucosamine, acetate, and thymidine incorporation were inhibited as well (Fig. 1).
Fig 1.

Inhibition of precursor incorporation into H. influenzae acrB::cat by rifampin (left), compound 1 (middle), and compound 7 (right). Inhibition of synthesis of protein (leucine incorporation; closed diamonds), RNA (uridine incorporation, open diamonds), fatty acids (acetate incorporation; triangles), peptidoglycan (N-acetylglucosamine incorporation; squares), and DNA (thymidine incorporation; circles) was measured as described in Materials and Methods. The incorporation rates of added precursors into uninhibited cells were 26, 974, 13,200, 5.7, and 18 μmol/h/OD, respectively. Data are averages from two independent experiments; standard deviations are approximately 7%.
Resistant mutants against compound 1 were isolated at a frequency of approximately 10−7 when the compound was present at 1 to 2 times its MIC. Although higher compound concentrations were not tried for mutant selection, MIC determination in broth showed resistance of up to 16-fold of the MIC observed in the parental strain (Table 2). No cross-resistance with rifampin was detected, suggesting that the binding site for compound 1 is different from the rifampin binding site. The genes encoding the RNAP core enzyme, rpoA-rpoC, were sequenced for 20 resistant strains. All mutants were found to contain a mutation in either rpoB or rpoC, among which 9 different mutations in 8 amino acids were identified. All were located in the carboxyl-terminal domain of either RpoB or RpoC, which form the so-called switch region (Fig. 2). Two of these mutations, affecting E1280 and L1327 in RpoB (E. coli numbering), cause resistance to myxopyronin, corallopyronin, and ripostatin in both S. aureus and E. coli, with the E1280K mutation being in common with mutations described in two previous studies (18, 21). Four mutations, affecting Q1258 and D1297 in RpoB and A1323 and K1349 in RpoC, had not been described before. Most mutants showed cross-resistance to compounds 2 to 6, but no cross-resistance to compounds 7 to 9 was observed (Table 2). The lack of cross-resistance to compounds 7 to 9 is consistent with compound 7 not preferably inhibiting RNA and protein synthesis. Since myxopyronin, corallopyronin, and ripostatin are not commercially available, cross-resistance against these compounds could not be tested.
Fig 2.

Amino acid alignments of the carboxyl-terminal end of RpoB (top) and RpoC isozymes (bottom) and the resistance mutations identified therein. Resistance mutations against compound 1 isolated in H. influenzae are indicated by an asterisk, against myxopyronin, corallopyronin, and ripostatin in E. coli by “o” (21), and fidaxomycin in Enterococcus faecalis by “f” (13). Mutation of rpoB-E1280 and rpoB-L1327 were found in H. influenzae, E. coli, and Staphylococcus aureus (18); rpoB-V1326 in E. coli, E. faecalis, and Mycobacterium tuberculosis (17); rpoB-Q1257 in E. faecalis and M. tuberculosis; rpoB-P1321 in both E. coli and H. influenzae; rpoC-L1333 in both Staphylococcus aureus and H. influenzae; and rpoB-S1323 in both E. coli and S. aureus. Numbering is according to the sequences of the E. coli isozymes. Switch regions (SW) (12) are underlined: SW1 (RpoC1310-1331), SW3 (RpoB1251-1268), SW4 (RpoB1293-1298), and SW5 (RpoC1355-1357); SW2 (RpoC327-334) is not shown. Eco, E. coli; Kpn, Klebsiella pneumoniae; Hin, H. influenzae; Pae, Pseudomonas aeruginosa; Aba, Acinetobacter baumannii; Mtb, M. tuberculosis; Efa, E. faecalis; Sau, S. aureus; and Spn, Streptococcus pneumoniae.
In the absence of an X-ray crystal structure of myxopyronin bound to E. coli RNAP, the X-ray crystal structure of the RNAP-myxopyronin complex structure from T. thermophilus (PDB 3DXJ [21]) was used in conjunction with switches 1 to 5 (SW1 to SW5) as assigned by Gnatt et al. (12) to provide a possible provisional model of the binding of the squaramide compounds. A comparison of the predicted binding mode for the squaramides to that of myxopyronin found in cocrystals revealed several important similarities in the protein-compound interactions of both classes of compounds (Fig. 3 and 4). The carbamate chain of myxopyronin forms significant contacts with the helix-turn-helix structure flanked by SW3 and SW4 (data not shown). The alpha-pyrone ring contacts SW2 and the dienone chain forms extensive contacts with SW1 and SW2, but the myxopyronins do not directly interact with SW5. The squaramides form similar interactions; the phenyl tail overlays with the dienone chain of myxopyronin, forming extensive interactions with SW1 and SW2. The squaramide core interacts with SW2, and the oxazole ring interacts with SW3 and SW4, although the squaramides form a smaller interface with SW3 and SW4 than the myxopyronins do.
Fig 4.

Docking of compound 1 into the switch region of RNAP. Shown is a top-scoring pose from the docking of the squaramide compound 1 (orange) to the myxopyronin binding site to illustrate the binding mode predicted by the docking compared to that of myxopyronin (green). The piperidine-linked hydrophobic tail occupies the dienone pocket, and the squaramide core appears to mimic the pyrone ring. The position of the oxazole ring, shown in this pose partially occupying the carbamate pocket, was highly variable in the docked poses, suggesting that it does not make specific, conserved interactions with this conformation of the target. For other details, see the legend to Fig. 3.
All nine squaramides docked with fairly equivalents scores; none of the compounds scored clearly better or worse than the others. This is not altogether unexpected, however, since myxopyronin allosterically stabilizes the RNAP structure, preventing proper DNA loading and initiation of transcription (3). Therefore, the potency of the compounds may correlate more strongly with their effect on the conformational motion than with their interactions with the crystallographic conformational state of the switch region.
DISCUSSION
Many of the antibacterial agents used in the clinical setting mediate their growth-inhibitory effect by interfering with the bacterial transcription and translation machinery. All of these drugs, or their precursors, were discovered in cell-based screening approaches (8). Inhibitors that were unable to traverse the cell wall would have gone undetected. The effort described here aimed to identify novel transcription or translation inhibitors without the necessity of cell penetration. Subsequently, chemical modifications would, in some cases, be needed to create derivatives able to enter the cell, requiring a chemical starting point that could easily be modified. To that end, S30 extracts were prepared from E. coli and allowed to transcribe the luc gene and subsequently translate its mRNA into active protein. An attractive feature of this approach was that, in principle, inhibitors to any of the many steps involved in transcription and translation could be found. The disadvantage was that the use of largely undefined cell extracts precluded the estimation of sensitivity of the assay to inhibition of each of those steps or of adjusting the sensitivity.
The screen identified multiple inhibitor scaffolds, one of which stood out because replacement of the luc-encoding plasmid with luc mRNA abolished inhibition, suggesting that these so-called squaramides inhibited RNAP. This was confirmed with a purified RNAP assay. The reason for the lower potency of the compounds in this assay than in the transcription-coupled translation assay is not known. The RNAP assay measured consumption of ribonucleotides during the production of mRNA. The transcription-coupled translation assay measured the production of luciferase protein subsequent to the production of luciferase mRNA. It may be that the latter assay is more sensitive to transcription inhibition than the former assay because each mRNA is translated multiple times simultaneously by polysomes, so that a small change in mRNA is amplified. If this is the case, the transcription-coupled translation assay should be a very sensitive method of identifying novel transcription inhibitors.
Compound 1 had a high propensity to select resistant mutants, with a frequency of approximately 10−7 at 1 to 2 times its MIC (data not shown). The high degree of resistance of many isolates, up to 16-fold the MIC of the parental strain (Table 2), suggests that the frequency would not be much lower at higher compound concentrations. All resistance mutations were found in genes encoding the RpoB and RpoC subunits of RNAP, proving that inhibition of this enzyme is the mode of action of compound 1. Since compound 1-resistant mutants are cross-resistant to compounds 2 to 6, these compounds have the same mode of action. In contrast, compounds 7 to 9 do not show cross-resistance and do not show preferred inhibition of RNA and protein synthesis. Replacement of the dimethylisoxazole side chain resulted in compounds 7 to 9, which are bona fide RNAP inhibitors in vitro, but their biochemical potency is an order of magnitude weaker than those of compounds 1 to 6. Apparently, their inhibition of RNA polymerase in the cell is overshadowed by more potent impediment of another essential process in the cell which remains to be determined.
Compound 1-resistant mutants were not cross-resistant with rifampin even though compounds 1 to 6 showed, like rifampin, preferred inhibition of RNA and protein synthesis. Determination of the resistance mutations showed that although the target of both classes is RNAP, they vary in their mechanism of inhibition. Rifampin binds in the vicinity of the active site, allowing transcription to initiate but limiting RNA extension to 2 to 3 nucleotides (7). Squaramide resistance mutations were found in the so-called switch region, a recently identified binding site that is formed at the interface between RpoB and RpoC (Fig. 5). Upon binding of myxopyronin, the switch is fixed in an orientation that still allows binding of RNAP to the melted DNA but distorts the interaction between RNAP and the promoter so that it cannot initiate transcription (3, 21).
Fig 5.

Locations of mutations conferring resistance to squaramides. Shown is an all-atom representation of the binding site (RpoB and RpoC are shown in red and blue, respectively) to illustrate the location of the resistance mutations in relation to the docked pose of compound 1 (orange). The labels represent the mapping of the E. coli residue numbers to the T. thermophilus sequence and structure. The residue labeled L1327 in the T. thermophilus docking model corresponds to I1327 in the E. coli protein. Hydrogen bonds between the predicted binding mode of compound 1 and RpoC are shown in green.
Remarkably, all mutations were found in amino acid residues that are conserved across a wide range of bacterial species. One would expect this conservation to reflect a critical role for these residues in RNAP function and cellular growth. It has been speculated that this binding site is a molecular checkpoint for DNA loading in response to regulatory signals. However, a large fitness loss for the mutants was not seen when colonies were grown on agar plates (reference 18 and data not shown). It is unclear whether in infected tissues a growth defect would be more pronounced and a high resistance frequency would be less relevant.
The predicted binding modes of the squaramides may also help to rationalize the relative MIC shifts observed for the nine resistance mutations (Table 2). The docked poses show a strong preference for the dienone pocket and only partial occupation of the carbamate pocket. The few poses that do occupy the carbamate pocket are characterized by poor docking scores and qualitatively poor binding geometries. Interestingly, the two mutations that occur deep in the carbamate binding pocket, E1280K in RpoB and K1349Q in RpoC, display the smallest MIC shifts for compounds 1 to 6.
The resistance mutations isolated against compound 1 support the hypothesis that the squaramides interfere with the conformational mobility of the RNAP clamp domain in the same manner as the myxopyronins. Of the eight resistance mutation sites identified in H. influenzae RNAP, three are located within the switches (12) and an additional two are located near the switches (Fig. 2). Specifically, RpoC-A1323 is located in SW1 and RpoC-L1333 is located within three residues of SW1. RpoB-Q1258 is located in SW3, RpoB-D1297 is located in SW4, and RpoB-E1280 is located between SW3 and SW4. These five mutation sites could interfere with the allosteric clamping motions of RNAP. Two of the mutations that fall outside the switch regions, RpoB-P1321 and RpoB-I1327, modify residues that directly contact the myxopyronins and the predicted binding mode of the squaramides. These mutations might directly interfere with inhibitor binding to the switch region. No mutations in SW2 were found, despite the facts that the predicted binding mode for the squaramides makes significant contacts with SW2 and that myxopyronin-resistant mutations in this region have been described (21). However, since mutagenesis in the latter study was more saturating than that described here, these mutations may have been missed.
The squaramides described here have a mode of inhibition distinct from that of the CBR703 series of non-natural-product-related, small-molecule inhibitors described previously. These compounds inhibited nucleotide addition and not the translocation of DNA and RNA. In line with this, resistance mutations did not map to the switch region (2). A class of antimicrobial 2-ureidothiophene-3-carboxylate inhibitors of RNAP maintained its activity against rifampin-resistant S. aureus, but the location of the resistance mutations remained undetermined (1). Attempts to dock 2-ureidothiophene-3-carboxylates using the protocol used for squaramides failed to reveal a binding pose within the switch region or anywhere else in the RNAP crystal structure.
The clinical drug fidaxomycin (lipiarmycin) acts by binding to the RNAP switch region, validating the switch region as a target for antibacterial drugs. Its exquisite potency against Clostridium difficile and absence of oral bioavailability limit its use to the very narrow clinical indication of C. difficile-associated diarrhea (20). Alternative chemical scaffolds or chemical derivatization of fidaxomycin may lead to agents with a broader spectrum that are absorbed from the gastrointestinal tract and could serve a larger medical need. Our work shows that screening of small-compound libraries using a transcription-coupled translation assay may yield chemical starting points that are more easily diversified than the known natural product antibiotics that bind to the switch region, and they could provide a viable way forward toward that goal.
ACKNOWLEDGMENTS
We thank the AstraZeneca R&D Boston clinical susceptibility group for MIC determinations, Bob McLaughlin for sequence alignments and design of oligonucleotides, and Laurel Hajec for construction of pLH1824.
Footnotes
Published ahead of print 27 July 2012
REFERENCES
- 1. Arhin F, et al. 2006. A new class of small molecule RNA polymerase inhibitors with activity against rifampicin-resistant Staphylococcus aureus. Bioorg. Med. Chem. 14: 5812–5832 [DOI] [PubMed] [Google Scholar]
- 2. Artsimovitch I, Chu C, Lynch AS, Landick R. 2003. A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science 302: 650–654 [DOI] [PubMed] [Google Scholar]
- 3. Belogurov GA, et al. 2009. Transcription inactivation through local refolding of the RNA polymerase structure. Nature 457: 332–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgess RR, Jendrisak J. 1975. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry 14: 4634–4638 [DOI] [PubMed] [Google Scholar]
- 5. Buurman ET, et al. 2011. In vitro validation of acetyltransferase activity of GlmU as an antibacterial target in Haemophilus influenzae. J. Biol. Chem. 286: 40734–40742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buurman ET, Johnson KD, Kelly RK, MacCormack K. 2006. Different modes of action of naphthyridones in Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 50: 385–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell EA, et al. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104: 901–912 [DOI] [PubMed] [Google Scholar]
- 8. Clardy J, Fischbach MA, Walsh CT. 2006. New antibiotics from bacterial natural products. Nat. Biotechnol. 24: 1541–1550 [DOI] [PubMed] [Google Scholar]
- 9. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed, M07-A8, vol. 29, no. 2 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 10. Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325: 1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fralick JA. 1996. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J. Bacteriol. 178: 5803–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. 2001. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science 292: 1876–1882 [DOI] [PubMed] [Google Scholar]
- 13. Gualtieri M, Tupin A, Brodolin K, Leonetti JP. 2009. Frequency and characterisation of spontaneous lipiarmycin-resistant Enterococcus faecalis mutants selected in vitro. Int. J. Antimicrob. Agents 34: 605–606 [DOI] [PubMed] [Google Scholar]
- 14. Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, Bush K. 1999. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother. 43: 1693–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itaya K, Ui M. 1966. A new micromethod for the colorimetric determination of inorganic phosphate. Clin. Chim. Acta 14: 361–366 [DOI] [PubMed] [Google Scholar]
- 16. Johnson JC, Shanoff M, Bass ST, Boezi JA, Hansen RG. 1968. An enzymic method for determination of inorganic phosphate and its use as an assay for RNA polymerase. Anal. Biochem. 26: 137–145 [DOI] [PubMed] [Google Scholar]
- 17. Kurabachew M, et al. 2008. Lipiarmycin targets RNA polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 62: 713–719 [DOI] [PubMed] [Google Scholar]
- 18. Mariner K, et al. 2011. Activity of and development of resistance to corallopyronin A, an inhibitor of RNA polymerase. Antimicrob. Agents Chemother. 55: 2413–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Merritt JR, et al. 2006. Synthesis and structure-activity relationships of 3,4-diaminocyclobut-3-ene-1,2-dione CXCR2 antagonists. Bioorg. Med. Chem. Lett. 16: 4107–4110 [DOI] [PubMed] [Google Scholar]
- 20. Miller M. 2010. Fidaxomicin (OPT-80) for the treatment of Clostridium difficile infection. Expert Opin. Pharmacother. 11: 1569–1578 [DOI] [PubMed] [Google Scholar]
- 21. Mukhopadhyay J, et al. 2008. The RNA polymerase “switch region” is a target for inhibitors. Cell 135: 295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pratt JM. 1984. Coupled transcription-translation in prokaryotic cell-free systems, p 179–209 In Hames BD, Higgens SJ. (ed), Transcription and translation: a practical approach. IRL Press, Oxford, United Kingdom [Google Scholar]
- 23. Reck F, et al. 2011. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 54: 7834–7847 [DOI] [PubMed] [Google Scholar]
- 24. Sanchez L, Pan W, Vinas M, Nikaido H. 1997. The acrAB homolog of Haemophilus influenzae codes for a functional multidrug efflux pump. J. Bacteriol. 179: 6855–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shapiro AB, Walkup GK, Keating TA. 2009. Correction for interference by test samples in high-throughput assays. J. Biomol. Screen. 14: 1008–1016 [DOI] [PubMed] [Google Scholar]
- 26. Srivastava A, et al. 2011. New target for inhibition of bacterial RNA polymerase: ‘switch region.’ Curr. Opin. Microbiol. 14: 532–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Storer RI, Aciro C, Jones LH. 2011. Squaramides: physical properties, synthesis and applications. Chem. Soc. Rev. 40: 2330–2346 [DOI] [PubMed] [Google Scholar]
- 28. Zubay G. 1973. In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7: 267–287 [DOI] [PubMed] [Google Scholar]